
Diversity and Evolution of Mamiellophyceae: Early-Diverging Phytoplanktonic Green Algae Containing Many Cosmopolitan Species


 , and
, and
Abstract
:1. What Are the Mamiellophyceae?
1.1. Diagnoses and Species Diversity
1.2. Genomics and Cryptic Species Discovery
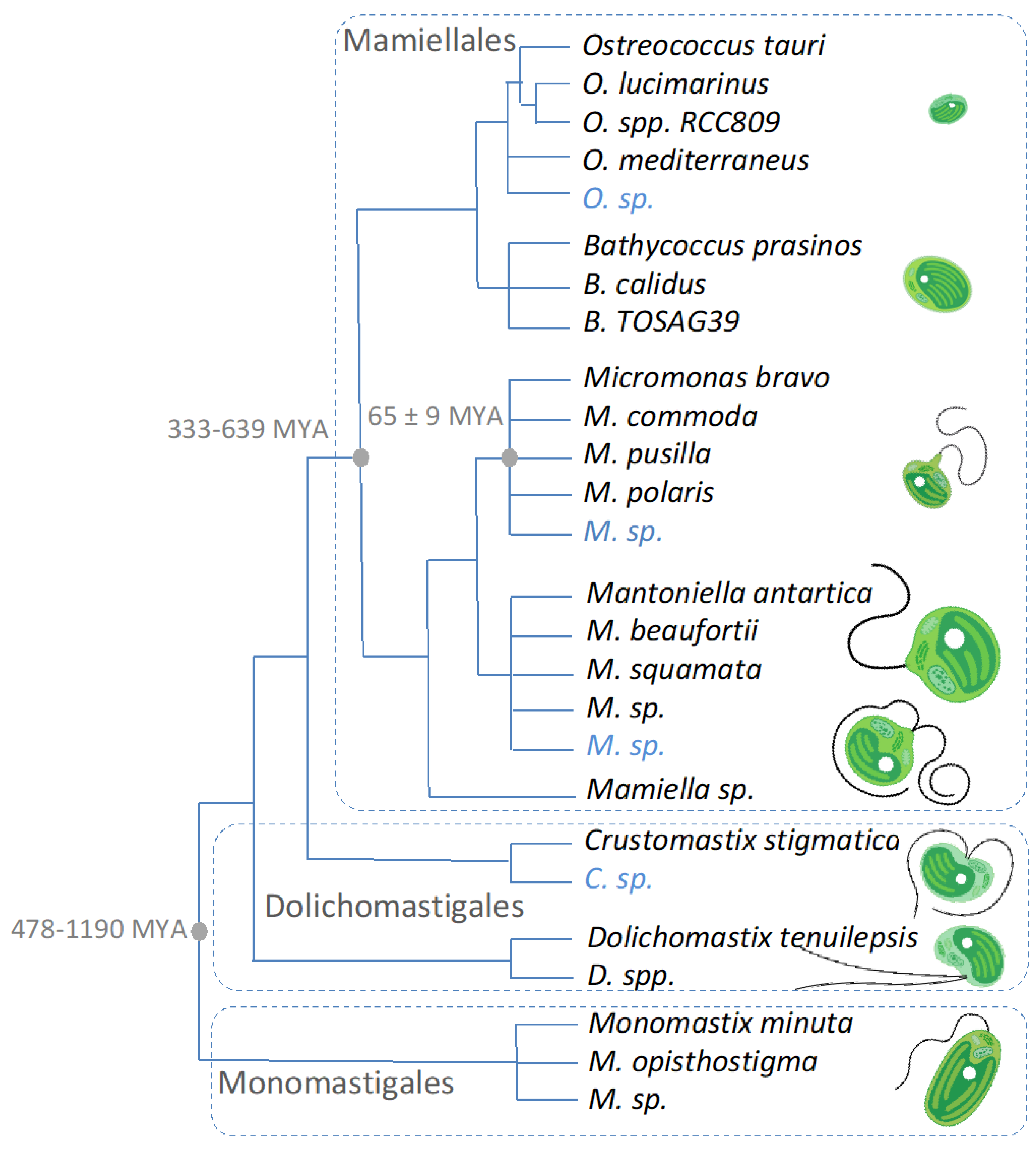
1.3. Timeline of Speciation and Population Coalescence Times
2. Genome Resources in Mamiellophyceae: From Reference Genomes to MAGs and SAGs

{kind=link}
{kind=link}
| Phylogeny | Source | Size (Mb) | Completeness | Reference |
|---|---|---|---|---|
 |  | 13 |  | [45,46] |
| | 13 | | [12] | |
| | 13 | | JGI | |
| | 13 | | [47] | |
| ≈ | - | - | [44] | |
| | 15 | | [48] | |
| | - | - | [9] | |
| o | 10 |  | [39] | |
| | - | - | [17] | |
| | 20 | | [15,16] | |
| | 21 | | [15,16] | |
| | - | - | [17] | |
| ≈ | 22 | | [43] | |
| | - | - | - | |
| | - | - | [49] | |
| | 65 * | ❖ | - | |
| | - | ❖ | - | |
| ≈ | 28 | | [43] | |
| | 97 * | ❖ | - | |
| | - | - | - | |
| ≈ | 21 | | [43] | |
| | - | - | - | |
| | 160 * | ❖ | - | |
| | 80 * | ❖ | - | |
| | 303 * | ❖ | - | |
| | 52 * | ❖ | - |
strain available in culture; o single-cell assembled genome; ≈ metagenomic origin; complete genome sequence with annotation; genome assembly available; ❖ genome sequencing in progress; - no data.3. Where Are the Mamiellophyceae in the Sunlit Ocean?
