Abstract
Tillage and stubble management play crucial roles in conservation agriculture, exerting a considerable influence on soil properties. This study aims to focus on the gaps in our understanding of how tillage and stubble management interact to affect the taxonomic and functional structure of the soil microbiome. Soil samples were collected from a long-term field trial implementing no tillage (NT) and conventional tillage (CT) with stubble retention and removal. Metagenomic sequencing facilitated the assembly of a gene catalog comprising 4.36 billion non-redundant genes. Stubble management markedly altered both the taxonomic and functional composition of the prokaryotic community, the addition of stubble caused a significant increase in Proteobacteria, but a decrease in Chloroflexi compared with no stubble. The key prokaryotic species and gene functions contributing most to the dissimilarity of the prokaryotic communities between the treatments with and without stubble were identified, including Rhodospirillum sp. Stubble retention increased the availability of carbon resources in the soil, resulting in a higher proportion of genes functional for metabolic activity and plant–pathogen interactions. However, tillage practice did not influence the structure or diversity of the soil prokaryote community. Our findings identify the target microbial species for future isolation, enabling the development of eco-friendly biofertilizers to promote sustainable agriculture.
1. Introduction
Agricultural practices play pivotal roles in shaping the soil micro-environment and microbial communities. Among the various parameters, tillage and stubble management are critical components that significantly affect soil physical and chemical properties, resulting in impacts on the taxonomic and functional structure of the soil microbiome [1]. Understanding the interaction of tillage and stubble practice on soil microbiota is essential for optimizing agricultural management and enhancing ecosystem health.
The mechanisms through which tillage and stubble management exert their effects on soil properties are multifaceted and distinct. Tillage involves the mechanical disruption of soil through actions including plowing, harrowing, cultivating, subsoiling, chiseling, discing, hoeing, ridging, and rolling [2], which are highly associated with increasing particle density, diminishing water infiltration, and escalating erosion risks [3,4,5]. By keeping the crop residues on site, stubble retention forms protective layers on the soil surface [6] that lead to improved soil structure and porosity, increased water infiltration, and decreased soil erosion [7]. These alterations in soil physical properties due to stubble and tillage management also influence the soil chemical properties, such as soil organic carbon (SOC) content, nutrient availability, electrical conductivity (EC), pH, and cation exchange capacity (CEC) [8,9,10,11].
The changes in soil properties, especially soil organic carbon, reshape the taxonomic structure of the soil microbiome [12,13,14]. The soil microbiome plays crucial roles in supporting crop health and development by improving nutrient availability [15,16] and inhibiting the growth of pathogens [17]. Stubble retention and no-tillage (NT) practices can keep the soil structure stable and facilitate soil organic matter (SOM) accumulation [18]. The process of crop residue degradation involves a diverse array of microorganisms, such as certain saprophytic bacteria and fungi, that produce enzymes to break down complex organic molecules into fundamental C compounds, thereby increasing SOM [19,20,21]. Tillage typically changed the activities [22] and structure of the soil microbial community [23,24], potentially because the soils under conventional tillage (CT) with low infiltration were dryer compared with those under NT, so organic residues were more difficult to break down and provide an available C source for microbes. Compared with stubble removal, the soil microbiome composition under stubble retention was more similar to that of the neighboring grassland ecosystems, leading to an increase in microbial diversity [25]. Stubble retention influenced soil microbiome structure by increasing the relative abundance of Acidobacteria, a widely distributed soil bacterial phylum that is able to utilize diverse sources of carbon [26]. In addition, stubble retention also interacts with the soil fungal community [27,28], e.g., members of the Ascomycota can break down complex C substrates, while mycorrhizal fungi can mobilize nutrients in the stubble [29,30]. However, another finding for semiarid cropping systems revealed that stubble had no effect on the bacterial and fungal diversity in the soil [31], possibly due to the low rainfall environment limiting the contribution of crop residues to SOM storage.
Stubble effects appear to be more pronounced than tillage effects in modifying the diversity and composition of the soil microbiome. For example, organic additives were found to be far more important than tillage to change soil microbe numbers, community structure, and enzyme (invertase and urease) activities [23]. Similar findings were reported that stubble retention increased both bacterial and fungal diversities, regardless of tillage management, possibly due to the stubble-driven changes in nutrient availability [32,33].
