A Critical Review on Soil Chemical Processes that Control How Soil pH Affects Phosphorus Availability to Plants



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Review and Discussion
2.1. The Need to Consider Context of Observations and Soil-Solution Dynamics
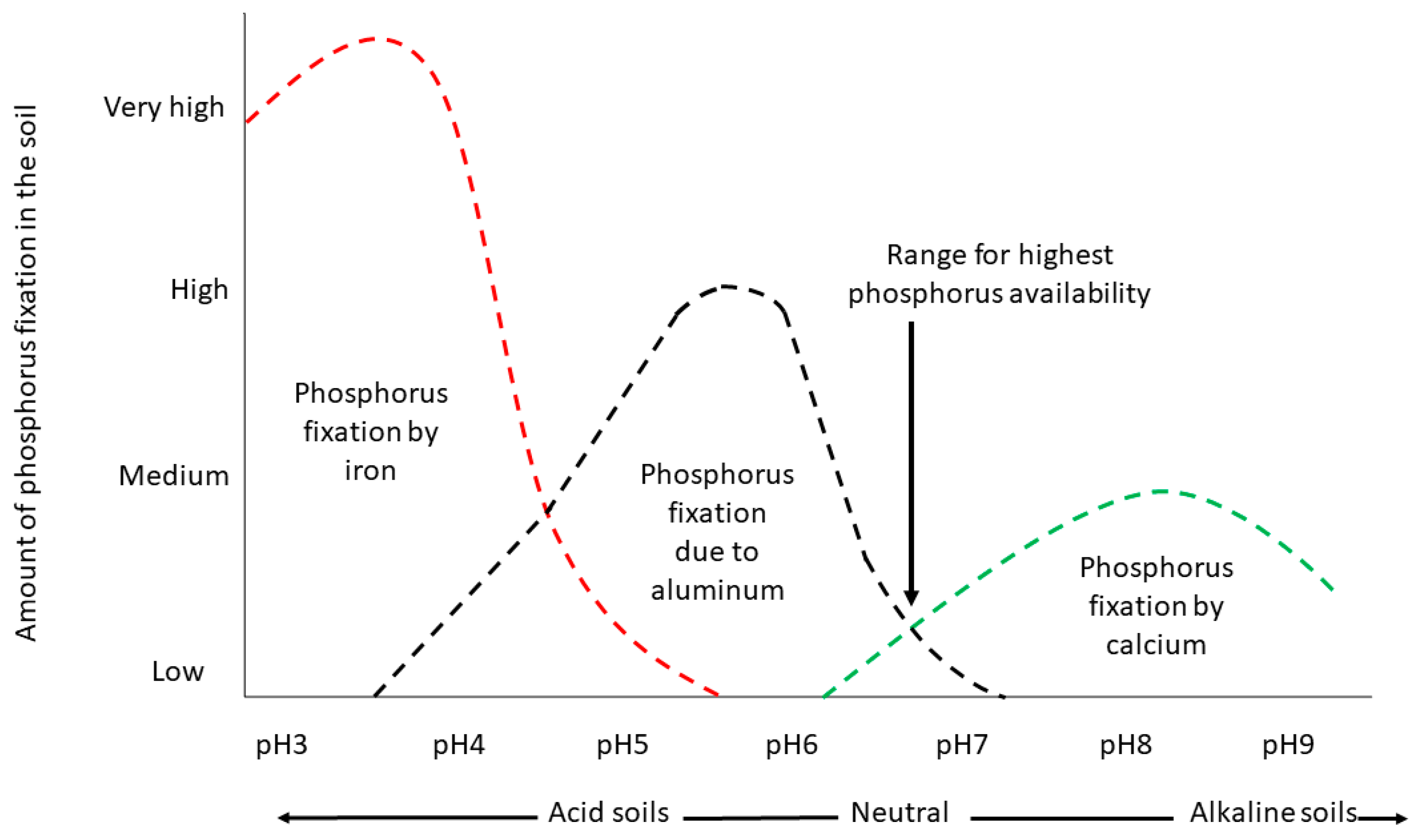
2.2. How Are Different Individual P Sorption Mechanisms Impacted by pH?
2.2.1. Anion Exchange
2.2.2. Precipitation of Ca Phosphates
2.2.3. Ligand Exchange (Adsorption) to Al and Fe Oxides/Hydroxides and Edges of Alumino-Silicate Minerals
2.2.4. Precipitation of Al and Fe Phosphates
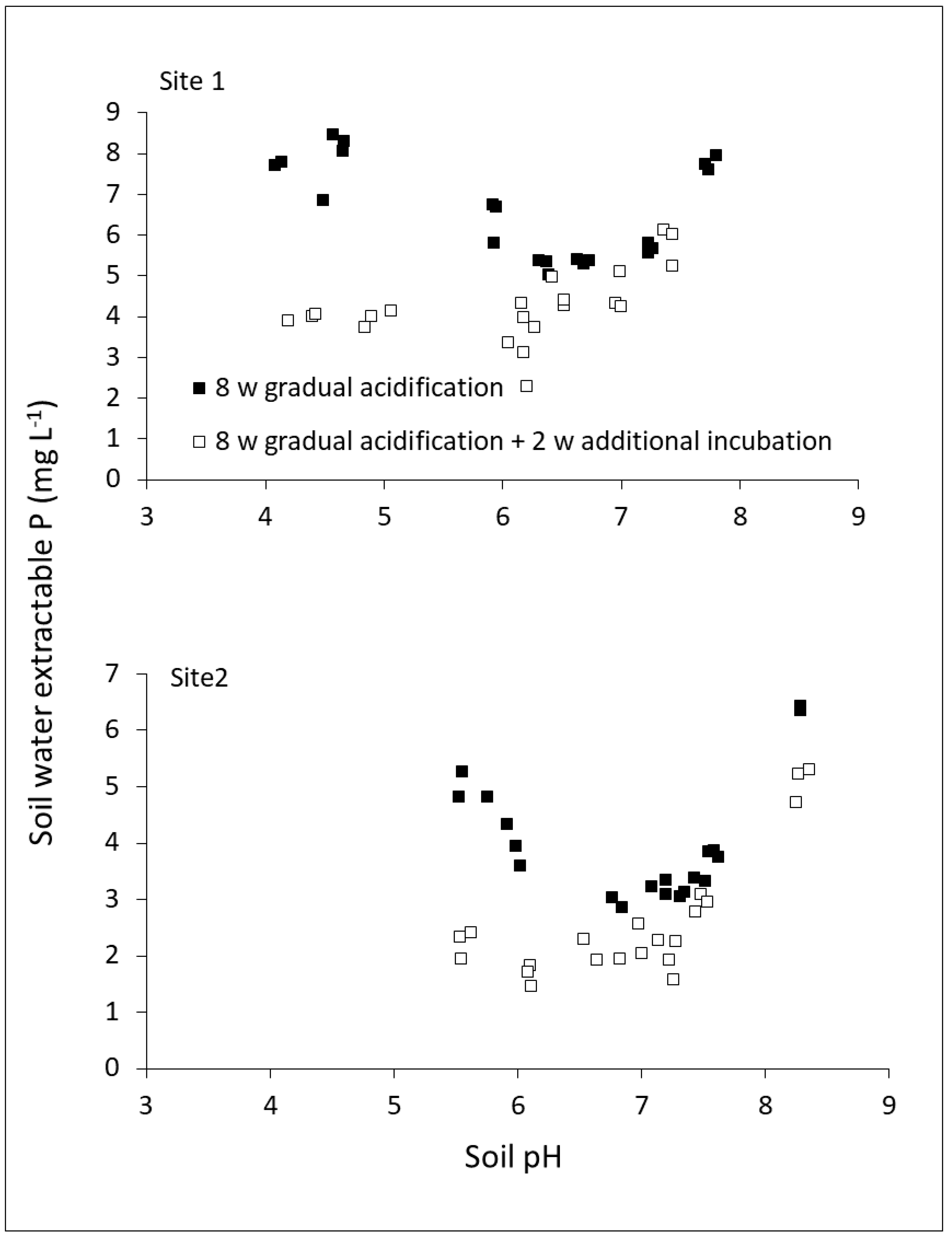
2.3. Dynamics among P Reactions in Soil
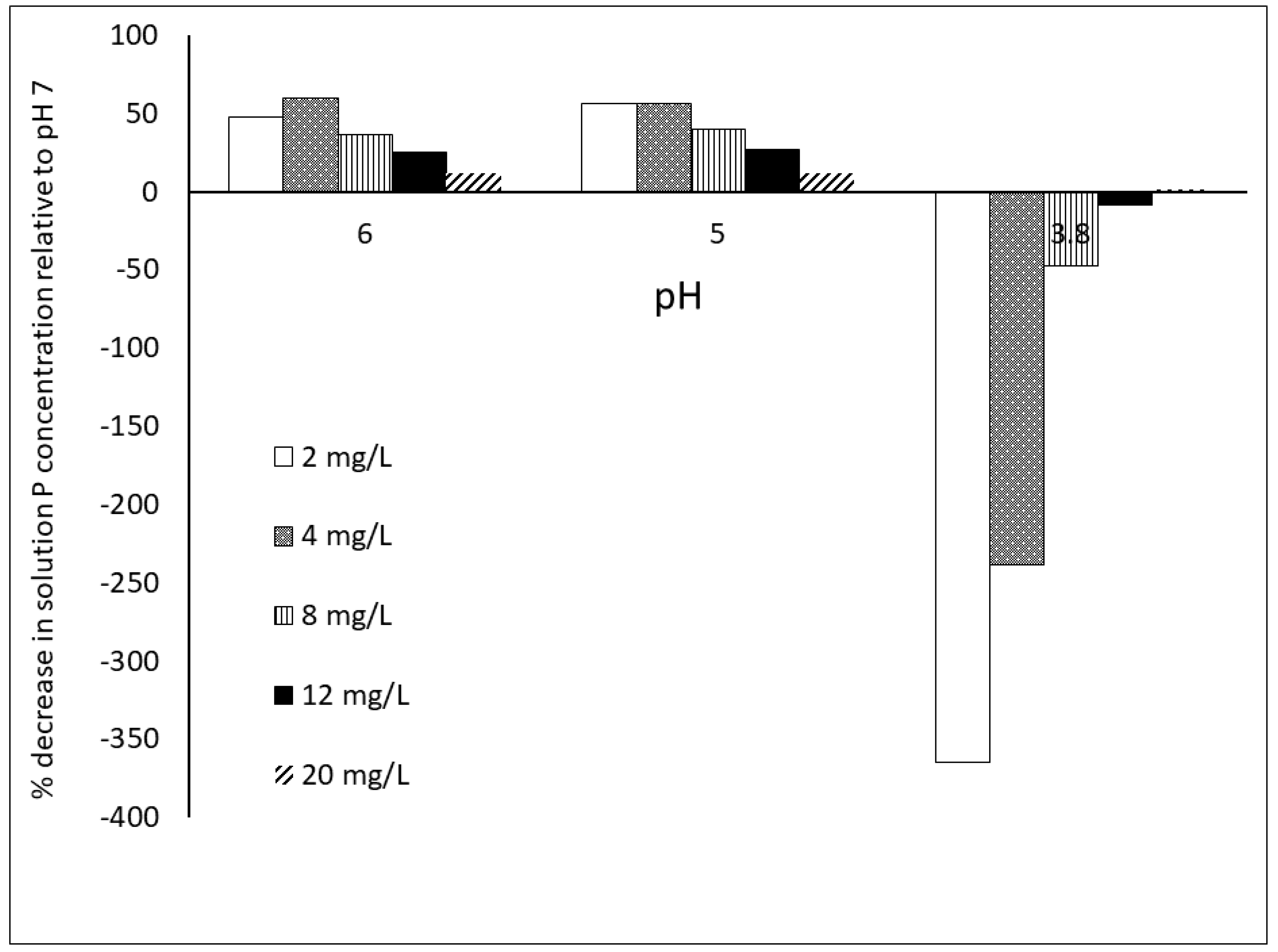
2.4. Impact of Methodology: Time of Equilibration and P Extraction Solution
2.5. Measuring P Uptake in Soil with Changing pH and the Interaction between Yield, Plant P Uptake, and Other Soil Properties Affected by pH
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CEC | cation exchange capacity |
| K | thermodynamic solubility constant |
| PZC | point of zero charge |
| AER | anion exchange resin |
| CER | cation exchange resin |
| d | days |
| w | weeks |
References
- Price, G. Australian Soil Fertility Manual; Fertilizer Industry Federation of Australia, Inc. & CSIRO: Collingwood, Australia, 2006. [Google Scholar]
- Barrow, N.J. The effects of pH on phosphate uptake from the soil. Plant Soil 2017, 410, 401–410. [Google Scholar] [CrossRef]
- Amarasiri, S.L.; Olsen, S.R. Liming as Related to Solubility of P and Plant Growth in an Acid Tropical Soil. Soil Sci. Soc. Am. J. 1973, 37, 716–721. [Google Scholar] [CrossRef]
- MacLean, A.J.; Cook, R.L. The Effect of Soil Reaction on the Availability of Phosphorus for Alfalfa in Some Eastern Ontario Soils. Soil Sci. Soc. Am. J. 1955, 19, 311–314. [Google Scholar] [CrossRef]
- Paton, D.F.; Loneragan, J.F. An effect of lime of residual phosphorus in soil. Aust. J. Agric. Res. 1960, 11, 524–529. [Google Scholar] [CrossRef]
- Neller, J.R. Effect of Lime on Availability of Labeled Phosphorus of Phosphates in Rutlege Fine Sand and Marlboro and Carnegie Fine Sandy Loams. Soil Sci. 1953, 75, 103–108. [Google Scholar] [CrossRef]
- Ensminger, L.E.; Pearson, R.W. Residual Effects of Various Phosphates as Measured by Yields, P32 Uptake, and Extractable Phosphorus. Soil Sci. Soc. Am. J. 1957, 21, 80–84. [Google Scholar] [CrossRef]
