Transcriptomics Uncovers Pathways Mediating Low-Nitrogen Stress Tolerance in Two Foxtail Millet Varieties

Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Materials
2.2. Experimental Design
2.2.1. Field Experiment
2.2.2. Seedling Hydroponic Experiment
2.2.3. Evaluation of Foxtail Millet Variety Tolerance to Low Nitrogen
2.2.4. Transcriptome Sequencing
2.2.5. Validation of DEGs Using qRT-PCR
2.3. Statistical Analysis
3. Results
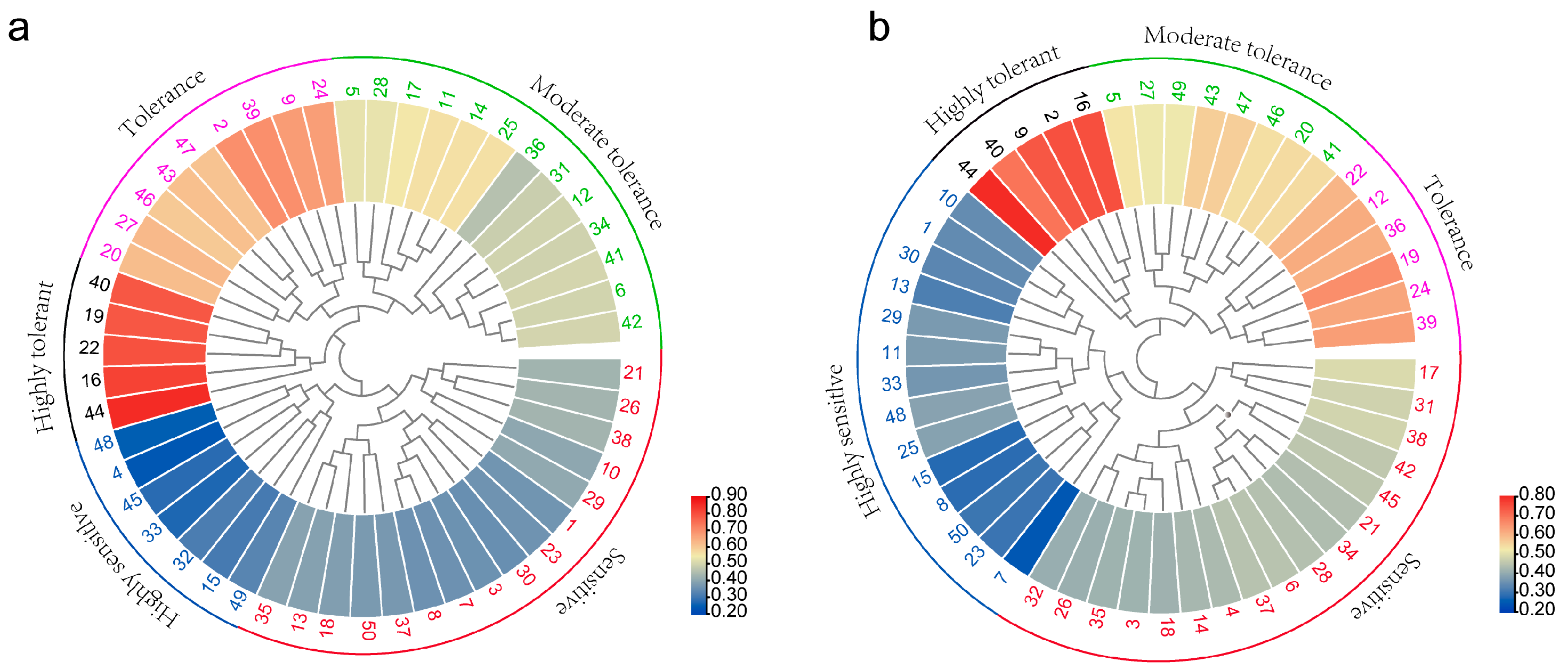
3.1. Analysis of Foxtail Millet Tolerance to Low Nitrogen at the Field and Seeding Hydroponic Stage
3.2. Correlation Analysis Between Investigated Traits
3.3. Transcriptome Analysis of Foxtail Millet Under Control and Low Nitrogen Conditions
3.4. GO and KEGG Enrichment Analyses of DEGs
3.5. Transcription Factors Among DEGs
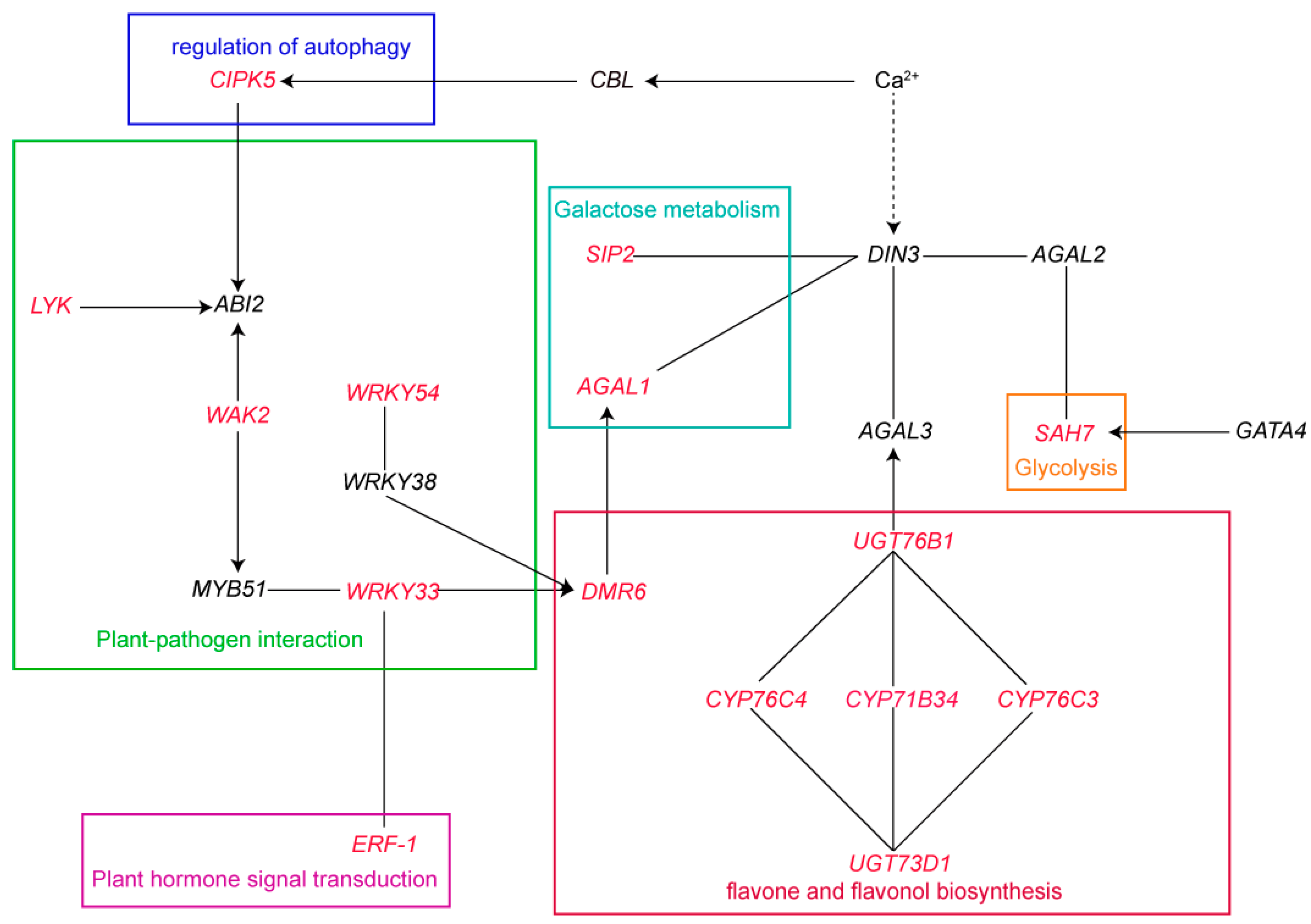
3.6. DEGs Are Involved in Important Pathways
3.7. Validation of Some Important DEGs Using qRT-PCR
4. Discussion
4.1. Morphological Trait Indexes for the Evaluation of NUE
4.2. Multiple Pathways Contribute to Regulating the Low Nitrogen Stress Response in Maotigu and Dahuanggu
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, G.; Fan, X.; Miller, A.J. Plant nitrogen assimilation and use efficiency. Annu. Rev. Plant Biol. 2012, 63, 153–182. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Ma, T.; Xian, W.; Hu, B.; Chu, C. Interplay between ethylene and nitrogen nutrition: How ethylene orchestrates nitrogen responses in plants. J. Integr. Plant Biol. 2023, 65, 399–407. [Google Scholar] [CrossRef]
- Frels, K.; Guttieri, M.; Joyce, B.; Leavitt, B.; Baenziger, P.S. Evaluating canopy spectral reflectance vegetation indices to estimate nitrogen use traits in hard winter wheat. Field Crops Res. 2018, 217, 82–92. [Google Scholar] [CrossRef]
- Lam, H.M.; Coschigano, K.T.; Oliveira, I.C.; Melo-Oliveira, R.; Coruzzi, G.M. The molecular-genetics of nitrogen assimilation into amino acids in higher plants. Annu. Rev. Plant Biol. 1996, 47, 569–593. [Google Scholar] [CrossRef]
- Islam, S.; Zhang, J.; Zhao, Y.; She, M.; Ma, W. Genetic regulation of the traits contributing to wheat nitrogen use efficiency. Plant Sci. Int. J. Exp. Plant Biol. 2021, 303, 110759. [Google Scholar] [CrossRef]
