Abstract
Researchers focused on assessing differences in gene diversity within and between populations, whether cosmopolitan or local. However, the identification of patterns of variation in non-random heterozygous genomic stretches, known as Heterozygosity-Rich regions (HRRs), has not yet been determined in European local pig breeds. A total of 23 pig breeds (20 local and 3 cosmopolitan) were assessed and compared in terms of heterozygosity-rich regions. The breeds with the highest number of HRRs were Large White, Lithuanian Old type, and Landrace, followed by Lithuanian Native, Mora Romagnola, and Duroc. The breeds with the lowest number were Alentejana, Iberian, and Majorcan Black. No shared HRR islands were found in all breeds, but gene enrichment analysis performed in the most common HRRs revealed several biologically important genes that cluster together and play significant roles, primarily related to the immune system. Permutation analysis indicated that some local breeds serve as true reservoirs of genetic diversity, displaying distinct and unique characteristics in terms of heterozygosity. This study suggests the importance of investigating heterozygosity to develop a comprehensive picture of pig breeds, regardless of the production system, country of origin, or population size.
1. Introduction
The genetic landscape of the domestic pig is globally dominated by the prevalence of highly improved cosmopolitan pig breeds or lines, regardless of continent or country. However, many local domestic pig breeds still exist and are expected to act as genetic reservoirs for the species [1,2]. Along with the existence of large morphological differences, there are wide differences in genetic diversity both among and within cosmopolitan and local domestic pig populations [3,4]. These genetic differences stem from a complex domestication process involving different geographical populations of the wild ancestor [5]. Furthermore, human-mediated dispersion; adaptation to different environments; artificial selection for different needs; isolation; or migration between populations, including recurring introgression events from local wild boar populations [6,7,8], have undoubtedly contributed to shaping the domestic pig diversity [6]. Although researchers have paid attention to assessing differences in gene diversity within and between pig populations, whether cosmopolitan or local, there are still gaps in understanding certain genomic features. Identifying patterns of variation in non-random heterozygous genomic stretches, known as Heterozygosity-Rich regions (HRRs), has yet to be comprehensively examined on a European level. Understanding these HRR patterns of variation is important because heterozygosity can provide insights into population structure and demographic history [9]. The heterozygous advantage is also expected to play important adaptative roles [10] and provide information on population diversity and evolutionary history [11]. The selective advantage of heterozygous genotypes showing overdominance leads to balancing selection. However, the proportion of loci where polymorphism is conserved by a heterozygote advantage is typically low [11]. An HRR is a short genomic area where polymorphic loci cluster non-randomly [12]. There is a growing interest in characterizing HRRs in livestock species for conservation purposes [12,13,14,15,16]. Compared to runs of homozygosity (ROH), HRRs are observed to be smaller and less frequent across species [14,17,18,19]. While some sites of heterozygous advantage have important adaptive functions, their role in general evolutionary change may be more of an unusual phenomenon than a significant contributor to adaptation, explaining their lower proportion [9]. HRRs could reflect the effect of divergent selection and provide insights into livestock populations’ structure and breeding history [2]. Moreover, the distribution of HRRs within species appears to be population-specific [12,15,16], even if candidate genes located in the significant HRRs are commonly classified as mainly related to the immune system, adaptation, and reproduction, independently from the species under study [20,21]. For instance, significant heterozygous regions detected in Maremmana cattle contain GABRB1, TARSL2, TM2D3, PCSK6, and SNRPA1 genes, which are associated with fitness and reproductive traits [14]. In another four Italian beef breeds, candidate genes related to male fertility, immune response, and survival were detected by Fabbri et al. (2024) [16]. In pigs, heterozygosity investigation remains nowadays understudied, especially in local populations.
This research aims to identify HRRs in a sample of local European and several cosmopolitan pig breeds. The goal is to describe their distribution patterns across the pig genome and conduct enrichment and functional annotation analyses to identify sets of candidate genes that could provide new insights into the importance of HRR in the species. Through the investigation of HRRs, the genetic characterization of pig breeds included in the study (20 local and 3 cosmopolitan breeds) was completed; indeed, the Runs of Homozygosity analysis was performed by Schiavo et al. [22] and Zorc et al. [23]. Additionally, three selection signatures analyses were performed: on the same SNP- chip data by Munoz et al. [22] and on whole-genome sequencing data by Bovo et al. [23] and by Poklukar et al. [24].
2. Materials and Methods
2.1. Animal Sampling
This study included 1144 individual pigs belonging to 23 different breeds collected within the European project TREASURE (https://treasure.kis.si/). The number of animals per breed and per country is reported in Table 1.

Table 1.
Number of animals per country included in the study.
Details on sampling methods, population structure, and differentiation (e.g., Admixture analysis, Multidimensional scaling Analysis) are reported in previous studies [25,26]. All individuals were genotyped with the GGP-70K HD porcine genotyping array containing 68,516 SNPs. Only autosomal SNPs with minor allele frequency (MAF) ≥ 0.01 and individuals with less than 10% missing genotypes were retained for population structure analyses.