- Shoop, G.J.; Brooks, C.R.; Blaser, R.E.; Thomas, G.W. Differential Responses of Grasses and Legumes to Liming and Phosphorus Fertilization. Agron. J. 1961, 53, 111–115. [Google Scholar] [CrossRef]
- Abruna, F.; Vicente-Chandler, J.; Pearson, R.W. Effects of Liming on Yields and Composition of Heavily Fertilized Grasses and on Soil Properties Under Humid Tropical Conditions. Soil Sci. Soc. Am. J. 1964, 28, 657–661. [Google Scholar] [CrossRef]
- Sumner, M.E.; Farina, M.P.W. Phosphorus interactions with other nutrients and lime in field cropping systems. In Advances in Soil Science; Springer: Heidelberg, Germany, 1986; pp. 201–236. [Google Scholar]
- Curtin, D.; Syers, J.K. Lime-induced changes in indices of soil phosphate availability. Soil Sci. Soc. Am. J. 2001, 65, 147–152. [Google Scholar] [CrossRef]
- MacKenzie, A.F. Inorganic soil phosphorus fractions of some Ontario soils as studied using isotopic exchange and solubility criteria. Can. J. Soil Sci. 1962, 42, 150–156. [Google Scholar] [CrossRef]
- Talibudeen, O. Isotopically exchangeable phosphorus in soils: Part II. Factors influencing the estimation of ‘labile’phosphorus. J. Soil Sci. 1957, 8, 86–96. [Google Scholar] [CrossRef]
- Murrmann, R.P.; Peech, M. Effect of pH on Labile and Soluble Phosphate in Soils. Soil Sci. Soc. Am. J. 1969, 33, 205–210. [Google Scholar] [CrossRef]
- Riley, D.; Barber, S.A. Effect of Ammonium and Nitrate Fertilization on Phosphorus Uptake as Related to Root-Induced pH Changes at the Root-Soil Interface. Soil Sci. Soc. Am. J. 1971, 35, 301–306. [Google Scholar] [CrossRef]
- Gustafsson, J.P.; Mwamila, L.B.; Kergoat, K. The pH dependence of phosphate sorption and desorption in Swedish agricultural soils. Geoderma 2012, 189, 304–311. [Google Scholar] [CrossRef]
- Curtin, D.; Syers, J.K.; Smillie, G.W. The importance of exchangeable cations and resin-sink characteristics in the release of soil phosphorus. J. Soil Sci. 1987, 38, 711–716. [Google Scholar] [CrossRef]
- Vaz, M.D.R.; Edwards, A.C.; Shand, C.A.; Cresser, M.S. Phosphorus fractions in soil solution: Influence of soil acidity and fertiliser additions. Plant Soil 1993, 148, 175–183. [Google Scholar]
- Hsu, P.H.; Rennie, D.A. Reactions of phosphate in aluminum systems.: I. adsorption of phosphate by X-ray amorphous” aluminum hydroxide”. Can. J. Soil Sci. 1962, 42, 197–209. [Google Scholar] [CrossRef]