- Vidal, E.A.; Alvarez, J.M.; Araus, V.; Riveras, E.; Brooks, M.D.; Krouk, G.; Ruffel, S.; Lejay, L.; Crawford, N.M.; Coruzzi, G.M. Nitrate in 2020: Thirty Years from Transport to Signaling Networks. Plant Cell 2020, 32, 2094–2119. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Tan, H.; Shan, M.; Duan, M.; Ye, L.; Yang, Y.; He, L.; Shen, H.; Yang, Z.; Wang, X. Genome-wide identification and characterization of the NPF genes provide new insight into low nitrogen tolerance in Setaria. Front. Plant Sci. 2022, 13, 1043832. [Google Scholar] [CrossRef]
- Yanagisawa, S. Transcription factors involved in controlling the expression of nitrate reductase genes in higher plants. Plant Sci. 2014, 229, 167–171. [Google Scholar] [CrossRef]
- Uauy, C.; Distelfeld, A.; Fahima, T.; Blechl, A.; Dubcovsky, J. A NAC Gene Regulating Senescence Improves Grain Protein, Zinc, and Iron Content in Wheat. Science 2006, 314, 1298–1301. [Google Scholar] [CrossRef]
- Jiading, Y.; Eric, W.; Torres-Jerez, I.; Randall, M.; Mingyi, W.; Chunxiang, F.; Zeng-Yu, W.; Yuhong, T.; Michael, U. PvNAC1 and PvNAC2 are associated with leaf senescence and nitrogen use efficiency in switchgrass. BioEnergy Res. 2014, 8, 868–880. [Google Scholar] [CrossRef]
- Ristova, D.; Carré, C.; Pervent, M.; Medici, A.; Kim, G.J.; Scalia, D.; Ruffel, S.; Birnbaum, K.D.; Lacombe, B.; Busch, W.; et al. Combinatorial interaction network of transcriptomic and phenotypic responses to nitrogen and hormones in the Arabidopsis thaliana root. Sci. Signal. 2016, 9, rs13. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Wu, D.; Li, Y.; Tao, W.; Chao, L.; Zhang, Y. The role of auxin in nitrogen-modulated shoot branching. Plant Signal. Behav. 2021, 16, 1885888. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, H. Cytokinin biosynthesis and transport for systemic nitrogen signaling. Plant J. Cell Mol. Biol. 2021, 105, 421–430. [Google Scholar] [CrossRef]