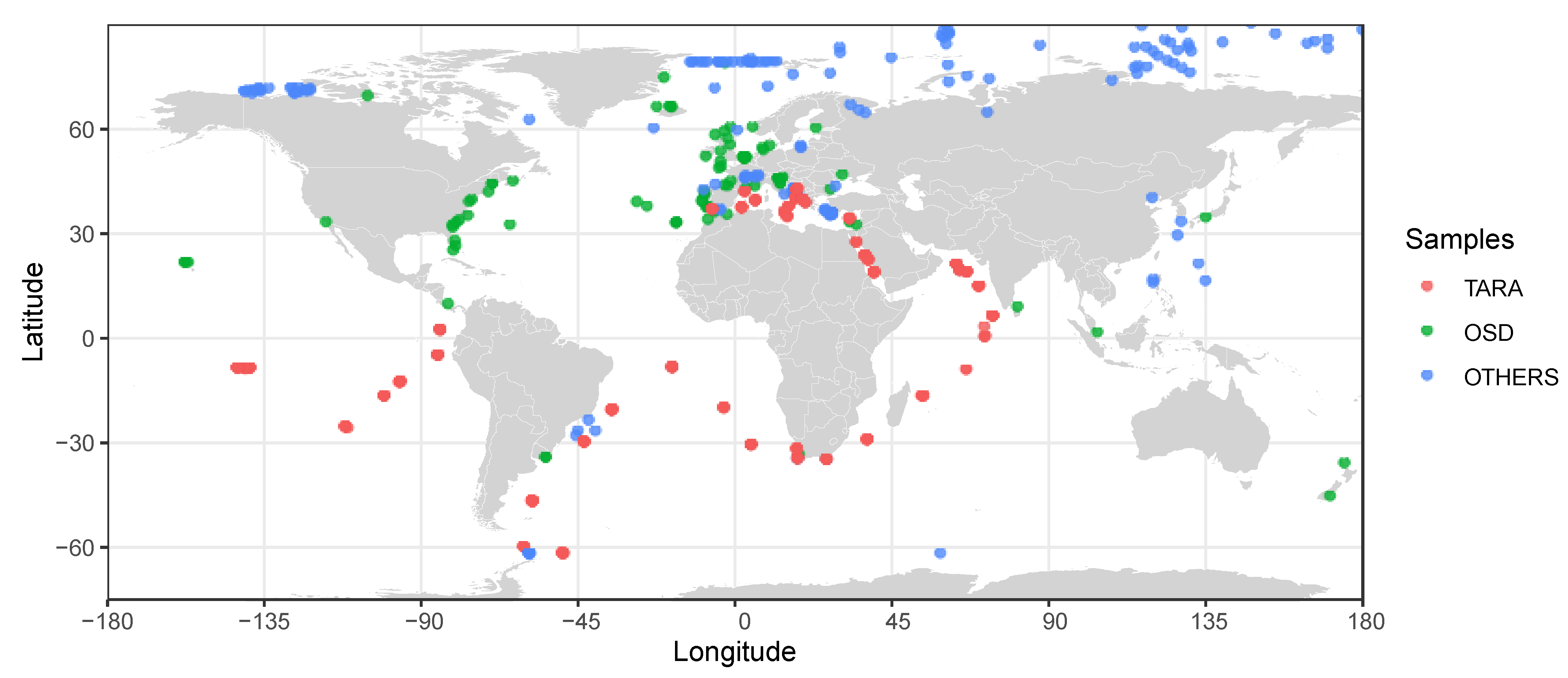
3.1. Metabarcoding Insights
3.2. Metagenomics and Genome-Informed Biogeography
4. Can Viruses of Mamiellophyceae Inform Their Diversity and Evolution?
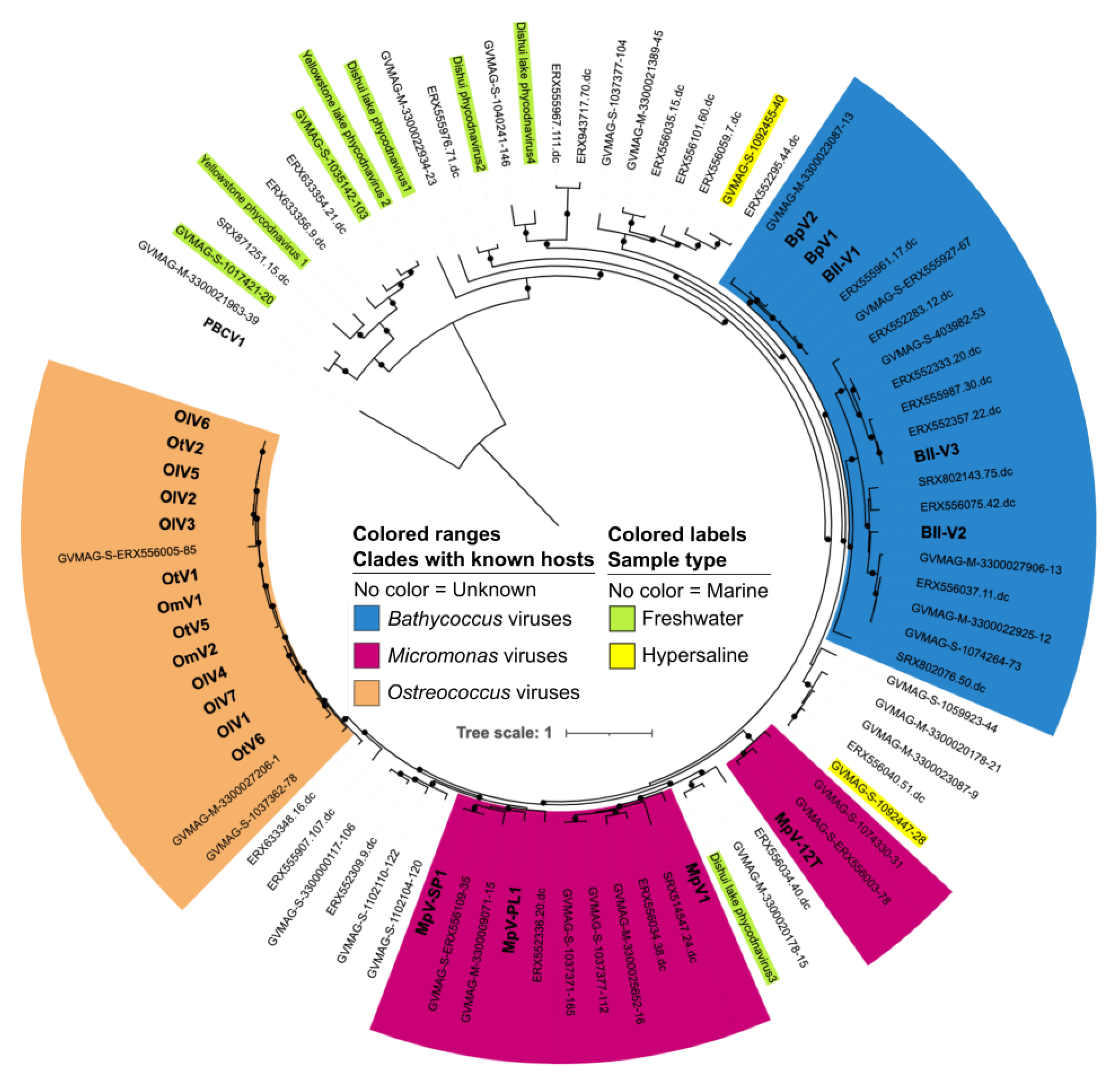
4.1. Prasinoviruses Are Key Regulators of Mamiellales Populations
4.2. Host Range of Available Isolated Prasinoviruses
4.3. Do Prasinoviruses Co-Occur with Their Host?
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boenigk, J.; Ereshefsky, M.; Hoef-Emden, K.; Mallet, J.; Bass, D. Concepts in Protistology: Species Definitions and Boundaries. Eur. J. Protistol. 2012, 48, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.; Cox, E. What Constitutes Gomphonema Parvulum? Long-Term Culture Studies Show That Some Varieties of G. Parvulum Belong with Other Gomphonema Species. Plant Ecol. Evol. 2014, 147, 366–373. [Google Scholar] [CrossRef]
- Moestrup, Ø.; Throndsen, J. Light and Electron Microscopical Studies on Pseudoscourfieldia marina, a Primitive Scaly Green Flagellate (Prasinophyceae) with Posterior Flagella. Can. J. Bot. 1988, 66, 1415–1434. [Google Scholar] [CrossRef]
- Steinkötter, J.; Bhattacharya, D.; Semmelroth, I.; Bibeau, C.; Melkonian, M. Prasinophytes form independent lineages within the Chlorophyta: Evidence from ribosomal RNA sequence comparisons. J. Phycol. 1994, 30, 340–345. [Google Scholar] [CrossRef]
- Foss, P.; Guillard, R.R.L.; Liaaen-Jensen, S. Prasinoxanthin: A Chemosystematic Marker for Algae. Phytochemistry 1984, 23, 1629–1633. [Google Scholar] [CrossRef]
- Marin, B.; Melkonian, M. Molecular Phylogeny and Classification of the Mamiellophyceae Class. Nov. (Chlorophyta) Based on Sequence Comparisons of the Nuclear- and Plastid-Encoded RRNA Operons. Protist 2010, 161, 304–336. [Google Scholar] [CrossRef]
- Guiry, M.D.; Guiry, G.M. AlgaeBase; World-Wide Electronic Publication, National University of Ireland: Galway, Ireland, 2021. [Google Scholar]
- Scherffel, A. Zwei Neue, Trichocystenartige Bildungen Führende Flagellaten. Arch. Protistenkd. 1912, 27, 94–128. [Google Scholar]
- Bachy, C.; Yung, C.C.M.; Needham, D.M.; Gazitúa, M.C.; Roux, S.; Limardo, A.J.; Choi, C.J.; Jorgens, D.M.; Sullivan, M.B.; Worden, A.Z. Viruses Infecting a Warm Water Picoeukaryote Shed Light on Spatial Co-Occurrence Dynamics of Marine Viruses and Their Hosts. ISME J. 2021, 15, 3129–3147. [Google Scholar] [CrossRef]
- Courties, C.; Vaquer, A.; Troussellier, M.; Lautier, J.; Chrétiennot-Dinet, M.J.; Neveux, J.; Machado, C.; Claustre, H. Smallest Eukaryotic Organism. Nature 1994, 370, 255. [Google Scholar] [CrossRef]
- Chrétiennot-Dinet, M.-J.; Courties, C.; Vaquer, A.; Neveux, J.; Claustre, H.; Lautier, J.; Machado, M.C. A New Marine Picoeucaryote: Ostreococcus tauri Gen. et Sp. Nov. (Chlorophyta, Prasinophyceae). Phycologia 1995, 34, 285–292. [Google Scholar] [CrossRef]
- Palenik, B.; Grimwood, J.; Aerts, A.; Rouze, P.; Salamov, A.; Putnam, N.; Dupont, C.; Jorgensen, R.; Derelle, E.; Rombauts, S.; et al. The Tiny Eukaryote Ostreococcus Provides Genomic Insights into the Paradox of Plankton Speciation. Proc. Natl. Acad. Sci. USA 2007, 104, 7705–7710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jancek, S.; Gourbière, S.; Moreau, H.; Piganeau, G. Clues about the Genetic Basis of Adaptation Emerge from Comparing the Proteomes of Two Ostreococcus Ecotypes (Chlorophyta, Prasinophyceae). Mol. Biol. Evol. 2008, 25, 2293–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piganeau, G.; Eyre-Walker, A.; Jancek, S.; Grimsley, N.; Moreau, H. How and Why DNA Barcodes Underestimate the Diversity of Microbial Eukaryotes. PLoS ONE 2011, 6, e16342. [Google Scholar] [CrossRef] [PubMed]
- Worden, A.Z.; Lee, J.H.; Mock, T.; Rouze, P.; Simmons, M.P.; Aerts, A.L.; Allen, A.E.; Cuvelier, M.L.; Derelle, E.; Everett, M.V.; et al. Green Evolution and Dynamic Adaptations Revealed by Genomes of the Marine Picoeukaryotes Micromonas. Science 2009, 324, 268–272. [Google Scholar] [CrossRef] [Green Version]
- Van Baren, M.J.; Bachy, C.; Reistetter, E.N.; Purvine, S.O.; Grimwood, J.; Sudek, S.; Yu, H.; Poirier, C.; Deerinck, T.J.; Kuo, A.; et al. Evidence-Based Green Algal Genomics Reveals Marine Diversity and Ancestral Characteristics of Land Plants. BMC Genom. 2016, 17, 267. [Google Scholar] [CrossRef] [Green Version]