- Scanlan, C.; Brennan, R.; Sarre, G.A. Effect of soil pH and crop sequence on the response of wheat (Triticum aestivum) to phosphorus fertiliser. Crop Pasture Sci. 2015, 66, 23–31. [Google Scholar] [CrossRef]
- Von Tucher, S.; Hörndl, D.; Schmidhalter, U. Interaction of soil pH and phosphorus efficacy: Long-term effects of P fertilizer and lime applications on wheat, barley, and sugar beet. Ambio 2018, 47, 41–49. [Google Scholar] [CrossRef]
- Murrmann, R.P.; Peech, M. Relative significance of labile and crystalline phosphates in soil. Soil Sci. 1969, 107, 249–255. [Google Scholar] [CrossRef]
- Chen, C.R.; Sinaj, S.; Condron, L.M.; Frossard, E.; Sherlock, R.R.; Davis, M.R. Characterization of phosphorus availability in selected New Zealand grassland soils. Nutr. Cycl. Agroecosyst. 2003, 65, 89–100. [Google Scholar] [CrossRef]
- De Smet, J.; Vanderdeelen, J.; Hofman, G. Effect of soil properties on the kinetics of phosphate release. Commun. Soil Sci. Plant Anal. 1998, 29, 2135–2147. [Google Scholar] [CrossRef]
- Penn, C.J.; Rutter, E.B.; Arnall, D.B.; Camberato, J.; Williams, M.; Watkins, P. A discussion on mehlich-3 phosphorus extraction from the perspective of governing chemical reactions and phases: Impact of soil pH. Agriculture 2018, 8, 106. [Google Scholar] [CrossRef]
- Penn, C.; Bryant, R. Incubation of dried and sieved soils can induce calcium phosphate precipitation/adsorption. Commun. Soil Sci. Plant Anal. 2006, 37, 1437–1449. [Google Scholar] [CrossRef]
- Pardo, M.T.; Guadalix, M.E.; Garcia-Gonzalez, M.T. Effect of pH and background electrolyte on P sorption by variable charge soils. Geoderma 1992, 54, 275–284. [Google Scholar] [CrossRef]
- Xu, R.; Jiang, J.; Cheng, C. Effect of Ionic Strength on Specific Adsorption of Ions by Variable Charge Soils: Experimental Testification on the Adsorption Model of Bowden et al. In Molecular Environmental Soil Science at the Interfaces in the Earth’s Critical Zone; Springer: Heidelberg, Germany, 2010; pp. 78–80. [Google Scholar]
- Bolan, N.S.; Syers, J.K.; Tillman, R.W. Ionic strength effects on surface charge and adsorption of phosphate and sulphate by soils. J. Soil Sci. 1986, 37, 379–388. [Google Scholar] [CrossRef]
- Darch, T.; Blackwell, M.S.A.; Hawkins, J.M.B.; Haygarth, P.M.; Chadwick, D. A meta-analysis of organic and inorganic phosphorus in organic fertilizers, soils, and water: Implications for water quality. Crit. Rev. Environ. Sci. Technol. 2014, 44, 2172–2202. [Google Scholar] [CrossRef]