The alteration of soil micro-environments triggered by tillage and stubble management have different impacts on the prokaryotic and fungal communities [34]. Prokaryotes have diverse metabolic pathways through which they are capable of adapting to various abiotic stresses in soil, such as drought, water logging, heat, and deficits of oxygen and nutrients induced by cropping practices [35,36]. With their rapid reproduction and growth rates, prokaryotes can acclimate to the changes in the soil microenvironment within a short period of time [37]. Fungi, on the other hand, obtain nutrients mainly through the decomposition of organic matter [38]; therefore, they can be more responsive to an increase in organic inputs, such as from stubble retention.
However, past research examining the interplay of tillage and stubble management on soil microbiomes has predominantly concentrated on microbial taxonomy, often overlooking functional structure and its correlation with taxonomic structure. Furthermore, there has been limited exploration into structure interactions among various microbiome kingdoms. This study aims to address these gaps by investigating the complex interplay between tillage and stubble management on the taxonomic and functional structure of bacterial, archaeal, and fungi communities as a means to assess sustainable agriculture and ecosystem management. Our hypothesis is that there is significant interplay between tillage and stubble management on soil microbiomes, alongside a strong correlation between the taxonomic and functional structures of various microbial kingdom communities.
2. Materials and Methods
2.1. Field Experiment
The field experiment was conducted in Linyi County, Shandong Province, in the northern region of China (GPS coordinates 37.46709° N, 116.94031° E) since its establishment in 2015. Throughout the course of the experiment, the region received average annual precipitation of 536.9 mm, with over 67% of this rainfall occurring between June and August. The average annual temperature recorded was 12.6 °C. The soil in this region is categorized as Cambisols [39].
This field experiment was conducted with alternating cultivation of wheat and maize each year. The experimental design followed a randomized complete block layout with four replicates, where each treatment plot measured 20 m in length and 4 m in width. Both tillage and stubble retention practices were implemented, where tillage treatments included no tillage (NT) and conventional tillage (CT), and stubble treatments included stubble-retained (+stubble) and stubble-removed (−stubble). CT was conducted using a moldboard plow to a depth of 20 cm. Stubble retention was conducted by direct application of crop residue onto the soil surface for NT treatment (NT +stubble) and by mixing crop residue into the soil for CT treatment (CT +stubble). During the experimental period, the winter wheat variety Jimai-22 was sown in October and harvested in June of the following year, and the maize variety Dika-1210 was sown after wheat harvest. The wheat crop was fertilized with 225 kg N ha−1, 52.4 kg P ha−1, and 87.1 kg K ha−1, while the maize crop was fertilized with 240 kg N ha−1, 52.4 kg P ha−1, and 74.7 kg K ha−1. In March every year, 130–160 mm of irrigation water was applied to the wheat crop depending on the rainfall, and no irrigation was used for the rest duration of the season. Herbicides and pesticides were applied when weeds and pests appeared.
The soils were collected after maize harvest in 2021, which was 6 years after the treatments. This study focused on the soil microbiome in the topsoil, where the microbes were more active and sensitive to the agricultural practice. Soils at 0–10 cm depth were collected from eight random spots in a single plot and mixed thoroughly to form a single soil sample representing the plot. A 10 g portion was taken off each soil sample and immediately transferred to a −80 °C freezer in the laboratory until DNA extraction. The remainder of the soil sample was air-dried and processed for physical and chemical analyses using the protocol of Rayment and Lyons [40]. The measured soil characteristics included soil texture, pH, electrical conductivity (EC), and the contents of organic C, ammonium N, nitrate N, total N, available P (based on the Colwell P test), total P, and total K.
2.2. Metagenome Sequencing and Bioinformatics
The DNA from 16 samples, consisting of 4 replicates of each tillage–stubble treatment, was extracted using a PowerSoil DNA isolation kit (Mo Bio, Carlsbad, CA, USA) in accordance with the manufacturer’s instructions. DNA sequencing was conducted based on the guidelines provided by Quince et al. [41]. To set up the library, we utilized the Hieff NGS® MaxUp II DNA Library Prep Kit from Illumina® San Diego, CA, USA, as per the manufacturer’s instructions. Adaptors were added to group different sequences from the same sample. We quantified and combined the libraries, performed paired-end sequencing on the NovaSeq 6000 sequencers from Illumina, USA, and then implemented certain criteria to filter out reads during the sequencing process. Reads were discarded if they: (1) contained ≥ 20% low-quality bases, or (2) exhibited adapter contamination, meaning that they had at most 15 bases of overlap between reads and adapters, allowing for a maximum of 3 base mismatches, (3) contained “N” indicating low quality, or (4) demonstrated low complexity, having more than 10 consecutive reads of the same base.