- Simon, N.; Foulon, E.; Grulois, D.; Six, C.; Desdevises, Y.; Latimier, M.; Le Gall, F.; Tragin, M.; Houdan, A.; Derelle, E.; et al. Revision of the Genus Micromonas Manton et Parke (Chlorophyta, Mamiellophyceae), of the Type Species M. pusilla (Butcher) Manton & Parke and of the Species M. commoda van Baren, Bachy and Worden and Description of Two New Species Based on the Genetic and Phenotypic Characterization of Cultured Isolates. Protist 2017, 168, 612–635. [Google Scholar] [CrossRef] [Green Version]
- Villac, C.; Harwood, D.; Wittkowski, J.; Medlin, L.K. Diatom Evolution from Fossils to Modern from the Benthos into the Plankton. JMSE, 2022; in press. [Google Scholar]
- Henderiks, J.; Strum, D.; Supraha, L. Evolutionary Rates in the Haptophyta: Exploring Molecular and Phenotypic Diversity. JMSE, 2022; in press. [Google Scholar]
- Riding, J.B.; Fensome, R.A.; Dale, B.; Gobillard, M.-O.; Medlin, L.K. An Overview of Dinoflagellate Evolution with Comments on Their Adaptation to the Plankton. JMSE, 2022; in press. [Google Scholar]
- Falkowski, P.G.; Katz, M.E.; Knoll, A.H.; Quigg, A.; Raven, J.A.; Schofield, O.; Taylor, F.J.R. The Evolution of Modern Eukaryotic Phytoplankton. Science 2004, 305, 354–360. [Google Scholar] [CrossRef] [Green Version]
- De Clerck, O.; Bogaert, K.A.; Leliaert, F. Chapter Two-Diversity and Evolution of Algae: Primary Endosymbiosis. In Advances in Botanical Research; Piganeau, G., Ed.; Genomic Insights into the Biology of Algae; Academic Press: London, UK, 2012; Volume 64, pp. 55–86. [Google Scholar]
- Blank, C.E. Origin and Early Evolution of Photosynthetic Eukaryotes in Freshwater Environments: Reinterpreting Proterozoic Paleobiology and Biogeochemical Processes in Light of Trait Evolution. J. Phycol. 2013, 49, 1040–1055. [Google Scholar] [CrossRef] [PubMed]
- Leliaert, F.; Tronholm, A.; Lemieux, C.; Turmel, M.; DePriest, M.S.; Bhattacharya, D.; Karol, K.G.; Fredericq, S.; Zechman, F.W.; Lopez-Bautista, J.M. Chloroplast Phylogenomic Analyses Reveal the Deepest-Branching Lineage of the Chlorophyta, Palmophyllophyceae Class. Nov. Sci. Rep. 2016, 6, 25367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, D.; Weiche, B.; Timmerhaus, G.; Richardt, S.; Riaño-Pachón, D.M.; Corrêa, L.G.G.; Reski, R.; Mueller-Roeber, B.; Rensing, S.A. Genome-Wide Phylogenetic Comparative Analysis of Plant Transcriptional Regulation: A Timeline of Loss, Gain, Expansion, and Correlation with Complexity. Genome Biol. Evol. 2010, 2, 488–503. [Google Scholar] [CrossRef] [PubMed]
- Parfrey, L.W.; Lahr, D.J.G.; Knoll, A.H.; Katz, L.A. Estimating the Timing of Early Eukaryotic Diversification with Multigene Molecular Clocks. Proc. Natl. Acad. Sci. USA 2011, 108, 13624–13629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, M.J. R8s: Inferring Absolute Rates of Molecular Evolution and Divergence Times in the Absence of a Molecular Clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef] [Green Version]
- Slapeta, J.; Lopez-Garcia, P.; Moreira, D. Global Dispersal and Ancient Cryptic Species in the Smallest Marine Eukaryotes. Mol. Biol. Evol. 2006, 23, 23–29. [Google Scholar] [CrossRef]
- Loeblich, A.R. Protistan Phylogeny as Indicated by the Fossil Record. Taxon 1974, 23, 277–290. [Google Scholar] [CrossRef]
- Colbath, G.K. Fossil Prasinophycean Phycomata (Chlorophyta) from the Silurian Bainbridge Formation, Missouri, U.S.A. Phycologia 1983, 22, 249–265. [Google Scholar] [CrossRef]
- Boalch, G.; Guyohlson, D. Tasmanites, The Correct Name For Pachysphaera (Prasinophyceae, Pterospermataceae). Taxon 1992, 41, 529–531. [Google Scholar] [CrossRef]
- Blanc-Mathieu, R.; Sanchez-Ferandin, S.; Eyre-Walker, A.; Piganeau, G. Organellar Inheritance in the Green Lineage: Insights from Ostreococcus tauri. Genome Biol. Evol. 2013, 5, 1503–1511. [Google Scholar] [CrossRef] [Green Version]
- Blanc-Mathieu, R.; Krasovec, M.; Hebrard, M.; Yau, S.; Desgranges, E.; Martin, J.; Schackwitz, W.; Kuo, A.; Salin, G.; Donnadieu, C.; et al. Population Genomics of Picophytoplankton Unveils Novel Chromosome Hypervariability. Sci. Adv. 2017, 3, e1700239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasovec, M.; Eyre-Walker, A.; Sanchez-Ferandin, S.; Piganeau, G. Spontaneous Mutation Rate in the Smallest Photosynthetic Eukaryotes. Mol. Biol. Evol. 2017, 34, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Benites, L.F.; Bucchini, F.; Sanchez-Brosseau, S.; Grimsley, N.; Vandepoele, K.; Piganeau, G. Evolutionary Genomics of Sex-Related Chromosomes at the Base of the Green Lineage. Genome Biol. Evol. 2021, 13, evab216. [Google Scholar] [CrossRef] [PubMed]