- Vange, M.S.; Holmern, K.; Nissen, P.E.R. Multiphasic uptake of sulfate by barley roots I. Effects of analogues, phosphate, and pH. Physiol. Plant. 1974, 31, 292–301. [Google Scholar] [CrossRef]
- Sentenac, H.; Grignon, C. Effect of pH on orthophosphate uptake by corn roots. Plant Physiol. 1985, 77, 136–141. [Google Scholar] [CrossRef]
- McDowell, R.W.; Sharpley, A.N. Phosphorus solubility and release kinetics as a function of soil test P concentration. Geoderma 2003, 112, 143–154. [Google Scholar] [CrossRef]
- Coleman, N.T.; Thorup, J.T.; Jackson, W.A. Phosphate-sorption Reactions That Involve Exchangeable A1. Soil Sci. 1960, 90, 1–7. [Google Scholar] [CrossRef]
- Guedes, R.S.; Melo, L.C.A.; Vergütz, L.; Rodríguez-Vila, A.; Covelo, E.F.; Fernandes, A.R. Adsorption and desorption kinetics and phosphorus hysteresis in highly weathered soil by stirred flow chamber experiments. Soil Tillage Res. 2016, 162, 46–54. [Google Scholar] [CrossRef]
- McDowell, R.W.; Condron, L.M.; Mahieu, N.; Brookes, P.C.; Poulton, P.R.; Sharpley, A.N. Analysis of potentially mobile phosphorus in arable soils using solid state nuclear magnetic resonance. J. Environ. Qual. 2002, 31, 450–456. [Google Scholar] [CrossRef]
- Hunger, S.; Sims, J.T.; Sparks, D.L. How accurate is the assessment of phosphorus pools in poultry litter by sequential extraction? J. Environ. Qual. 2005, 34, 382–389. [Google Scholar] [CrossRef]
- Prietzel, J.; Dümig, A.; Wu, Y.; Zhou, J.; Klysubun, W. Synchrotron-based P K-edge XANES spectroscopy reveals rapid changes of phosphorus speciation in the topsoil of two glacier foreland chronosequences. Geochim. Cosmochim. Acta 2013, 108, 154–171. [Google Scholar] [CrossRef]
- Beauchemin, S.; Hesterberg, D.; Chou, J.; Beauchemin, M.; Simard, R.R.; Sayers, D.E. Speciation of phosphorus in phosphorus-enriched agricultural soils using X-ray absorption near-edge structure spectroscopy and chemical fractionation. J. Environ. Qual. 2003, 32, 1809–1819. [Google Scholar] [CrossRef]
- Sato, S.; Solomon, D.; Hyland, C.; Ketterings, Q.M.; Lehmann, J. Phosphorus speciation in manure and manure-amended soils using XANES spectroscopy. Environ. Sci. Technol. 2005, 39, 7485–7491. [Google Scholar] [CrossRef]