- Smoczynska, A.; Pacak, A.; Grabowska, A.; Bielewicz, D.; Zadworny, M.; Singh, K.; Dolata, J.; Bajczyk, M.; Nuc, P.; Kesy, J.; et al. Excess nitrogen responsive HvMADS27 transcription factor controls barley root architecture by regulating abscisic acid level. Front. Plant Sci. 2022, 13, 950796. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Wang, S.; Song, W.; Zhang, J.; Wang, Y.; Liu, Q.; Yu, J.; Ye, Y.; Li, S.; Chen, J.; et al. Enhanced sustainable green revolution yield via nitrogen-responsive chromatin modulation in rice. Science 2020, 367, 6478. [Google Scholar] [CrossRef]
- Souza, L.C.; Monteiro, G.; Marinho, R.K.M.; Souza, E.F.L.; Oliveira, S.C.F.; Ferreira, A.C.S.; Oliveira Neto, C.F.; Okumura, R.S.; Souza, L.C. Nitrogen metabolism in maize plants submitted to drought, brassinosteroids and azospirillum. Braz. J. Biol. 2023, 83, e276264. [Google Scholar] [CrossRef]
- Wu, X.; Ding, C.; Baerson, S.R.; Lian, F.; Lin, X.; Zhang, L.; Wu, C.; Hwang, S.Y.; Zeng, R.; Song, Y. The roles of jasmonate signalling in nitrogen uptake and allocation in rice (Oryza sativa L.). Plant Cell Environ. 2019, 42, 659–672. [Google Scholar] [CrossRef]
- Conesa, C.M.; Saez, A.; Navarro-Neila, S.; de Lorenzo, L.; Hunt, A.G.; Sepúlveda, E.B.; Baigorri, R.; Garcia-Mina, J.M.; Zamarreño, A.M.; Sacristán, S.; et al. Alternative Polyadenylation and Salicylic Acid Modulate Root Responses to Low Nitrogen Availability. Plants 2020, 9, 251. [Google Scholar] [CrossRef]
- Sigalas, P.P.; Buchner, P.; Thomas, S.G.; Jamois, F.; Arkoun, M.; Yvin, J.-C.; Bennett, M.J.; Hawkesford, M.J. Nutritional and tissue-specific regulation of cytochrome P450 CYP711A MAX1 homologues and strigolactone biosynthesis in wheat. J. Exp. Bot. 2023, 74, 1890–1910. [Google Scholar] [CrossRef]
- Signora, L.; De Smet, I.; Foyer, C.H.; Zhang, H. ABA plays a central role in mediating the regulatory effects of nitrate on root branching in Arabidopsis. Plant J. 2001, 28, 655–662. [Google Scholar] [CrossRef]
- Gou, J.; Strauss, S.H.; Tsai, C.J.; Fang, K.; Chen, Y.; Jiang, X.; Busov, V.B. Gibberellins regulate lateral root formation in Populus through interactions with auxin and other hormones. Plant Cell 2010, 22, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Liu, F.; Crawford, N.M.; Wang, Y. Molecular Regulation of Nitrate Responses in Plants. Int. J. Mol. Sci. 2018, 19, 2039. [Google Scholar] [CrossRef]