- Hanschen, E.R.; Starkenburg, S.R. The State of Algal Genome Quality and Diversity. Algal Res. 2020, 50, 101968. [Google Scholar] [CrossRef]
- Yilmaz, S.; Singh, A.K. Single Cell Genome Sequencing. Curr. Opin. Biotechnol. 2012, 23, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Benites, L.F.; Poulton, N.; Labadie, K.; Sieracki, M.E.; Grimsley, N.; Piganeau, G. Single Cell Ecogenomics Reveals Mating Types of Individual Cells and SsDNA Viral Infections in the Smallest Photosynthetic Eukaryotes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20190089. [Google Scholar] [CrossRef] [Green Version]
- Vannier, T.; Leconte, J.; Seeleuthner, Y.; Mondy, S.; Pelletier, E.; Aury, J.-M.; de Vargas, C.; Sieracki, M.; Iudicone, D.; Vaulot, D.; et al. Survey of the Green Picoalga Bathycoccus Genomes in the Global Ocean. Sci. Rep. 2016, 6, 37900. [Google Scholar] [CrossRef] [Green Version]
- Rusch, D.B.; Halpern, A.L.; Sutton, G.; Heidelberg, K.B.; Williamson, S.; Yooseph, S.; Wu, D.Y.; Eisen, J.A.; Hoffman, J.M.; Remington, K.; et al. The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS. Biol. 2007, 5, 398–431. [Google Scholar] [CrossRef]
- Piganeau, G.; Moreau, H. Screening the Sargasso Sea Metagenome for Data to Investigate Genome Evolution in Ostreococcus (Prasinophyceae, Chlorophyta). Gene 2007, 406, 184–190. [Google Scholar] [CrossRef]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Ocean Plankton. Structure and Function of the Global Ocean Microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [Green Version]
- Delmont, T.O.; Gaia, M.; Hinsinger, D.D.; Fremont, P.; Vanni, C.; Guerra, A.F.; Eren, A.M.; Kourlaiev, A.; d’Agata, L.; Clayssen, Q.; et al. Functional Repertoire Convergence of Distantly Related Eukaryotic Plankton Lineages Revealed by Genome-Resolved Metagenomics. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tragin, M.; Vaulot, D. Novel Diversity within Marine Mamiellophyceae (Chlorophyta) Unveiled by Metabarcoding. Sci. Rep. 2019, 9, 5190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derelle, E.; Ferraz, C.; Rombauts, S.; Rouzé, P.; Worden, A.Z.; Robbens, S.; Partensky, F.; Degroeve, S.; Echeynié, S.; Cooke, R.; et al. Genome Analysis of the Smallest Free-Living Eukaryote Ostreococcus tauri Unveils Many Unique Features. Proc. Natl. Acad. Sci. USA 2006, 103, 11647–11652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanc-Mathieu, R.; Verhelst, B.; Derelle, E.; Rombauts, S.; Bouget, F.-Y.; Carré, I.; Château, A.; Eyre-Walker, A.; Grimsley, N.; Moreau, H.; et al. An Improved Genome of the Model Marine Alga Ostreococcus tauri Unfolds by Assessing Illumina de Novo Assemblies. BMC Genom. 2014, 15, 1103. [Google Scholar] [CrossRef] [Green Version]
- Yau, S.; Krasovec, M.; Benites, L.F.; Rombauts, S.; Groussin, M.; Vancaester, E.; Aury, J.-M.; Derelle, E.; Desdevises, Y.; Escande, M.-L.; et al. Virus-Host Coexistence in Phytoplankton through the Genomic Lens. Sci. Adv. 2020, 6, eaay2587. [Google Scholar] [CrossRef] [Green Version]
- Moreau, H.; Verhelst, B.; Couloux, A.; Derelle, E.; Rombauts, S.; Grimsley, N.; Van Bel, M.; Poulain, J.; Katinka, M.; Hohmann-Marriott, M.F.; et al. Gene Functionalities and Genome Structure in Bathycoccus prasinos Reflect Cellular Specializations at the Base of the Green Lineage. Genome Biol. 2012, 13, R74. [Google Scholar] [CrossRef] [Green Version]
- Yau, S.; dos Santos, A.L.; Eikrem, W.; Ribero, C.G.; Gourvil, P.; Balzano, S.; Escande, M.-L.; Moreau, H.; Vaulot, D. Mantoniella beaufortii and Mantoniella baffinensis sp. nov. (Mamiellales, Mamiellophyceae), Two New Green Algal Species from the High Arctic. J. Phycol. 2019, 56, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Surek, B.; Melkonian, M. CCAC-Culture Collection of Algae at the University of Cologne: A New Collection of Axenic Algae with Emphasis on Flagellates. Nova Hedwig. 2004, 79, 77–92. [Google Scholar] [CrossRef]
- Gachon, C.; Day, J.; Campbell, C.; Proschold, T.; Saxon, R.; Kupper, F. The Culture Collection of Algae and Protozoa (CCAP): A Biological Resource for Protistan Genomics. Gene 2007, 406, 51–57. [Google Scholar] [CrossRef]