2.2. Detection of Heterozygosity-Rich Regions and Gene Annotation
Analysis of HRRs was conducted using the R package detectRUNS v. 0.9.5 [27]. The consecutive method was used, meaning that a scan of the genome, SNP by SNP, was directly carried out. A sensitivity analysis was performed on specific parameters known to affect results in HRR detection, such as the number of opposite and/or missing genotypes allowed [19]. As a result, three arbitrary different scenarios, defined summarizing information on chip density and HRRs parameters commonly found in the bibliography, were applied to the dataset, ordered from the most conservative to the most liberal. Setting parameters a priori could over- or underestimate the number of runs, without knowing the “entity” of this over/underestimation. To find an intermediate restriction in runs detection should be preferable when results fluctuate among scenarios. In Scenario 1 (SC1), no missing or homozygous genotypes were allowed. Scenario 2 (SC2) had intermediate restrictions, where missing and homozygous SNPs were set to 1. Scenario 3 (SC3) was the most relaxed, allowing for 1 missing SNP and 2 homozygous SNPs. A minimum number of 15 SNPs was required for all three scenarios. The minimum length of an HRR was set to 10 kb, and the maximum gap between consecutive homozygous SNPs was 1 Mb for all scenarios.
The HRR length classes were calculated considering five intervals for each scenario: 0 to 0.5 Mb, >0.5 Mb and ≤1 Mb, >1 Mb and ≤2 Mb, >2 Mb and ≤4 Mb, and >4 Mb.
Highly heterozygous genomic regions (HRR islands) were identified by selecting SNPs with an in-HRR frequency > 25%, calculated separately for each breed.
Gene annotation analysis was conducted using the Biomart R package [28]. Each breed gene located within HRR islands was identified, and the most common genes (namely, the genes included in the most recurrent HRR islands) were then searched in the literature. It was determined that HRR islands were considered the most common if they were identified in 10 breeds or more. For the Gene set enrichment analysis, the lists of protein-coding genes were uploaded to STRING 12 [29].
2.3. Between-Breeds Heterozygosity Islands
Principal Components Analysis (PCA) was performed on the entire SNP dataset and then specifically on the SNPs included in the HRR islands to assess between-breed variability captured by heterozygosity segments. This step was useful to understand the power of heterozygous segments in terms of clustering capacity. The PCA was conducted with PLINK v1.9 [30].
A permutation analysis was performed to determine empirical thresholds (p < 0.05) for the similarity in terms of heterozygosity. Each gene within the HRR islands identified through the previously described method was converted into a binomial variable: 1 was assigned if the gene was part of any identified HRR islands and 0 if the HRR was not present in that individual. This transformation was repeated for each breed separately. Subsequently, the values were combined when breed 1 and breed 2 were coded as 1, and a permutation analysis was utilized (N = 100,000) to establish the significance of the pairwise heterozygosity pattern.
The Iberian and Alentejana breeds were excluded from this analysis for their low number of HRRs.
3. Results
3.1. Detection of Heterozygosity-Rich Regions and Gene Annotation
After data quality control, 52,647 SNPs and 1144 individuals were retained to identify HRRs.
The total number of HRRs for each scenario was 19,834; 64,470; and 132,282, respectively, for SC1, SC2, and SC3. Figure 1 illustrates the number of HRRs shared among scenarios (i.e., 1488 common to all three approaches).
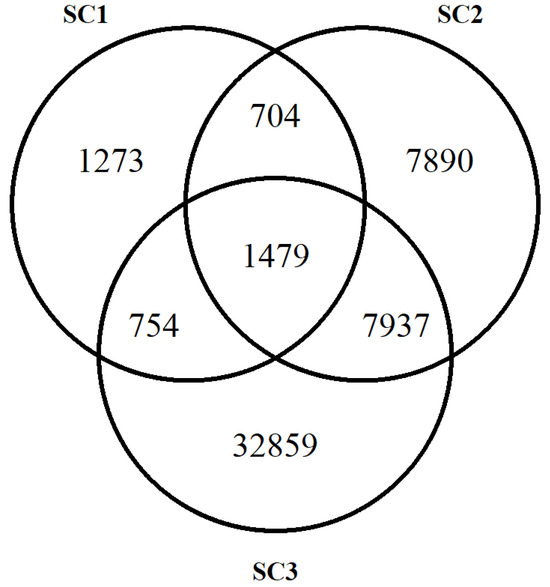
Figure 1.
Total number of HRRs identified in each scenario.
The distribution of HRRs per autosome is reported in Supplementary Figure S1. As we move from SC1 to SC3, the variability in the average number of HRRs per individual among breeds decreases.