- Mauriat, M.; Petterle, A.; Bellini, C.; Moritz, T. Gibberellins inhibit adventitious rooting in hybrid aspen and Arabidopsis by affecting auxin transport. Plant J. 2014, 78, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Nacry, P.; Bouguyon, E.; Gojon, A. Nitrogen acquisition by roots: Physiological and developmental mechanisms ensuring plant adaptation to a fluctuating resource. Plant Soil 2013, 370, 1–29. [Google Scholar] [CrossRef]
- Xing, J.; Cao, X.; Zhang, M.; Wei, X.; Zhang, J.; Wan, X. Plant nitrogen availability and crosstalk with phytohormones signallings and their biotechnology breeding application in crops. Plant Biotechnol. J. 2023, 21, 1320–1342. [Google Scholar] [CrossRef]
- Kjeldahl, J. Neue Methode zur Bestimmung des Stickstoffs in organischen Körpern. Zeitschrift für Analytische Chemie 1883, 22, 366–382. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Qin, L.; Li, F.; Yang, Y.; Liu, Z.; Wang, R.; Yu, X.; Niu, J.; Zhang, H.; Wang, H.; et al. Physiological and Transcriptomic Analysis Provides Insights into Low Nitrogen Stress in Foxtail Millet (Setaria italica L.). Int. J. Mol. Sci. 2023, 24, 16321. [Google Scholar] [CrossRef]
- Bandyopadhyay, T.; Maurya, J.; Bentley, A.R.; Griffiths, H.; Swarbreck, S.M.; Prasad, M. Identification of the mechanistic basis of nitrogen responsiveness in two contrasting Setaria italica accessions. J. Exp. Bot. 2024, 75, 5008–5020. [Google Scholar] [CrossRef]
- Brasier, K.; Ward, B.; Smith, J.; Seago, J.; Oakes, J.; Balota, M.; Davis, P.; Fountain, M.; Brown-Guedira, G.; Sneller, C.; et al. Identification of quantitative trait loci associated with nitrogen use efficiency in winter wheat. PLoS ONE 2020, 15, e0228775. [Google Scholar] [CrossRef]
- Yan, H.; Shi, H.; Hu, C.; Luo, M.; Xu, C.; Wang, S.; Li, N.; Tang, W.; Zhou, Y.; Wang, C.; et al. Transcriptome Differences in Response Mechanisms to Low-Nitrogen Stress in Two Wheat Varieties. Int. J. Mol. Sci. 2021, 22, 12278. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Yang, J.; Yu, N.; Luo, L.; Wang, E. Biological nitrogen fixation in cereal crops: Progress, strategies, and perspectives. Plant Commun. 2023, 4, 100499. [Google Scholar] [CrossRef]