To perform de novo metagenomic assembly, all the clean reads from the 16 samples were pooled. Initially, mixed assembly of multiple samples was applied using the de Bruijn graph method with Megahit version 1.2.9 [42]. The clean reads were then mapped back to the assembled contigs using bowtie2 version 2.1.0 [43]. Unmapped reads were extracted and reassembled using SPAdes version 3.13 [44] to obtain low-abundance contigs. We determined the assembly rate of each sample by mapping all clean reads to the assembled contigs, considering a 90% identity threshold, using SoapAligner software [45].
For gene prediction, Prodigal version 2.60 [46] was used to identify open reading frames (ORFs) from the assembly results, where the genes with a length of 100 bp or greater were selected and converted into amino acid sequences. Predicted genes were clustered into non-redundant genes using CD-HIT version 2.60 [47]. In order to accurately quantify gene abundance in each sample, we utilized a dual-phase parallel inference algorithm with Salmon version 1.5.0 [48], taking into account both the number of mapped sequences and the gene length.
The non-redundant protein sequences were annotated using the NCBI microbial NR database that covered bacteria, fungi, archaea, and viruses, and the functional databases of KEGG [49,50]. This annotation was carried out using the DIAMOND software version 0.8.20 [51] with an e value threshold of ≤ 1 × 10−5. Taxonomic annotation for each gene was performed using the lowest common ancestor method (LCA) with MEGAN [52]. The best-aligned hit based on the lowest blast e value was used as the functional annotation of the metagenomic genes in the KEGG database.
2.3. Statistics
Initially, the low-abundance genes were filtered out based on the criteria of at least 4 samples with over one read. Hellinger transformation [53] was applied to normalize the library size across all samples. The whole gene catalog was divided into three groups based on the taxonomic annotation at the kingdom level, specifically bacteria, archaea, and fungi. Within each of these groups, genes were categorized into taxonomic species or KEGG Orthology (KO); then, species and KO were used as the basic unit for the analysis. Multivariate analysis was conducted for species and KO. Non-metric multidimensional scaling (NMDS) based on Bray–Curtis distance were calculated by metaMDS function from the Vegan Package in R. Permutational multivariate analysis of variance (PERMANOVA) with a maximum of 999 permutations using the adonis function was conducted to test the effects of tillage and stubble on beta-diversity. A similarity percentages breakdown (SIMPER) analysis from the Vegan package was conducted to identify the species/KO that contributed most to the dissimilarity of the microbial communities between + and − stubble treatments. The identified species/KO were considered as the key species/KO. The relative abundance of sequenced reads grouped into species and KO was compared in response to tillage and stubble treatment using the Statistical Analysis of Metagenomic Profiles (STAMP) package and Benjamini–Hochberg FDR method to correct for the p-value.
The Shannon index was calculated to indicate the alpha diversity for taxonomic (at the species level) and functional diversity (at the KO level) in each of the microbiome communities, including bacteria, fungi, and archaea, using the Vegan package in R based on rarefied species and KO table. Two-way ANOVA was used to test the effect of tillage and stubble treatment on the soil sand and clay content, pH, EC, the contents of organic C, ammonium N, nitrate N, total N, available P, total P, and total K, and Shannon index in Minitab 15 (Minitab Inc., State College, PA, USA). The normal distribution of the data was checked before the analysis. Mantel analysis was conducted to test the association between the taxonomic and functional composition of different microbial kingdoms. Pearson correlation was performed among the Shannon index values of the taxonomic and functional diversity of microbial kingdoms. Permutational multivariate ANOVA was used to test the correlation of the soil traits with the taxonomic and functional composition of different microbial kingdoms.
3. Results
3.1. Soil Physical and Chemical Properties
The analysis of soil physical and chemical properties (Table 1) showed that stubble retention significantly increased SOC, total N, Colwell P, total P, and total K by 14%, 12%, 45%, 25% and 4%, respectively, compared with the stubble removal. The only significant tillage impact (p < 0.05) was found on Colwell P, where CT was 22% higher than NT.

Table 1.
Soil physical and chemical properties under tillage and stubble management.
3.2. Soil Microbiome Diversity
We sequenced 2.08 billion raw reads (71.65 M–152.36 M per sample) and obtained 2.08 billion clean reads (71.53 M–151.75 M per sample) after quality control (Table S1). By pooling all 16 samples, we assembled a gene catalog with 4.36 billion non-redundant genes (2.08 M–2.18 M per sample). The gene catalog was divided into three groups, including bacteria, archaea, and fungi. The taxonomic diversity and functional diversity of bacterial, archaeal, and fungal communities were evaluated under different tillage and stubble management treatments.