Similar results regarding the length classes have been found: the most relaxed approach reduced the variability between breeds (Supplementary Figure S2). In summary, the shortest-length classes were predominant, accounting for approximately 80% of all runs identified (40% of classes from 0 to 0.5 Mb and 40% of classes from 0.5 to 1 Mb). On average, the 1 to 2 Mb class accounted for 15% of the total, and the 2 to 4 Mb class was around 5% of the total. The longest class (>4 Mb) was barely represented, with a maximum of 1%.
Applying the 25% occurrence threshold, 242, 721, and 1477 HRR islands were identified in SC1, SC2, and SC3, respectively. The distribution of HRR islands is reported in Figure 2 for each breed and scenario. Across all three scenarios, the trend among breeds appeared to be consistent, with no fluctuations observed. The breeds with the highest number of HRRs were Italian Large White, Lithuanian Old type, and Italian Landrace, followed by Lithuanian Native, Mora Romagnola, and Italian Duroc. Conversely, the breeds with the lowest number of HRRs were Alentejana, Iberian, and Majorcan Black.
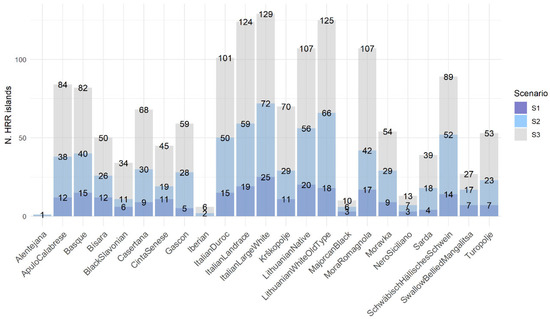
Figure 2.
The number of HRR islands per breed and scenario.
The results of SC2 were selected for further analyses due to the intermediate and more realistic HRR values it provided, taking into account the density of the SNP chip used and the variability previously described that SC2 captured among breeds compared to SC3.
Figure 3 illustrates the number of HRR islands per chromosome in each breed in Scenario 2. HRRs are primarily located in SSC1, SSC2, SSC6, and SSC14. Specifically, Italian Landrace had 17 HRR islands in SSC14, followed by Lithuanian Old Type with 15 HRRs in SSC1, and Large White with 10 in SSC14, along with Swallow-Bellied Mangalitsa and Lithuanian Native, both having 10 HRR islands in SSC6.
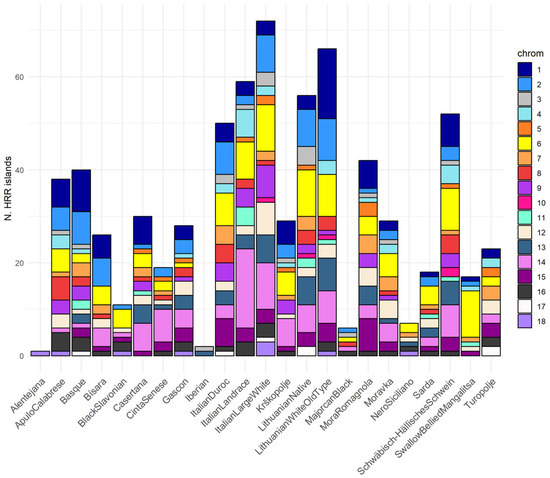
Figure 3.
HRR islands distribution among breeds, divided by autosomes, as determined with Scenario 2.
To avoid underestimating heterozygous regions, it was decided to compare heterozygosity based on genes within the segments rather than solely on the complete overlap of bp positions. An HRR could start at the same bp as an HRR in another breed but end a few bp before it. This lack of overlap could lead to underestimating the heterozygosity shared between the two breeds in the example provided.
There were 2321 unique genes found within the 721 HRR islands, with no regions being recurrent across breeds. Therefore, each breed was analyzed separately, and the percentage of breed-specific genes within HRRs, overlapping with the total of 2321 genes, was calculated and reported in Table 2. The Italian Large White breed had the highest number of genes (916, almost 40% of the total) present in all regions of heterozygosity. The Lithuanian White Old type had the second highest number of genes present in all regions of heterozygosity (31%). The Nero Siciliano and Black Slavonian breeds had the lowest percentage of overlapped heterozygosity compared to the other breeds.
Table 2.
Number of genes per breed within HRR islands and percentage of overlap with the total number of genes identified (2321 genes).
Genes found within the HRR islands shared by at least 10 breeds were uploaded to the STRING software (the complete list is reported in Supplementary Table S1), and the resulting clusters were then investigated in the literature. Figure 4 shows some clusters formed by the genes located in heterozygous regions.

Figure 4.
Clusters of genes found in HRRs islands shared by at least 10 breeds.
Up to 12 genes strongly linked to the TRIM28 gene were identified. Of these, 11 were Zinc Finger genes. This first cluster also included the CDC27 (Cell Division Cycle 27) gene. Two other clusters gathered four genes, namely, ISG15, RPS5, MRPL20, and AURKAIP1, in one cluster and UBE2M, UBE2J2, B3GALT6, and AGRN in another cluster. Finally, NOC2L, PES1, and PUSL1 constituted another cluster besides a pair of genes (ZNF444 and ZNF487) constituting the smallest cluster.