- Scheible, W.R.; Morcuende, R.; Czechowski, T.; Fritz, C.; Osuna, D.; Palacios-Rojas, N.; Schindelasch, D.; Thimm, O.; Udvardi, M.K.; Stitt, M. Genome-wide reprogramming of primary and secondary metabolism, protein synthesis, cellular growth processes, and the regulatory infrastructure of Arabidopsis in response to nitrogen. Plant Physiol. 2004, 136, 2483–2499. [Google Scholar] [CrossRef]
- Léran, S.; Edel, K.H.; Pervent, M.; Hashimoto, K.; Corratgé-Faillie, C.; Offenborn, J.N.; Tillard, P.; Gojon, A.; Kudla, J.; Lacombe, B. Nitrate sensing and uptake in Arabidopsis are enhanced by ABI2, a phosphatase inactivated by the stress hormone abscisic acid. Sci. Signal. 2015, 8, ra43. [Google Scholar] [CrossRef] [PubMed]
- Gaudinier, A.; Rodriguez-Medina, J.; Zhang, L.; Olson, A.; Liseron-Monfils, C.; Bågman, A.-M.; Foret, J.; Abbitt, S.; Tang, M.; Li, B.; et al. Transcriptional regulation of nitrogen-associated metabolism and growth. Nature 2018, 563, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Wada, S.; Hayashida, Y.; Izumi, M.; Kurusu, T.; Hanamata, S.; Kanno, K.; Kojima, S.; Yamaya, T.; Kuchitsu, K.; Makino, A.; et al. Autophagy supports biomass production and nitrogen use efficiency at the vegetative stage in rice. Plant Physiol. 2015, 168, 60–73. [Google Scholar] [CrossRef]
- Di Berardino, J.; Marmagne, A.; Berger, A.; Yoshimoto, K.; Cueff, G.; Chardon, F.; Masclaux-Daubresse, C.; Reisdorf-Cren, M. Autophagy controls resource allocation and protein storage accumulation in Arabidopsis seeds. J. Exp. Bot. 2018, 69, 1403–1414. [Google Scholar] [CrossRef]
- Li, W.; Chen, M.; Wang, E.; Hu, L.; Hawkesford, M.J.; Zhong, L.; Chen, Z.; Xu, Z.; Li, L.; Zhou, Y.; et al. Genome-wide analysis of autophagy-associated genes in foxtail millet (Setaria italica L.) and characterization of the function of SiATG8a in conferring tolerance to nitrogen starvation in rice. BMC Genom. 2016, 17, 797. [Google Scholar] [CrossRef]