- Ler, A.; Stanforth, R. Evidence for surface precipitation of phosphate on goethite. Environ. Sci. Technol. 2003, 37, 2694–2700. [Google Scholar] [CrossRef]
- Kim, Y.; Kirkpatrick, R.J. An investigation of phosphate adsorbed on aluminium oxyhydroxide and oxide phases by nuclear magnetic resonance. Eur. J. Soil Sci. 2004, 55, 243–251. [Google Scholar] [CrossRef]
- Kittrick, J.A.; Jackson, M.L. Rate of Phosphate Reaction with Soil Minerals and Electron Microscope Observations on the Reaction Mechanism. Soil Sci. Soc. Am. J. 1955, 19, 292–295. [Google Scholar] [CrossRef]
- Barrow, N.J.; Ellis, A.S. Testing a mechanistic model. V. The points of zero salt effect for phosphate retention, for zinc retention and for acid/alkali titration of a soil. J. Soil Sci. 1986, 37, 303–310. [Google Scholar] [CrossRef]
- Barrow, N.J.; Debnath, A. Effect of phosphate status on the sorption and desorption properties of some soils of northern India. Plant Soil 2014, 378, 383–395. [Google Scholar] [CrossRef]
- Strauss, R.; Brümmer, G.W.; Barrow, N.J. Effects of crystallinity of goethite: II. Rates of sorption and desorption of phosphate. Eur. J. Soil Sci. 1997, 48, 101–114. [Google Scholar] [CrossRef]
- Lindsay, W.L. Chemical Equilibria in Soils; John Wiley and Sons Ltd.: Hoboken, NJ, USA, 1979; ISBN 0471027049. [Google Scholar]
- Essington, M.E. Soil and Water Chemistry: An Integrative Approach, 2nd ed.; Routledge: Abingdon, UK, 2015; ISBN 9781466573154. [Google Scholar]
- Haseman, J.F.; Brown, E.H.; Whitt, C.D. Some reactions of phosphate with clays and hydrous oxides of iron and aluminum. Soil Sci. 1950, 70, 257–272. [Google Scholar] [CrossRef]
- Huffman, E.O. Reactions of Phosphate in Soils: Recent Research by TVA; Fertiliser Society: Colchester, UK, 1962. [Google Scholar]
- Lindsay, W.L.; Stephenson, H.F. Nature of the Reactions of Monocalcium Phosphate Monohydrate in Soils: II. Dissolution and Precipitation Reactions Involving Iron, Aluminum, Manganese, and Calcium. Soil Sci. Soc. Am. J. 1959, 23, 18–22. [Google Scholar] [CrossRef]
- Lindsay, W.L.; Lehr, J.R.; Stephenson, H.F. Nature of the Reactions of Monocalcium Phosphate Monohydrate in Soils: III. Studies with Metastable Triple-Point Solution. Soil Sci. Soc. Am. J. 1959, 23, 342–345. [Google Scholar] [CrossRef]
- Eriksson, A.K.; Gustafsson, J.P.; Hesterberg, D. Phosphorus speciation of clay fractions from long-term fertility experiments in Sweden. Geoderma 2015, 241, 68–74. [Google Scholar] [CrossRef]