- Vaulot, D.; Le Gall, F.; Marie, D.; Guillou, L.; Partensky, F. The Roscoff Culture Collection (RCC): A Collection Dedicated to Marine Picoplankton. Nova Hedwig. 2004, 79, 49–70. [Google Scholar] [CrossRef]
- Starr, R.; Zeikus, J. Utex-The Culture Collection Of Algae At The University-Of-Texas At Austin 1993 List of Cultures. J. Phycol. 1993, 29, 1–106. [Google Scholar] [CrossRef]
- Not, F.; Siano, R.; Kooistra, W.H.C.F.; Simon, N.; Vaulot, D.; Probert, I. Chapter One-Diversity and Ecology of Eukaryotic Marine Phytoplankton. In Advances in Botanical Research; Piganeau, G., Ed.; Genomic Insights into the Biology of Algae; Academic Press: London, UK, 2012; Volume 64, pp. 1–53. [Google Scholar]
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-Generation Sequencing Technologies for Environmental DNA Research. Mol. Ecol. 2012, 21, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Belevich, T.A.; Milyutina, I.A.; Abyzova, G.A.; Troitsky, A.V. The Pico-Sized Mamiellophyceae and a Novel Bathycoccus Clade from the Summer Plankton of Russian Arctic Seas and Adjacent Waters. FEMS Microb. Ecol. 2021, 97, fiaa251. [Google Scholar] [CrossRef] [PubMed]
- Monier, A.; Worden, A.Z.; Richards, T.A. Phylogenetic Diversity and Biogeography of the Mamiellophyceae Lineage of Eukaryotic Phytoplankton across the Oceans: Global Diversity of Marine Class II Prasinophytes. Environ. Microbiol. Rep. 2016, 8, 461–469. [Google Scholar] [CrossRef]
- Ribeiro, C.G.; dos Santos, A.L.; Marie, D.; Brandini, F.P.; Vaulot, D. Small Eukaryotic Phytoplankton Communities in Tropical Waters off Brazil Are Dominated by Symbioses between Haptophyta and Nitrogen-Fixing Cyanobacteria. ISME J. 2018, 12, 1360–1374. [Google Scholar] [CrossRef] [Green Version]
- Majaneva, M.; Enberg, S.; Autio, R.; Blomster, J.; Rintala, J. Mamiellophyceae Shift in Seasonal Predominance in the Baltic Sea. Aquat. Microb. Ecol. 2019, 83, 181–187. [Google Scholar] [CrossRef]
- De Vargas, C.; Audic, S.; Henry, N.; Decelle, J.; Mahe, F.; Logares, R.; Lara, E.; Berney, C.; Le Bescot, N.; Probert, I.; et al. Eukaryotic Plankton Diversity in the Sunlit Ocean. Science 2015, 348, 1261605. [Google Scholar] [CrossRef] [Green Version]
- Taib, N.; Mangot, J.-F.; Domaizon, I.; Bronner, G.; Debroas, D. Phylogenetic Affiliation of SSU RRNA Genes Generated by Massively Parallel Sequencing: New Insights into the Freshwater Protist Diversity. PLoS ONE 2013, 8, e58950. [Google Scholar] [CrossRef]
- Piwosz, K.; Całkiewicz, J.; Gołębiewski, M.; Creer, S. Diversity and Community Composition of Pico- and Nanoplanktonic Protists in the Vistula River Estuary (Gulf of Gdańsk, Baltic Sea). Estuar. Coast. Shelf Sci. 2018, 207, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.H.; An, S.M.; Chun, S.; Yang, E.C.; Selph, K.E.; Lee, C.M.; Noh, J.H. Dynamic Changes in the Composition of Photosynthetic Picoeukaryotes in the Northwestern Pacific Ocean Revealed by High-Throughput Tag Sequencing of Plastid 16S RRNA Genes. FEMS Microbiol. Ecol. 2016, 92, fiv170. [Google Scholar] [CrossRef]
- Simmons, M.P.; Bachy, C.; Sudek, S.; van Baren, M.J.; Sudek, L.; Ares, M., Jr.; Worden, A.Z. Intron Invasions Trace Algal Speciation and Reveal Nearly Identical Arctic and Antarctic Micromonas Populations. Mol. Biol. Evol. 2015, 32, 2219–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dela Peña, L.B.R.O.; Tejada, A.J.P.; Quijano, J.B.; Alonzo, K.H.; Gernato, E.G.; Caril, A.; Dela Cruz, M.A.M.; Onda, D.F.L. Diversity of Marine Eukaryotic Picophytoplankton Communities with Emphasis on Mamiellophyceae in Northwestern Philippines. Philipp. J. Sci. 2021, 150, 27–42. [Google Scholar]
- Pearman, J.K.; Kürten, S.; Sarma, Y.V.B.; Jones, B.H.; Carvalho, S. Biodiversity Patterns of Plankton Assemblages at the Extremes of the Red Sea. FEMS Microbiol. Ecol. 2016, 92, fiw002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Cao, S.; Gao, Y.; He, J. Distribution and Environmental Correlations of Picoeukaryotes in an Arctic Fjord (Kongsfjorden, Svalbard) during the Summer. Polar Res. 2019, 38, 3390. [Google Scholar] [CrossRef]
- SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools|Nucleic Acids Research|Oxford Academic. Available online: https://academic.oup.com/nar/article/41/D1/D590/1069277 (accessed on 9 November 2021).