3.2. Between-Breeds Heterozygosity Islands
A total of 721 HRR islands gathering 6288 unique SNPs were used as variables to perform a PCA. Figure 5A illustrates the PCA performed on the entire dataset. Previous papers have largely discussed the genetic distances among breeds [25,26]. It is remarkable that with the whole dataset, some breeds (e.g., Mora Romagnola, Italian Duroc, Basque, Lithuanian Old type, Italian Large White, Gascon) exhibited very low within-breed variability, with individuals plotting in well-defined clusters. When only SNPs of HRR islands were included in the PCA, it was clear how the clustering changed (Figure 5B). The breeds that maintained a similar cluster as previously defined were Mora Romagnola and Italian Duroc. The within-breed variability was extended in almost all other pig breeds.
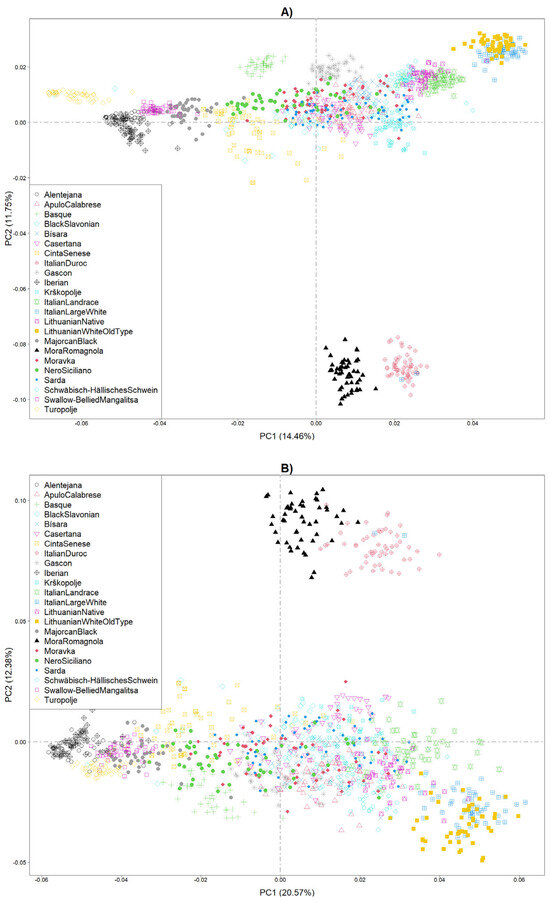
Figure 5.
Principal Components Analysis (PCA) using (A) the entire genomic dataset (n. SNPs = 52,647) or using (B) only SNPs included in HRR islands (n. SNPs = 6228).
Figure 6 visualizes the permutation analysis results on pairwise breeds heterozygosity. In light sea green, the p < 0.05; in grey, p ≥ 0.05 (not significant). Turopolje, Basque, Italian Duroc, Cinta Senese, and Mora Romagnola were the breeds with the highest levels of breed-specific heterozygosity. They showed almost all pairwise comparisons as being dissimilar in terms of heterozygosity. Conversely, Bísara, Black Slavonian, Casertana, Sarda, Nero Siciliano, Swallow-Bellied Mangalitsa, and Schwäbisch Hällisches Schwein showed a high overall degree of similarity in their heterozygosity pattern (p < 0.05).

Figure 6.
Heatmap of p values obtained by permutation analysis for each pairwise breed combination related to heterozygosity patterns (light sea green = p < 0.05; grey = p ≥ 0.05).
4. Discussion
This study is the most comprehensive research on heterozygosity in European pig breeds. Heterozygosity in pigs should be thoroughly studied, particularly due to the interest in cross-breeding and heterosis. It is expected that common heterozygosity patterns will be found, regardless of whether they are investigated in 20 local or 3 cosmopolitan populations, because of the connection between heterozygosity and fitness, as well as heterozygosity and balancing selection. In this study, heterozygosity was analyzed extensively in 23 pig breeds and examined from various perspectives.
4.1. Detection of Heterozygosity-Rich Regions and Gene Annotation
The parameters used in HRR analysis, such as in ROH analysis, can influence results, as demonstrated in previous studies [16], especially regarding the number of detected regions. Testing different scenarios and changing parameters could be a good strategy to observe the consistency and accuracy of results or at least to control potential bias. It has been observed that modifying the number of allowed homozygous and missing SNPs influenced the total number of runs detected but did not alter the trend among breeds.
It is important to note that some breeds did not show HRRs in the three scenarios (Alentejana and Iberian) or had a very limited number (Majorcan Black, Nero Siciliano, Mangalitsa, and Sarda). Increasing the sample size may help address this issue. It is worth mentioning the high degree of admixture in some breeds, such as Nero Siciliano and Sarda [25], which could explain why numerous consecutive heterozygous segments were not perfectly overlapping and are found in more than 25% of the populations.