For the alpha diversity, the impact of stubble was only found to be significant (p < 0.05) on the functional diversity of the soil bacterial community, where stubble retention treatment caused a higher Shannon index (Table 2). The impact of tillage on both the species diversity and functional diversity of the soil fungal community was significant (p < 0.05), where higher diversity occurred under NT treatment (Table 2). However, a significant interaction between tillage and stubble treatments also occurred (p < 0.05) in terms of the soil fungal functional diversity.
Table 2.
Shannon index of the soil microbiome taxonomic diversity at species level and functional diversity under tillage and stubble management.
For the beta-diversity, the impact of stubble was significant (p < 0.05, Table 3) on both the species composition and the functional composition of bacterial and archaeal communities, as shown by the stubble-dependent clusters of the samples in the biplots (Figure 1a,b,e,f). The impact of tillage was only significant (p < 0.05, Table 3) on the functional composition of the archaeal community. No significance was detected in the fungal community’s response to soil management.
Table 3.
Beta-diversity analysis showing the impact of tillage and stubble on soil microbiome taxonomic composition at species level and functional composition.
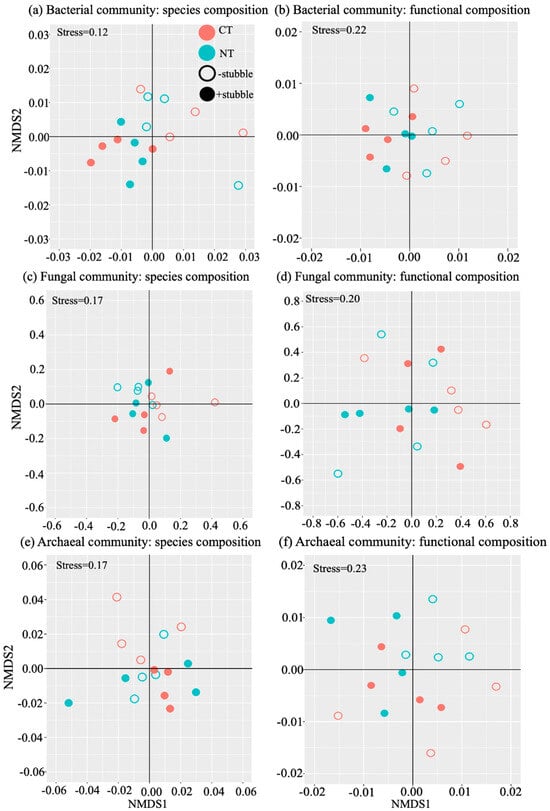
Figure 1.
Beta-diversity analysis of the species composition and functional composition of bacterial (a,b), fungal (c,d), and archaeal (e,f) communities. Each dot represents a single sample of treatments, including conventional tillage (CT), no tillage (NT), no stubble (−stubble), and stubble-retained (+stubble).
Despite the significant stubble impact detected at the species level (p < 0.05, Table 3), the bacterial phyla Proteobacteria and Chloroflexi were the only taxa affected by stubble management across the soil microbiome, including bacterial, archaeal, and fungal communities. Stubble retention treatment caused a significant increase in Proteobacteria (p < 0.05), but a significant decrease in Chloroflexi (p < 0.05). Proteobacteria and Actinobacteria were the most abundant bacterial phyla, accounting for one-third and one-quarter of the community, respectively, followed by Acidobacteria, which only accounted for about 12% (Figure 2a). In the fungal community (Figure 2b), Ascomycota was the most abundant phylum, accounting for ca. 80% of the total, followed by Mucoromycota (ca. 15%). Regarding the archaeal community (Figure 2c), Thaumarchaeota was dominant, accounting for ca. 98% of the total, where more than half of Thaumarchaeota members were in the class Nitrososphaeria.
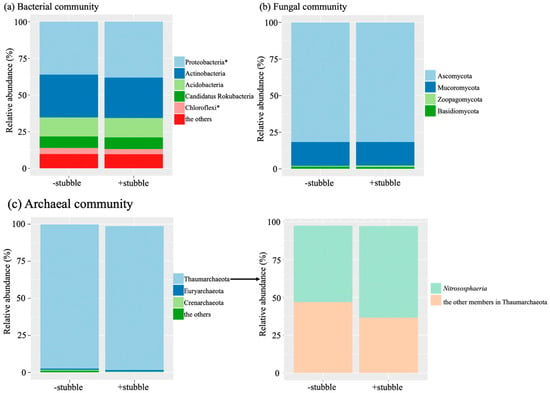
Figure 2.