- Guillou, L.; Bachar, D.; Audic, S.; Bass, D.; Berney, C.; Bittner, L.; Boutte, C.; Burgaud, G.; de Vargas, C.; Decelle, J.; et al. The Protist Ribosomal Reference Database (PR2): A Catalog of Unicellular Eukaryote Small Sub-Unit RRNA Sequences with Curated Taxonomy. Nucleic Acids Res. 2013, 41, D597–D604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, I.M.; Pinto, A.J.; Guest, J.S. Design and Evaluation of Illumina MiSeq-Compatible, 18S RRNA Gene-Specific Primers for Improved Characterization of Mixed Phototrophic Communities. Appl. Environ. Microbiol. 2016, 82, 5878–5891. [Google Scholar] [CrossRef] [Green Version]
- Tragin, M.; Zingone, A.; Vaulot, D. Comparison of Coastal Phytoplankton Composition Estimated from the V4 and V9 Regions of the 18S RRNA Gene with a Focus on Photosynthetic Groups and Especially Chlorophyta. Environ. Microbiol. 2018, 20, 506–520. [Google Scholar] [CrossRef] [Green Version]
- Leconte, J.; Benites, L.F.; Vannier, T.; Wincker, P.; Piganeau, G.; Jaillon, O. Genome Resolved Biogeography of Mamiellales. Genes 2020, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Cottrell, M.T.; Suttle, C.A. Wide-Spread Occurrence and Clonal Variation in Viruses Which Cause Lysis of a Cosmopolitan, Eukaryotic Marine Phytoplankter, Micromonas pusilla. Mar. Ecol. Prog. Ser. 1991, 78, 1–9. [Google Scholar] [CrossRef]
- Mayer, J.A.; Taylor, F.J.R. A Virus Which Lyses the Marine Nanoflagellate Micromonas pusilla. Nature 1979, 281, 299–301. [Google Scholar] [CrossRef]
- Evans, C.; Archer, S.D.; Jacquet, S.; Wilson, W.H. Direct Estimates of the Contribution of Viral Lysis and Microzooplankton Grazing to the Decline of a Micromonas spp. Population. Aquat. Microb. Ecol. 2003, 30, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Cottrell, M.T.; Suttle, C.A. Dynamics of Lytic Virus Infecting the Photosynthetic Marine Picoflagellate Micromonas pusilla. Limnol. Oceanogr. 1995, 40, 730–739. [Google Scholar] [CrossRef]
- Hingamp, P.; Grimsley, N.; Acinas, S.G.; Clerissi, C.; Subirana, L.; Poulain, J.; Ferrera, I.; Sarmento, H.; Villar, E.; Lima-Mendez, G.; et al. Exploring Nucleo-Cytoplasmic Large DNA Viruses in Tara Oceans Microbial Metagenomes. ISME J. 2013, 7, 1678–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, F.; Roux, S.; Paez-Espino, D.; Jungbluth, S.; Walsh, D.A.; Denef, V.J.; McMahon, K.D.; Konstantinidis, K.T.; Eloe-Fadrosh, E.A.; Kyrpides, N.C.; et al. Giant Virus Diversity and Host Interactions through Global Metagenomics. Nature 2020, 578, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Pringle, C.R. Virus Taxonomy-San Diego 1998. Arch. Virol. 1998, 143, 1449–1459. [Google Scholar] [CrossRef]
- Short, S.M. The Ecology of Viruses That Infect Eukaryotic Algae. Environ. Microbiol. 2012, 14, 2253–2271. [Google Scholar] [CrossRef]
- Bellec, L.; Grimsley, N.; Moreau, H.; Desdevises, Y. Phylogenetic Analysis of New Prasinoviruses (Phycodnaviridae) That Infect the Green Unicellular Algae Ostreococcus, Bathycoccus and Micromonas. Environ. Microbiol. Rep. 2009, 1, 114–123. [Google Scholar] [CrossRef]
- Moreau, H.; Piganeau, G.; Desdevises, Y.; Cooke, R.; Derelle, E.; Grimsley, N. Marine Prasinovirus Genomes Show Low Evolutionary Divergence and Acquisition of Protein Metabolism Genes by Horizontal Gene Transfer. J. Virol. 2010, 84, 12555–12563. [Google Scholar] [CrossRef] [Green Version]
- Martínez, J.M.; Boere, A.; Gilg, I.; van Lent, J.W.M.; Witte, H.J.; van Bleijswijk, J.D.L.; Brussaard, C.P.D. New Lipid Envelope-Containing DsDNA Virus Isolates Infecting Micromonas pusilla Reveal a Separate Phylogenetic Group. Aquat. Microb. Ecol. 2015, 74, 17–28. [Google Scholar] [CrossRef]
- Baudoux, A.-C.; Lebredonchel, H.; Dehmer, H.; Latimier, M.; Edern, R.; Rigaut-Jalabert, F.; Ge, P.; Guillou, L.; Foulon, E.; Bozec, Y.; et al. Interplay between the Genetic Clades of Micromonas and Their Viruses in the Western English Channel. Environ. Microbiol. Rep. 2015, 7, 765–773. [Google Scholar] [CrossRef] [Green Version]
- Zingone, A.; Natale, F.; Biffali, E.; Borra, M.; Forlani, G.; Sarno, D. Diversity in Morphology, Infectivity, Molecular Characteristics and Induced Host Resistance between Two Viruses Infecting Micromonas pusilla. Aquat. Microb. Ecol. 2006, 45, 1–14. [Google Scholar] [CrossRef]
- Maat, D.S.; Biggs, T.; Evans, C.; Van Bleijswijk, J.D.L.; Van der Wel, N.N.; Dutilh, B.E.; Brussaard, C.P.D. Characterization and Temperature Dependence of Arctic Micromonas polaris Viruses. Viruses 2017, 9, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellec, L.; Clerissi, C.; Edern, R.; Foulon, E.; Simon, N.; Grimsley, N.; Desdevises, Y. Cophylogenetic Interactions between Marine Viruses and Eukaryotic Picophytoplankton. BMC Evol. Biol. 2014, 14, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellec, L.; Grimsley, N.; Desdevises, Y. Isolation of Prasinoviruses of the Green Unicellular Algae Ostreococcus spp. on a Worldwide Geographical Scale. Appl. Environ. Microbiol. 2010, 76, 96–101. [Google Scholar] [CrossRef] [Green Version]
- Derelle, E.; Monier, A.; Cooke, R.; Worden, A.Z.; Grimsley, N.H.; Moreau, H. Diversity of Viruses Infecting the Green Microalga Ostreococcus lucimarinus. J. Virol. 2015, 89, 5812–5821. [Google Scholar] [CrossRef] [Green Version]