On the other hand, the breeds with the highest number of HRR islands were the cosmopolitan breeds (Italian Large White, Italian Landrace, and Italian Duroc), the Lithuanian breeds, and the Schwäbisch Hällisches Schwein.
The autosomes that showed a higher number of heterozygosity islands were some of the longest in pigs, as expected, namely, SSC1, SSC2, SSC6, and SSC14. This aligns with the findings reported by Schiavo et al. (2021) [22], as these autosomes were generally unaffected by homozygosity islands in the majority of the breeds. The exceptions were SSC2 and SSC14 for Alentejana; SSC6 and SSC14 for Iberian and Krškopolje; and SSC14 for Swallow-Bellied Mangalitsa, where homozygosity peaks were revealed.
Previous studies have found that HRRs in pigs are mainly related to reproductive traits [19,31] and immune response [20,21,31]. Bordonaro et al. [20] observed that HRRs between different pig breeds or populations (wild boar, Italian local breeds, and cosmopolitan breeds) have been previously identified as segments of fixed homozygosity, suggesting underlying diversity between breeds. The gene enrichment analysis performed in this study sheds light on several interesting genes that cluster together and play important roles, especially in the immune system, as identified in other pig populations [21]. The idea that genes related to the immune system could be the best candidates in terms of heterozygosity hotspots is here reinforced. Only genes in HRR islands found in more than half of the pig breeds included in this study (10 breeds) were tested for network interaction. Up to five clusters were identified (Figure 5). The cluster with the TRIM28 (Tripartite Motif Containing 28) gene positioned in the center seemed particularly interesting. TRIM28 has been identified as an essential regulatory factor in controlling innate antiviral immune responses [32]. This gene is linked to Zinc-finger proteins (ZNFs) that are involved in several cellular processes acting through different molecular mechanisms and playing key roles in the development and differentiation of several tissues; indeed, they have been defined as the guardians of genome stability [33].
Other genes linked to the immune system belonged to the other clusters identified, namely, the ISG15 (interferon-stimulated genes 15), MRPL20 (Mitochondrial Ribosomal Protein L20), and AURKAIP1 (Aurora Kinase A Interacting Protein 1) genes. ISG15 gene expression was found to be induced by Interferons (IFNs), and it is a central player in the host antiviral response (e.g., ISG15 is found to be upregulated during classical swine fever infection [34]). In the same cluster, a study on cattle found MRPL20 and AURKAIP1 within the genomic window where the SNP was significantly associated with Mycobacterium avium ssp. Paratuberculosis infection status [35].
The last cluster containing four genes was formed by B3GALT6, AGRN, and two Ubiquitin Conjugating Enzyme E2, M and J2 (UBE2M and UBE2J2, respectively). It is important to underline that the exploitation of the ubiquitin system by viruses represents a central theme, and several studies highlight the use of ubiquitin inhibitors as an antiviral approach [36].
Genes NOC2L (Nucleolar Associated Transcriptional Repressor) and PUSL1 (pseudouridine synthase-like 1) have also been identified in a genomic region as significantly associated with bacterial infection in cattle [35]. These genes should be further studied since local pig breeds are known to be more persistent and resilient to harsh environments.
It was interesting that all these aforementioned genes were found in significant HRRs in Apulo Calabrese, Bísara, Black Slavonian, Casertana, Large White, Lithuanian breeds, Majorcan Black, Moravka, Schwäbisch Hällisches Schwein, and Swallow-Bellied Mangalitsa. Some of those genes related to the immune system were found in heterozygous segments in Nero Siciliano, Sarda, and Krškopolje, making these genomic regions extremely interesting, as they are common to several European pig populations analyzed. Moreover, compared to cattle and sheep populations, few studies on HRRs islands have been performed on pigs, so similarities or comparisons are difficult to address within and between breeds, both in terms of numbers and in terms of genes identified.
4.2. Between-Breeds Heterozygosity Islands
Two approaches were used to define the heterozygosity patterns in these 23 pig breeds: Principal Components Analysis and correlation. In PCA, the variables were the total number of SNPs in all HRR islands across the 23 breeds investigated. In the correlation approach, genes were classified as 1 (presence of an HRR in that location) or 0 (no HRR revealed in that location) for each breed separately. The results from both approaches are complementary and serve different purposes. PCA aimed to determine if HRR islands could distinguish breeds based on genetic variability and, potentially, fitness indirectly associated with the level of heterozygosity. The correlation aimed to measure the similarity of heterozygosity patterns among breeds (considering only segments where genes are located).
PCA results based on heterozygous segments indicated that, for most pig breeds, heterozygosity regions did not perfectly define the genetic differences among breeds. While it did not drastically change the genetic diversity picture, it did not show that heterozygous islands reduced differences between breeds and increased variability within breeds. It would be beneficial to test the same approach using homozygosity.
Interestingly, none of the 23 European pig breeds had the same HRR islands, which could be expected due to the putative link between heterozygosity and fitness-related traits [37].