- Zhang, T.T.; Lin, Y.J.; Liu, H.F.; Liu, Y.Q.; Zeng, Z.F.; Lu, X.Y.; Li, X.W.; Zhang, Z.L.; Zhang, S.; You, C.X.; et al. The AP2/ERF transcription factor MdDREB2A regulates nitrogen utilisation and sucrose transport under drought stress. Plant Cell Environ. 2024, 47, 1668–1684. [Google Scholar] [CrossRef]
- Lian, X.; Zhao, X.; Zhao, Q.; Wang, G.; Li, Y.; Hao, Y. MdDREB2A in apple is involved in the regulation of multiple abiotic stress responses. Hortic. Plant J. 2021, 7, 197–208. [Google Scholar] [CrossRef]
- Shin, J.M.; Chung, K.; Sakamoto, S.; Kojima, S.; Yeh, C.M.; Ikeda, M.; Mitsuda, N.; Ohme-Takagi, M. The chimeric repressor for the GATA4 transcription factor improves tolerance to nitrogen deficiency in Arabidopsis. Plant Biotechnol. 2017, 34, 151–158. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Control Nitrogen Treatment | Low Nitrogen Treatment | Low Nitrogen Tolerance Coefficient | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | ±SD | CV (%) | Mean | ±SD | CV (%) | Mean | ±SD | CV (%) | |
| Seeding trait | |||||||||
| RL | 13.10 B | 2.06 | 15.70 | 14.57 A | 2.73 | 18.76 | 1.13 | 0.22 | 19.18 |
| SH | 35.40 A | 4.90 | 13.83 | 22.95 B | 5.26 | 22.93 | 0.65 | 0.12 | 17.89 |
| RN | 12.55 A | 1.98 | 15.83 | 8.83 B | 1.33 | 15.03 | 0.71 | 0.09 | 12.43 |
| TLL | 21.66 A | 3.06 | 14.14 | 11.71 B | 2.76 | 23.55 | 0.54 | 0.11 | 19.51 |
| TLW | 0.94 A | 0.14 | 14.34 | 0.63 B | 0.12 | 19.31 | 0.67 | 0.10 | 14.55 |
| TLN | 5.82 A | 0.42 | 7.22 | 4.60 B | 0.56 | 11.99 | 0.79 | 0.08 | 10.05 |
| RDW | 0.13 A | 0.04 | 26.25 | 0.06 B | 0.03 | 39.25 | 0.47 | 0.17 | 36.11 |
| SDW | 0.03 A | 0.01 | 34.44 | 0.02 B | 0.01 | 36.53 | 0.60 | 0.20 | 33.03 |
| RRD | 0.20 B | 0.04 | 20.78 | 0.25 A | 0.06 | 23.06 | 1.34 | 0.38 | 28.16 |
| PDW | 0.16 A | 0.04 | 26.58 | 0.08 B | 0.29 | 37.58 | 0.49 | 0.17 | 33.99 |
| SNC | 7.23 A | 1.42 | 19.67 | 3.56 B | 0.87 | 24.36 | 0.49 | 0.12 | 23.42 |
| RNC | 5.10 A | 1.01 | 19.85 | 3.19 B | 0.63 | 19.79 | 0.64 | 0.14 | 22.35 |
| PNC | 6.91 A | 1.32 | 19.14 | 3.48 B | 0.74 | 21.28 | 0.51 | 0.11 | 21.64 |
| SNA | 1.00 A | 0.34 | 33.94 | 0.23 B | 0.13 | 57.87 | 0.24 | 0.12 | 48.21 |
| RNAC | 0.14 A | 0.05 | 32.66 | 0.05 B | 0.02 | 37.85 | 0.38 | 0.14 | 38.14 |