- Luo, L.; Ma, Y.; Sanders, R.L.; Xu, C.; Li, J.; Myneni, S.C.B. Phosphorus speciation and transformation in long-term fertilized soil: Evidence from chemical fractionation and P K-edge XANES spectroscopy. Nutr. Cycl. Agroecosyst. 2017, 107, 215–226. [Google Scholar] [CrossRef]
- McBride, M.B. Environmental Soil Chemistry; Elsevier: Amsterdam, Netherlands, 1994. [Google Scholar]
- Zelazny, L.W.; He, L.; Vanwormhoudt, A.N. Charge analysis of soils and anion exchange. In Methods of Soil Analysis Part 3—Chemical Methods; Sparks, D.L., et al., Eds.; SSSA, ASA: Madison, WI, USA, 1996; pp. 1231–1253. [Google Scholar]
- Sparks, D.L. Environmental Soil Chemistry; Elsevier: Amsterdam, The Netherlands, 2003; ISBN 0080494803. [Google Scholar]
- Hingston, F.J. A review of anion adsorption. In Adsorption Inorganic Solid Liquids Interfaces; Ann Arbor Science: Ann Arbor, MI, USA, 1981; pp. 51–90. [Google Scholar]
- He, L.M.; Zelazny, L.W.; Martens, D.C.; Baligar, V.C.; Ritchey, K.D. Ionic strength effects on sulfate and phosphate adsorption on γ-alumina and kaolinite: Triple-layer model. Soil Sci. Soc. Am. J. 1997, 61, 784–793. [Google Scholar] [CrossRef]
- Penn, C.J.; Warren, J.G. Investigating phosphorus sorption onto kaolinite using isothermal titration calorimetry. Soil Sci. Soc. Am. J. 2009, 73, 560–568. [Google Scholar] [CrossRef]
- Penn, C.J.; Zhang, H. Isothermal titration calorimetry as an indicator of phosphorus sorption behavior. Soil Sci. Soc. Am. J. 2010, 74, 502–511. [Google Scholar] [CrossRef]
- Barrow, N.J. A mechanistic model for describing the sorption and desorption of phosphate by soil. J. Soil Sci. 1983, 34, 733–750. [Google Scholar] [CrossRef]
- Sample, E.C.; Soper, R.J.; Racz, G.J. Reactions of phosphate fertilizers in soils. In Role of Phosphorus in Agriculture; Khasawneh, F.E., et al., Eds.; SSSA: Madison, WI, USA, 1980; pp. 263–310. [Google Scholar]
- Kittrick, J.A.; Jackson, M.L. Electron-microscope observations of the reaction of phosphate with minerals, leading to a unified theory of phosphate fixation in soils. J. Soil Sci. 1956, 7, 81–89. [Google Scholar] [CrossRef]
- Van Riemsdijk, W.H.; Lyklema, J. Reaction of phosphate with gibbsite (Al (OH) 3) beyond the adsorption maximum. J. Colloid Interface Sci. 1980, 76, 55–66. [Google Scholar] [CrossRef]