- Weynberg, K.D.; Allen, M.J.; Gilg, I.C.; Scanlan, D.J.; Wilson, W.H. Genome Sequence of Ostreococcus tauri Virus OtV-2 Throws Light on the Role of Picoeukaryote Niche Separation in the Ocean. J. Virol. 2011, 85, 4520–4529. [Google Scholar] [CrossRef] [Green Version]
- Derelle, E.; Ferraz, C.; Escande, M.-L.; Eychenié, S.; Cooke, R.; Piganeau, G.; Desdevises, Y.; Bellec, L.; Moreau, H.; Grimsley, N. Life-Cycle and Genome of OtV5, a Large DNA Virus of the Pelagic Marine Unicellular Green Alga Ostreococcus tauri. PLoS ONE 2008, 3, e2250. [Google Scholar] [CrossRef]
- Weynberg, K.D.; Allen, M.J.; Ashelford, K.; Scanlan, D.J.; Wilson, W.H. From Small Hosts Come Big Viruses: The Complete Genome of a Second Ostreococcus tauri Virus, OtV-1. Environ. Microbiol. 2009, 11, 2821–2839. [Google Scholar] [CrossRef]
- Bellec, L.; Grimsley, N.; Derelle, E.; Moreau, H.; Desdevises, Y. Abundance, Spatial Distribution and Genetic Diversity of Ostreococcus tauri Viruses in Two Different Environments. Environ. Microbiol. Rep. 2010, 2, 313–321. [Google Scholar] [CrossRef]
- Clerissi, C.; Grimsley, N.; Ogata, H.; Hingamp, P.; Poulain, J.; Desdevises, Y. Unveiling of the Diversity of Prasinoviruses (Phycodnaviridae) in Marine Samples by Using High-Throughput Sequencing Analyses of PCR-Amplified DNA Polymerase and Major Capsid Protein Genes. Appl. Environ. Microbiol. 2014, 80, 3150–3160. [Google Scholar] [CrossRef] [Green Version]
- Aylward, F.O.; Moniruzzaman, M.; Ha, A.D.; Koonin, E.V. A Phylogenomic Framework for Charting the Diversity and Evolution of Giant Viruses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemoine, F.; Gascuel, O. Gotree/Goalign: Toolkit and Go API to Facilitate the Development of Phylogenetic Workflows. bioRxiv 2021. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v3: An Online Tool for the Display and Annotation of Phylogenetic and Other Trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Brussaard, C.P.D.; Noordeloos, A.A.M.; Sandaa, R.-A.; Heldal, M.; Bratbak, G. Discovery of a DsRNA Virus Infecting the Marine Photosynthetic Protist Micromonas pusilla. Virology 2004, 319, 280–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahlsten, E. Seasonal Abundance in Skagerrak-Kattegat Coastal Waters and Host Specificity of Viruses Infecting the Marine Photosynthetic Flagellate Micromonas pusilla. Aquat. Microb. Ecol. 1998, 16, 103–108. [Google Scholar] [CrossRef]
- Clerissi, C.; Desdevises, Y.; Grimsley, N. Prasinoviruses of the Marine Green Alga Ostreococcus tauri Are Mainly Species Specific. J. Virol. 2012, 86, 4611–4619. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhang, W.; Li, X.; Pan, Y.; Yan, S.; Wang, Y. The Genome of a Prasinoviruses-Related Freshwater Virus Reveals Unusual Diversity of Phycodnaviruses. BMC Genom. 2018, 19, 49. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Zhou, L.; Liang, X.; Zhou, Y.; Chen, H.; Yan, S.; Wang, Y. Novel Cell-Virus-Virophage Tripartite Infection Systems Discovered in the Freshwater Lake Dishui Lake in Shanghai, China. J. Virol. 2020, 94, e00149-20. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, J.; Liu, T.; Yu, Y.; Pan, Y.; Yan, S.; Wang, Y. Four Novel Algal Virus Genomes Discovered from Yellowstone Lake Metagenomes. Sci. Rep. 2015, 5, 15131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingone, A.; Sarno, D.; Forlani, G. Seasonal Dynamics in the Abundance of Micromonas pusilla (Prasinophyceae) and Its Viruses in the Gulf of Naples (Mediterranean Sea). J. Plankton Res. 1999, 21, 2143–2159. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Endo, H.; Blanc-Mathieu, R.; Chaffron, S.; Hernández-Velázquez, R.; Kaneko, H.; Ogata, H. Quantitative Assessment of NCLDV–Host Interactions Predicted by Co-Occurrence Analyses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Endo, H.; Blanc-Mathieu, R.; Li, Y.; Salazar, G.; Henry, N.; Labadie, K.; de Vargas, C.; Sullivan, M.B.; Bowler, C.; Wincker, P.; et al. Biogeography of Marine Giant Viruses Reveals Their Interplay with Eukaryotes and Ecological Functions. Nat. Ecol. Evol. 2020, 4, 1639–1649. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Blanc-Mathieu, R.; Endo, H.; Chaffron, S.; Delmont, T.O.; Gaia, M.; Henry, N.; Hernández-Velázquez, R.; Nguyen, C.H.; Mamitsuka, H.; et al. Eukaryotic Virus Composition Can Predict the Efficiency of Carbon Export in the Global Ocean. iScience 2021, 24, 102002. [Google Scholar] [CrossRef] [PubMed]
- Castillo, Y.M.; Forn, I.; Yau, S.; Morán, X.A.G.; Alonso-Sáez, L.; Arandia-Gorostidi, N.; Vaqué, D.; Sebastián, M. Seasonal Dynamics of Natural Ostreococcus Viral Infection at the Single Cell Level Using VirusFISH. Environ. Microbiol. 2021, 23, 3009–3019. [Google Scholar] [CrossRef]
- Xie, H.; Yang, C.; Sun, Y.; Igarashi, Y.; Jin, T.; Luo, F. PacBio Long Reads Improve Metagenomic Assemblies, Gene Catalogs, and Genome Binning. Front. Genet. 2020, 11, 1077. [Google Scholar] [CrossRef]
- Priest, T.; Orellana, L.H.; Huettel, B.; Fuchs, B.M.; Amann, R. Microbial Metagenome-Assembled Genomes of the Fram Strait from Short and Long Read Sequencing Platforms. PeerJ 2021, 9, e11721. [Google Scholar] [CrossRef]