| PNA | 1.14 A | 0.37 | 31.16 | 0.28 B | 0.15 | 52.97 | 0.26 | 0.11 | 44.34 |
| SNPE | 142.24 B | 29.73 | 20.48 | 290.98 A | 69.33 | 23.2 | 2.10 | 0.48 | 22.88 |
| RNPE | 206.18 B | 42.72 | 20.72 | 317.97 A | 59.08 | 18.58 | 1.58 | 0.33 | 20.85 |
| PNPE | 151.17 B | 29.86 | 19.76 | 301.63 A | 61.07 | 20.25 | 2.04 | 0.43 | 21.22 |
| SPAD | 30.19 A | 3.09 | 10.25 | 18.07 B | 3.06 | 16.92 | 0.60 | 0.09 | 15.59 |
| Field trait | |||||||||
| PH-2019 | 138.41A | 28.63 | 20.69 | 122.33 B | 24.63 | 20.14 | 0.89 | 0.04 | 4.97 |
| SL-2019 | 27.64 A | 5.34 | 19.30 | 23.79 B | 4.40 | 18.50 | 0.87 | 0.07 | 8.30 |
| LSN-2019 | 29.63 A | 4.98 | 16.82 | 25.71 B | 4.83 | 18.79 | 0.87 | 0.09 | 10.53 |
| SW-2019 | 113.08 A | 29.02 | 25.66 | 91.32 B | 24.67 | 27.05 | 0.81 | 0.09 | 11.22 |
| GW-2019 | 118.36 A | 52.59 | 44.43 | 89.96 B | 37.92 | 42.15 | 0.77 | 0.11 | 14.57 |
| GRW-2019 | 92.32 A | 25.06 | 27.14 | 70.41 B | 20.15 | 28.61 | 0.76 | 0.09 | 11.44 |
| HD-2019 | 62.88 A | 6.75 | 10.73 | 60.27 B | 6.75 | 11.20 | 0.96 | 0.03 | 3.08 |
| PH-2020 | 128.73 A | 25.45 | 19.37 | 118.02 B | 23.93 | 20.27 | 0.90 | 0.04 | 4.70 |
| SL-2020 | 26.75 A | 4.31 | 16.11 | 21.78 B | 4.46 | 20.48 | 0.81 | 0.08 | 9.49 |
| LSN-2020 | 26.49 A | 7.24 | 27.33 | 22.59 B | 6.51 | 28.82 | 0.85 | 0.08 | 9.23 |
| SW-2020 | 109.58 A | 35.57 | 30.94 | 85.43 B | 31.97 | 35.19 | 0.79 | 0.09 | 11.33 |
| GW-2020 | 104.40 A | 24.80 | 23.75 | 84.97 B | 22.26 | 26.19 | 0.81 | 0.08 | 10.36 |
| GRW-2020 | 86.62 A | 22.57 | 26.06 | 66.76 B | 16.80 | 25.16 | 0.78 | 0.07 | 9.04 |
| HD-2020 | 61.74 A | 6.96 | 11.27 | 58.96 B | 6.733 | 11.42 | 0.96 | 0.03 | 3.13 |
| Trait | PC1 | PC2 | PC3 | PC4 | PC5 |
|---|---|---|---|---|---|
| Seeding trait | |||||
| SPAD | 0.15 | −0.19 | 0.34 | 0.21 | 0.05 |
| RL | 0.13 | 0.18 | 0.26 | 0.45 | 0.26 |
| SH | 0.27 | 0.17 | 0.06 | −0.09 | −0.26 |
| RN | 0.24 | 0.14 | −0.05 | 0.09 | 0.15 |
| TLL | 0.23 | 0.11 | 0.28 | −0.04 | −0.50 |
| TLW | 0.30 | 0.07 | 0.01 | 0.09 | 0.10 |
| TLN | 0.18 | 0.06 | −0.28 | −0.17 | 0.66 |
| RDW | 0.29 | 0.22 | −0.07 | −0.11 | −0.03 |
| SDW | 0.19 | 0.23 | −0.08 | 0.00 | 0.02 |
| PDW | −0.15 | 0.06 | 0.20 | 0.59 | 0.13 |
| SNC | 0.28 | 0.25 | −0.05 | −0.02 | −0.05 |