- Chen, Y.S.R.; Butler, J.N.; Stumm, W. Kinetic study of phosphate reaction with aluminum oxide and kaolinite. Environ. Sci. Technol. 1973, 7, 327–332. [Google Scholar] [CrossRef]
- Lindsay, W.L.; Frazier, A.W.; Stephenson, H.F. Identification of reaction products from phosphate fertilizers in soils. Soil Sci. Soc. Am. J. 1962, 26, 446–452. [Google Scholar] [CrossRef]
- Philen, O.D.; Lehr, J.R. Reactions of Ammonium Polyphosphates with Soil Minerals. Soil Sci. Soc. Am. J. 1967, 31, 196–199. [Google Scholar] [CrossRef]
- Hashimoto, I.; Hughes, J.D.; Philen, O.D. Reactions of Triammonium Pyrophosphate with Soils and Soil Minerals. Soil Sci. Soc. Am. J. 1969, 33, 401–405. [Google Scholar] [CrossRef]
- Low, P.F.; Black, C.A. Phosphate-Induced Decomposition of Kaolinite. Soil Sci. Soc. Am. J. 1948, 12, 180–184. [Google Scholar] [CrossRef]
- Robarge, W.P.; Corey, R.B. Adsorption of phosphate by hydroxy-aluminum species on a cation exchange resin. Soil Sci. Soc. Am. J. 1979, 43, 481–487. [Google Scholar] [CrossRef]
- Hsu, P.H.; Rennie, D.A. Reactions of phosphate in aluminum systems: II. Precipitation of phosphate by exchangeable aluminum on a cation exchange resin. Can. J. Soil Sci. 1962, 42, 210–221. [Google Scholar] [CrossRef][Green Version]
- Woodruff, J.R.; Kamprath, E.J. Phosphorus Adsorption Maximum as Measured by the Langmuir Isotherm and Its Relationship to Phosphorus Availability. Soil Sci. Soc. Am. J. 1965, 29, 148–150. [Google Scholar] [CrossRef]
- Penn, C.J.; Bryant, R.B. Phosphorus solubility in response to acidification of dairy manure amended soils. Soil Sci. Soc. Am. J. 2008, 72, 238–243. [Google Scholar] [CrossRef]
- Devau, N.; Hinsinger, P.; Le Cadre, E.; Colomb, B.; Gérard, F. Fertilization and pH effects on processes and mechanisms controlling dissolved inorganic phosphorus in soils. Geochim. Cosmochim. Acta 2011, 75, 2980–2996. [Google Scholar] [CrossRef]
- Hinsinger, P.; Herrmann, L.; Lesueur, D.; Robin, A.; Trap, J.; Waithaisong, K.; Plassard, C. Impact of roots, microorganisms and microfauna on the fate of soil phosphorus in the rhizosphere. Annu. Plant Rev. Online 2018, 48, 377–407. [Google Scholar]
- Allison, J.D.; Brown, D.S.; Novo-Gradac, K.J. MINTEQA2/PRODEFA2, A Geochemical Assessment Model for Environmental Systems: Version 3.0 User’s Manual; Environmental Research Laboratory, Office of Research and Development, US Environmental Protection Agency: Washington, DC, USA, 1991.