Regarding the permutation results, patterns of heterozygosity shared among breeds were revealed (Figure 6), but several pairwise comparisons of breeds also identified breed-specific heterozygosity.
Schiavo et al. [22] reported the pairwise similarities between breeds based on ROH islands, and the results are consistent and complementary to what is reported in this study. Generally, breeds that showed low similarity in homozygosity patterns also maintained the dissimilarity in heterozygosity, representing a true heritage of genetic diversity. These local breeds include Turopolje, Basque, and Mora Romagnola, followed by Cinta Senese, Nero Siciliano, Sarda, Moravka, Bísara, and Black Slavonian. Despite being breeds found by Dadousis et al. [25] to be highly admixed, they are shown to have strong similar heterozygosity and homozygosity [22], as well as Swallow-Bellied Mangalitsa and Schwäbisch Hällisches Schwein.
5. Conclusions
This study is the first to investigate heterozygosity patterns in such a high number of European local pig breeds. The trends and distribution of heterozygosity differed among breeds, and no regions recurred in all 23 breeds, but some common patterns of heterozygosity have been revealed. The discriminant power of heterozygosity has been evaluated here, suggesting that a certain degree of breed-specific heterozygosity exists. Still, it is insufficient to characterize breeds if considered as the only evolutionary force applied. Interestingly, permutation analysis indicated that some local breeds serve as true reservoirs of genetic diversity, displaying distinct and unique characteristics in terms of heterozygosity. In addition, genes in heterozygous islands should be further studied due to their relationship with the immune response and, consequently, with the resilience trait, a unique feature of local breeds. Finally, heterozygosity has been confirmed to be an essential aspect to investigate to complete and clearly characterize populations.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/agriculture15070761/s1, Figure S1: Average number of HRRs per autosomes and chromosomes in each pig breed; Figure S2: Frequency of HRRs for classes of length in each pig breed; Table S1: Genes per breed.
Author Contributions
Conceptualization, M.C.F. and K.D.A.; methodology, M.C.F., K.D.A. and F.G.; software, M.C.F.; validation, F.T. and S.B. (Stefano Biffani); formal analysis, M.C.F.; investigation, K.D.A. and M.P.G.R.; resources, R.B., L.F. and M.Č.-P.; writing—original draft preparation, M.C.F., K.D.A. and M.P.G.R.; writing—review and editing, K.D.A., M.P.G.R., F.T., S.B. (Stefano Biffani), F.G., G.S., S.B. (Samuele Bovo), L.F., M.Č.-P., M.M., C.O., K.P., M.Š. and R.B.; supervision, F.T., S.B. (Stefano Biffani) and R.B.; funding acquisition, M.Č.-P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the European Union’s Horizon 2020 research and innovation program under grant agreement no. 634476 to C.O. (project acronym TREASURE).
Data Availability Statement
The data presented in this study are available on reasonable request from the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Megens, H.-J.; Crooijmans, R.P.M.A.; Cristobal, M.S.; Hui, X.; Li, N.; Groenen, M.A. Biodiversity of pig breeds from China and Europe estimated from pooled DNA samples: Differences in microsatellite variation between two areas of domestication. Genet. Sel. Evol. 2008, 40, 103–128. [Google Scholar] [CrossRef] [PubMed]
- Arias, K.D.; Fernández, I.; Gutiérrez, J.P.; Bozzi, R.; Álvarez, I.; Goyache, F. Characterizing local pig breeds as reservoirs for the domestic pig genetic variability worldwide via contributions to gene diversity and allelic richness. J. Anim. Sci. 2024, 102, skae329. [Google Scholar] [CrossRef] [PubMed]
- Buchanan Smith, A.D.; Robison, O.J.; Bryant, D.M. The Genetics of the Pig; Bibliographia Genetica: The Hague, The Netherlands, 2016; Available online: https://www.abebooks.com/Genetics-Pig-Buchanan-Smith-Robison-Bryant/30294075136/bd (accessed on 7 November 2024).