| RNC | 0.19 | −0.36 | 0.19 | −0.05 | 0.10 |
| PNC | 0.09 | −0.23 | −0.51 | 0.24 | −0.28 |
| SNA | 0.20 | −0.37 | 0.08 | −0.02 | −0.01 |
| RNAC | 0.32 | −0.01 | 0.03 | −0.15 | 0.08 |
| PNA | 0.23 | 0.12 | −0.26 | 0.45 | −0.15 |
| SNPE | 0.33 | 0.01 | −0.01 | −0.08 | 0.04 |
| RNPE | −0.19 | 0.36 | −0.19 | 0.02 | −0.07 |
| PNPE | −0.06 | 0.31 | 0.44 | −0.20 | 0.03 |
| RRD | −0.21 | 0.37 | −0.07 | −0.02 | 0.02 |
| Eigenvalue | 8.49 | 4.36 | 1.85 | 1.45 | 0.87 |
| Contribution rate (%) | 42.43 | 21.80 | 9.24 | 7.25 | 4.33 |
| Accumulated contribution (%) | 42.43 | 64.23 | 73.46 | 80.71 | 85.04 |
| Field trait | |||||
| PH | 0.41 | −0.41 | 0.04 | 0.32 | – |
| SL | 0.44 | −0.39 | 0.16 | 0.16 | – |
| LSN | 0.43 | −0.15 | −0.04 | 0.09 | – |
| SW | 0.46 | 0.20 | −0.08 | −0.41 | – |
| GW | 0.21 | 0.54 | −0.52 | 0.61 | – |
| GRW | 0.42 | 0.28 | −0.14 | −0.53 | – |
| HD | 0.16 | 0.50 | 0.82 | 0.21 | – |
| Eigenvalue | 3.25 | 1.09 | 0.90 | 0.77 | – |
| Contribution rate (%) | 46.46 | 15.55 | 12.82 | 11.06 | – |
| Accumulated contribution (%) | 46.46 | 62.01 | 74.83 | 85.89 | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Chen, L.; Yang, Z.; Lu, J.; Yang, J.; Li, N.; Shi, H. Transcriptomics Uncovers Pathways Mediating Low-Nitrogen Stress Tolerance in Two Foxtail Millet Varieties. Agriculture 2025, 15, 628. https://doi.org/10.3390/agriculture15060628
Wu J, Chen L, Yang Z, Lu J, Yang J, Li N, Shi H. Transcriptomics Uncovers Pathways Mediating Low-Nitrogen Stress Tolerance in Two Foxtail Millet Varieties. Agriculture. 2025; 15(6):628. https://doi.org/10.3390/agriculture15060628
Chicago/Turabian StyleWu, Jirong, Lu Chen, Zhenrong Yang, Juan Lu, Jinwen Yang, Ning Li, and Huawei Shi. 2025. "Transcriptomics Uncovers Pathways Mediating Low-Nitrogen Stress Tolerance in Two Foxtail Millet Varieties" Agriculture 15, no. 6: 628. https://doi.org/10.3390/agriculture15060628
APA StyleWu, J., Chen, L., Yang, Z., Lu, J., Yang, J., Li, N., & Shi, H. (2025). Transcriptomics Uncovers Pathways Mediating Low-Nitrogen Stress Tolerance in Two Foxtail Millet Varieties. Agriculture, 15(6), 628. https://doi.org/10.3390/agriculture15060628