- Racz, G.J.; Soper, R.J. Solubility of dimagnesium phosphate trihydrate and trimagnesium phosphate. Can. J. Soil Sci. 1968, 48, 265–269. [Google Scholar] [CrossRef][Green Version]
- Fuhrman, J.K.; Zhang, H.; Schroder, J.L.; Davis, R.L.; Payton, M.E. Water-Soluble Phosphorus as Affected by Soil to Extractant Ratios, Extraction Times, and Electrolyte. Commun. Soil Sci. Plant Anal. 2005, 36, 925–935. [Google Scholar] [CrossRef]
- Eriksson, A.K.; Ulén, B.; Berzina, L.; Iital, A.; Janssons, V.; Sileika, A.S.; Toomsoo, A. Phosphorus in agricultural soils around the Baltic Sea–comparison of laboratory methods as indices for phosphorus leaching to waters. Soil Use Manag. 2013, 29, 5–14. [Google Scholar] [CrossRef]
- Ryden, J.C.; Syers, J.K. Rationalization of ionic strength and cation effects on phosphate sorption by soils. J. Soil Sci. 1975, 26, 395–406. [Google Scholar] [CrossRef]
- Ryden, J.C.; Syers, J.K. Calcium Retention in Response to Phosphate Sorption by Soils. Soil Sci. Soc. Am. J. 1976, 40, 845–846. [Google Scholar] [CrossRef]
- Kuo, S. Phosphorus. In Methods of Soil Analysis Part 3; Chemical Methods; SSSA Book Series 5; Sparks, D.L., et al., Eds.; SSSA, ASA: Madison, WI, USA, 1996; pp. 869–919. [Google Scholar]
- Barrow, N.J. Further investigations on the use of lime on established pastures. Aust. J. Exp. Agric. 1965, 5, 442–449. [Google Scholar] [CrossRef]
- Merlin, A.; Rosolem, C.A.; He, Z. Non-labile phosphorus acquisition by Brachiaria. J. Plant Nutr. 2016, 39, 1319–1327. [Google Scholar] [CrossRef][Green Version]
- Almeida, D.S.; Penn, C.J.; Rosolem, C.A. Assessment of phosphorus availability in soil cultivated with ruzigrass. Geoderma 2018, 312, 64–73. [Google Scholar] [CrossRef]
- Osaki, M.; Watanabe, T.; Tadano, T. Beneficial effect of aluminum on growth of plants adapted to low pH soils. Soil Sci. Plant Nutr. 1997, 43, 551–563. [Google Scholar] [CrossRef]
- Chen, J.-H.; Barber, S.A. Soil pH and phosphorus and potassium uptake by maize evaluated with an uptake model. Soil Sci. Soc. Am. J. 1990, 54, 1032–1036. [Google Scholar] [CrossRef]
- Nadeem, M.; Mollier, A.; Morel, C.; Vives, A.; Prud’homme, L.; Pellerin, S. Relative contribution of seed phosphorus reserves and exogenous phosphorus uptake to maize (Zea mays L.) nutrition during early growth stages. Plant Soil 2011, 346, 231–244. [Google Scholar] [CrossRef]
- White, P.J.; Veneklaas, E.J. Nature and nurture: The importance of seed phosphorus content. Plant Soil 2012, 357, 1–8. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penn, C.J.; Camberato, J.J. A Critical Review on Soil Chemical Processes that Control How Soil pH Affects Phosphorus Availability to Plants. Agriculture 2019, 9, 120. https://doi.org/10.3390/agriculture9060120
Penn CJ, Camberato JJ. A Critical Review on Soil Chemical Processes that Control How Soil pH Affects Phosphorus Availability to Plants. Agriculture. 2019; 9(6):120. https://doi.org/10.3390/agriculture9060120
Chicago/Turabian StylePenn, Chad J., and James J. Camberato. 2019. "A Critical Review on Soil Chemical Processes that Control How Soil pH Affects Phosphorus Availability to Plants" Agriculture 9, no. 6: 120. https://doi.org/10.3390/agriculture9060120
APA StylePenn, C. J., & Camberato, J. J. (2019). A Critical Review on Soil Chemical Processes that Control How Soil pH Affects Phosphorus Availability to Plants. Agriculture, 9(6), 120. https://doi.org/10.3390/agriculture9060120