- Yang, S.-L.; Wang, Z.-G.; Liu, B.; Zhang, G.-X.; Zhao, S.-H.; Yu, M.; Fan, B.; Li, M.-H.; Xiong, T.-A.; Li, K. Genetic variation and relationships of eighteen Chinese indigenous pig breeds. Genet. Sel. Evol. 2003, 35, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Cui, L.; Perez-Enciso, M.; Traspov, A.; Crooijmans, R.P.M.A.; Zinovieva, N.; Schook, L.B.; Archibald, A.; Gatphayak, K.; Knorr, C.; et al. Genome-wide SNP data unveils the globalization of domesticated pigs. Genet. Sel. Evol. 2017, 49, 71. [Google Scholar] [CrossRef]
- Hague, M.T.J.; Routman, E.J. Does population size affect genetic diversity? A test with sympatric lizard species. Heredity 2015, 116, 92–98. [Google Scholar] [CrossRef]
- Larson, G.; Dobney, K.; Albarella, U.; Fang, M.; Matisoo-Smith, E.; Robins, J.; Lowden, S.; Finlayson, H.; Brand, T.; Willerslev, E.; et al. Worldwide Phylogeography of Wild Boar Reveals Multiple Centers of Pig Domestication. Science 2005, 307, 1618–1621. [Google Scholar] [CrossRef]
- Larson, G.; Albarella, U.; Dobney, K.; Rowley-Conwy, P.; Schibler, J.; Tresset, A.; Vigne, J.-D.; Edwards, C.J.; Schlumbaum, A.; Dinu, A.; et al. Ancient DNA, pig domestication, and the spread of the Neolithic into Europe. Proc. Natl. Acad. Sci. USA 2007, 104, 15276–15281. [Google Scholar] [CrossRef]
- Bizarria Dos Santos, W.; Pimenta Schettini, G.; Fonseca, M.G.; Pereira, G.L.; Loyola Chardulo, L.A.; Rodrigues Machado Neto, O.; Baldassini, W.A.; Nunes De Oliveira, H.; Abdallah Curi, R. Fine-scale estimation of inbreeding rates, runs of homozygosity and genome-wide heterozygosity levels in the Mangalarga Marchador horse breed. J. Anim. Breed. Genet. 2020, 138, 161–173. [Google Scholar] [CrossRef]
- Hedrick, P.W. What is the evidence for heterozygote advantage selection? Trends. Ecol. Evol. 2012, 27, 698–704. [Google Scholar] [CrossRef]
- Samuels, D.C.; Wang, J.; Ye, F.; He, J.; Levinson, R.T.; Sheng, Q.; Zhao, S.; A Capra, J.; Shyr, Y.; Zheng, W.; et al. Heterozygosity Ratio, a Robust Global Genomic Measure of Autozygosity and Its Association with Height and Disease Risk. Genetics 2016, 204, 893–904. [Google Scholar] [CrossRef]
- Williams, J.L.; Hall, S.J.G.; Del Corvo, M.; Ballingall, K.T.; Colli, L.; Ajmone Marsan, P.; Biscarini, F. Inbreeding and purging at the genomic Level: The Chillingham cattle reveal extensive, non-random SNP heterozygosity. Anim. Genet. 2015, 47, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Ferenčaković, M.; Banadinović, M.; Mercvajler, M.; Khayat-zadeh, N.; Mészáros, G.; Cubric-Curik, V.; Curik, I.; Sölkner, J. Mapping of heterozygosity rich regions in Austrian Pinzgauer cattle. Acta. Agric. Slov. 2016, 5, 41–44. [Google Scholar] [CrossRef]
- Biscarini, F.; Mastrangelo, S.; Catillo, G.; Senczuk, G.; Ciampolini, R. Insights into Genetic Diversity, Runs of Homozygosity and Heterozygosity-Rich Regions in Maremmana Semi-Feral Cattle Using Pedigree and Genomic Data. Animals 2020, 10, 2285. [Google Scholar] [CrossRef]
- Mulim, H.A.; Brito, L.F.; Batista Pinto, L.F.; Moletta, J.L.; Da Silva, L.R.; Pedrosa, V.B. Genetic and Genomic Characterization of a New Beef Cattle Composite Breed (Purunã) Developed for Production in Pasture-Based Systems. Front. Genet. 2022, 13, 858970. [Google Scholar] [CrossRef]
- Fabbri, M.C.; Tiezzi, F.; Crovetti, A.; Maltecca, C.; Bozzi, R. Investigation of cosmopolitan and local Italian beef cattle breeds uncover common patterns of heterozygosity. Animal 2024, 18, 101142. [Google Scholar] [CrossRef]
- Arias, K.D.; Gutiérrez, J.P.; Fernández, I.; Álvarez, I.; Goyache, F. Approaching autozygosity in a small pedigree of Gochu Asturcelta pigs. Genet. Sel. Evol. 2023, 55, 74. [Google Scholar] [CrossRef]
- Tsartsianidou, V.; Sánchez-Molano, E.; Kapsona, V.V.; Basdagianni, Z.; Chatziplis, D.; Arsenos, G.; Triantafyllidis, A.; Banos, G. A comprehensive genome-wide scan detects genomic regions related to local adaptation and climate resilience in Mediterranean domestic sheep. Genet. Sel. Evol. 2021, 53, 90. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, Z.; Wang, Z.; Zhang, Z.; Wang, Q.; Pan, Y. Heterozygosity and homozygosity regions affect reproductive success and the loss of reproduction: A case study with litter traits in pigs. Comput. Struct. Biotechnol. J. 2022, 20, 4060–4071. [Google Scholar] [CrossRef]