- Marbouty, M.; Cournac, A.; Flot, J.-F.; Marie-Nelly, H.; Mozziconacci, J.; Koszul, R. Metagenomic Chromosome Conformation Capture (Meta3C) Unveils the Diversity of Chromosome Organization in Microorganisms. eLife 2014, 3, e03318. [Google Scholar] [CrossRef]
- DeMaere, M.Z.; Darling, A.E. Bin3C: Exploiting Hi-C Sequencing Data to Accurately Resolve Metagenome-Assembled Genomes. Genome Biol. 2019, 20, 46. [Google Scholar] [CrossRef] [Green Version]
- Burton, J.N.; Liachko, I.; Dunham, M.J.; Shendure, J. Species-Level Deconvolution of Metagenome Assemblies with Hi-C–Based Contact Probability Maps. G3 Genes|Genomes|Genetics 2014, 4, 1339–1346. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Gao, P.; Li, B.; Xing, P.; Wu, Q.L. Tracking Double-Stranded DNA Bacteriophages and Their Hosts in a Deep Freshwater Lake by Integrating Metagenomics and The Hi-C Technique. Res. Sq. Prepr. 2020. [Google Scholar] [CrossRef]
- Bickhart, D.M.; Watson, M.; Koren, S.; Panke-Buisse, K.; Cersosimo, L.M.; Press, M.O.; Van Tassell, C.P.; Van Kessel, J.A.S.; Haley, B.J.; Kim, S.W.; et al. Assignment of Virus and Antimicrobial Resistance Genes to Microbial Hosts in a Complex Microbial Community by Combined Long-Read Assembly and Proximity Ligation. Genome Biol. 2019, 20, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignacio-Espinoza, J.C.; Laperriere, S.M.; Yeh, Y.-C.; Weissman, J.; Hou, S.; Long, A.M.; Fuhrman, J.A. Ribosome-Linked mRNA-rRNA Chimeras Reveal Active Novel Virus Host Associations. bioRxiv 2020. [Google Scholar] [CrossRef]
- Moran, M.A.; Satinsky, B.; Gifford, S.M.; Luo, H.; Rivers, A.; Chan, L.-K.; Meng, J.; Durham, B.P.; Shen, C.; Varaljay, V.A.; et al. Sizing up Metatranscriptomics. ISME J. 2013, 7, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Ji, N.; Lin, L.; Li, L.; Yu, L.; Zhang, Y.; Luo, H.; Li, M.; Shi, X.; Wang, D.-Z.; Lin, S. Metatranscriptome Analysis Reveals Environmental and Diel Regulation of a Heterosigma Akashiwo (Raphidophyceae) Bloom. Environ. Microbiol. 2018, 20, 1078–1094. [Google Scholar] [CrossRef]
- Lampe, R.H.; Cohen, N.R.; Ellis, K.A.; Bruland, K.W.; Maldonado, M.T.; Peterson, T.D.; Till, C.P.; Brzezinski, M.A.; Bargu, S.; Thamatrakoln, K.; et al. Divergent Gene Expression among Phytoplankton Taxa in Response to Upwelling. Environ. Microbiol. 2018, 20, 3069–3082. [Google Scholar] [CrossRef]
- Salazar, G.; Paoli, L.; Alberti, A.; Huerta-Cepas, J.; Ruscheweyh, H.-J.; Cuenca, M.; Field, C.M.; Coelho, L.P.; Cruaud, C.; Engelen, S.; et al. Gene Expression Changes and Community Turnover Differentially Shape the Global Ocean Metatranscriptome. Cell 2019, 179, 1068–1083.e21. [Google Scholar] [CrossRef] [Green Version]
| Host Genus | Host Species | Host Clade * | Genome-Sequenced Viruses | Prasinovirus Isolates | Isolation Sites | References |
|---|---|---|---|---|---|---|
| Bathycoccus | prasinos | BI | BpV1, BpV2 | 9 | Mediterranean Sea, North Atlantic | [81,82] |
| calidus | BII | BIIV-1, BIIV-2, BIIV-3 | 3 | North Atlantic | [9] | |
| Micromonas | commoda | A.ABC.12 | MpV-12T | 31 | North Atlantic, North Sea | [83,84] |
| bravo | B.E.3 | - | 7 | Mediterranean Sea, North Atlantic | [84,85] | |
| pusilla | C.D.5 | MpV-SP1, MpV-P1 | 35 | Mediterranean Sea, North Atlantic, North Pacific, North Sea | [73,83,84] | |
| polaris | Ea | - | 4 | Barents and Greenland Seas (Spitsbergen) | [86] | |
| candidate sp. 2 | unknown | MpV1 | 16 | Mediterranean Sea, North Atlantic | [87,84] | |
| Unclassified Mamiellales † | unknown | unknown | - | 11 | North Atlantic (English Channel) | [87,84] |
| Ostreococcus | lucimarinus | A | OlV1, OlV2, OlV3, OlV4, OlV5, OlV6, OlV7 | 27 | Mediterranean Sea, North Atlantic, North Pacific, South Pacific | [88,89] |
| sp. | B | OtV2 | 1 | North Atlantic | [90] | |
| tauri | C | OtV1, OtV5, OtV6 | 54 | Mediterranean lagoon, North Atlantic | [91,92,93,94] | |
| mediterraneus | D | OmV1, OmV2 | 7 | Mediterranean lagoon | [47,81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yung, C.C.M.; Rey Redondo, E.; Sanchez, F.; Yau, S.; Piganeau, G. Diversity and Evolution of Mamiellophyceae: Early-Diverging Phytoplanktonic Green Algae Containing Many Cosmopolitan Species. J. Mar. Sci. Eng. 2022, 10, 240. https://doi.org/10.3390/jmse10020240
Yung CCM, Rey Redondo E, Sanchez F, Yau S, Piganeau G. Diversity and Evolution of Mamiellophyceae: Early-Diverging Phytoplanktonic Green Algae Containing Many Cosmopolitan Species. Journal of Marine Science and Engineering. 2022; 10(2):240. https://doi.org/10.3390/jmse10020240
Chicago/Turabian StyleYung, Charmaine C. M., Elvira Rey Redondo, Frederic Sanchez, Sheree Yau, and Gwenael Piganeau. 2022. "Diversity and Evolution of Mamiellophyceae: Early-Diverging Phytoplanktonic Green Algae Containing Many Cosmopolitan Species" Journal of Marine Science and Engineering 10, no. 2: 240. https://doi.org/10.3390/jmse10020240
APA StyleYung, C. C. M., Rey Redondo, E., Sanchez, F., Yau, S., & Piganeau, G. (2022). Diversity and Evolution of Mamiellophyceae: Early-Diverging Phytoplanktonic Green Algae Containing Many Cosmopolitan Species. Journal of Marine Science and Engineering, 10(2), 240. https://doi.org/10.3390/jmse10020240