- Bordonaro, S.; Chessari, G.; Mastrangelo, S.; Senczuk, G.; Chessa, S.; Castiglioni, B.; Tumino, S.; Marletta, D.; Criscione, A. Genome-wide population structure, homozygosity, and heterozygosity patterns of Nero Siciliano pig in the framework of Italian and cosmopolitan breeds. Anim. Genet. 2023, 54, 591–605. [Google Scholar] [CrossRef]
- Liu, S.Q.; Xu, Y.J.; Chen, Z.T.; Li, H.; Zhang, Z.; Wang, Q.S.; Pan, Y.C. Genome-Wide Detection of Runs of Homozygosity and Heterozygosity in Tunchang pigs. Animal 2024, 18, 101236. [Google Scholar] [CrossRef]
- Schiavo, G.; Bovo, S.; Muñoz, M.; Ribani, A.; Alves, E.; Araújo, J.P.; Bozzi, R.; Čandek-Potokar, M.; Charneca, R.; Fernandez, A.I.; et al. Runs of homozygosity provide a genome landscape picture of inbreeding and genetic history of European autochthonous and commercial pig breeds. Anim. Genet. 2021, 52, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Zorc, M.; Škorput, D.; Gvozdanović, K.; Margeta, P.; Karolyi, D.; Luković, Z.; Salajpal, K.; Savić, R.; Muñoz, M.; Bovo, S.; et al. Genetic diversity and population structure of six autochthonous pig breeds from Croatia, Serbia, and Slovenia. Genet. Sel. Evol. 2022, 54, 30. [Google Scholar] [CrossRef] [PubMed]
- Poklukar, K.; Mestre, C.; Škrlep, M.; Čandek-Potokar, M.; Ovilo, C.; Fontanesi, L.; Riquet, J.; Bovo, S.; Schiavo, G.; Ribani, A.; et al. A meta-analysis of genetic and phenotypic diversity of European local pig breeds reveals genomic regions associated with breed differentiation for production traits. Genet. Sel. Evol. 2023, 55, 88. [Google Scholar] [CrossRef] [PubMed]
- Dadousis, C.; Muñoz, M.; Óvilo, C.; Fabbri, M.C.; Araújo, J.P.; Bovo, S.; Potokar, M.Č.; Charneca, R.; Crovetti, A.; Gallo, M.; et al. Admixture and breed traceability in European indigenous pig breeds and wild boar using genome-wide SNP data. Sci. Rep. 2022, 12, 7346. [Google Scholar] [CrossRef]
- Muñoz, M.; Bozzi, R.; García, F.; Núñez, Y.; Geraci, C.; Crovetti, A.; García-Casco, J.; Alves, E.; Škrlep, M.; Charneca, R.; et al. Diversity across major and candidate genes in European local pig breeds. PLOS ONE 2018, 13, e0207475. [Google Scholar] [CrossRef]
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes; CRAN: Vienna, Austria, 2019. [Google Scholar]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Ruan, D.; Yang, J.; Zhuang, Z.; Ding, R.; Huang, J.; Quan, J.; Gu, T.; Hong, L.; Zheng, E.; Li, Z.; et al. Assessment of Heterozygosity and Genome-Wide Analysis of Heterozygosity Regions in Two Duroc Pig Populations. Front. Genet. 2022, 12, 812456. [Google Scholar] [CrossRef]
- Hua, F.; Nass, T.; Parvatiyar, K. TRIM28 facilitates type I interferon activation by targeting TBK1. Front. Immunol. 2024, 15, 1279920. [Google Scholar] [CrossRef]
- Kamaliyan, Z.; Clarke, T.L. Zinc finger proteins: Guardians of genome stability. Front. Cell Dev. Biol. 2024, 12, 1448789. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Zheng, H.; Dong, W.; Lv, H.; Lin, J.; Guo, K.; Zhang, Y. Antiviral activity of ISG15 against classical swine fever virus replication in porcine alveolar macrophages via inhibition of autophagy by ISGylating BECN1. Vet. Res. 2020, 51, 22. [Google Scholar] [CrossRef] [PubMed]
- Mallikarjunappa, S.; Sargolzaei, M.; Brito, L.F.; Meade, K.G.; Karrow, N.A.; Pant, S.D. Short communication: Uncovering quantitative trait loci associated with resistance to Mycobacterium avium ssp. paratuberculosis infection in Holstein cattle using a high-density single nucleotide polymorphism panel. J. Dairy Sci. 2018, 101, 7280–7286. [Google Scholar] [CrossRef] [PubMed]
- Freitas, F.B.; Frouco, G.; Martins, C.; Ferreira, F. African swine fever virus encodes for an E2-ubiquitin conjugating enzyme that is mono- and di-ubiquitinated and required for viral replication cycle. Sci. Rep. 2018, 8, 3471. [Google Scholar] [CrossRef]
- Chessari, G.; Criscione, A.; Marletta, D.; Crepaldi, P.; Portolano, B.; Manunza, A.; Cesarani, A.; Biscarini, F.; Mastrangelo, S. Characterization of heterozygosity-rich regions in Italian and worldwide goat breeds. Sci. Rep. 2024, 14, 3. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).