Sequence of relative abundance in taxonomic phylum groups in bacterial (a), fungal (b), and archaeal (c) communities in response to −stubble and +stubble treatments. The class composition of the dominant archaeal phylum Thaumarchaeota is also shown (c). The symbol * indicates significance at p < 0.05 in response to stubble treatment based on the p-value corrected by Benjamini–Hochberg FDR.
3.3. Key Species and Key Functional Units
To follow up on the significant impact of stubble (p < 0.05, Table 3) on both species composition and functional composition, the key bacterial and archaeal species that accounted for more than 1% of contribution to the differentiation of the soil microbiome between stubble retention and stubble removal treatments were identified using similarity percentages analysis (Figure 3). The relative abundance of the identified key species had an inconsistent pattern with the contribution to the associated community; however, each key species received a significant impact of stubble (p < 0.05), as shown in Figure 3a,b.
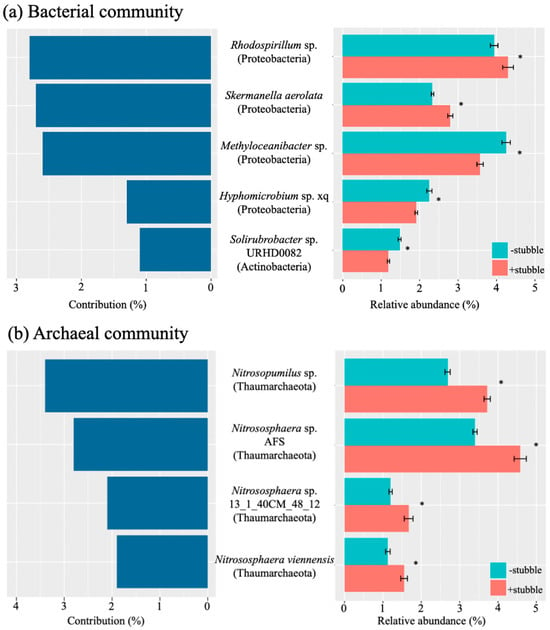
Figure 3.
Key species accounting for more than 1% of the contribution to the differentiation of soil bacterial (a) and archaeal (b) composition by stubble management, and the relative abundance of the key species under −stubble and +stubble treatments. Similarity percentages breakdown analysis was performed to determine the contribution to the differentiation of microbiome composition. The identified microbes were annotated at the species level. The symbol * indicates significance at p < 0.05 in response to stubble treatment, based on the p-value corrected by Benjamini–Hochberg FDR.
Five key bacterial species were identified, Rhodospirillum sp., Skermanella aerolata, Methyloceanibacter sp., Hyphomicrobium sp. xq, and Solirubrobacter sp. URHD0082, where all of the top four key species were the members of Proteobacteria. For the top three key species (i.e., Rhodospirillum sp., S. aerolata, and Methyloceanibacter sp.) each accounted for an at least twice greater contribution (above 2.6%) than the other two species. Despite making the second greatest contribution, S. aerolata had noticeably lower relative abundance than Methyloceanibacter sp. Rhodospirillum sp. and S. aerolata were the only bacterial species showing significantly higher relative abundance under stubble retention compared with stubble removal (Figure 3a).
In the archaeal community (Figure 3b), four key species included Nitrosopumilus sp., Nitrososphaera sp. AFS, Nitrososphaera sp. 131_40CM_48_12, and Nitrososphaera viennensis, and all were members of the Thaumarchaeota. Despite accounting for the greatest contribution (ca. 3.4%), the relative abundance of Nitrosopumilus sp. was noticeably lower than that of Nitrososphaera sp. AFS, which accounted for the greatest part of the community (Figure 3b). Significantly greater relative abundance was detected under stubble retention treatment for all species (p < 0.05, Figure 3b).
Six key annotated KOs were identified in the soil bacterial community (Figure 4a) and in the soil archaeal community, respectively (Figure 4b), in which two of the six KOs were identified to have the same function. An inconsistent pattern was observed for the relative abundance of the identified key KOs against each contribution to the associated community, but the impact of stubble was significant (p < 0.05) on each of the identified key KOs, regardless of community. The greatest contributor in the soil bacterial community (Figure 4a) was the function “RNA polymerase” (K03043), which was approximately twice as great as the function “Plant–pathogen interaction” (K02358), but had a similar relative abundance. Both “RNA polymerase” (K03043) and “Plant–pathogen interaction” (K02358) had a relative abundance nearly triple that of the other key functions in the bacterial community, and both received a significantly negative impact from stubble retention treatment (p < 0.05, Figure 4a). The treatment of stubble retention was found to only significantly favor (p < 0.05) “ABC transporters” (K01996 and K01997) and “Oxidative phosphorylation” (K00340).
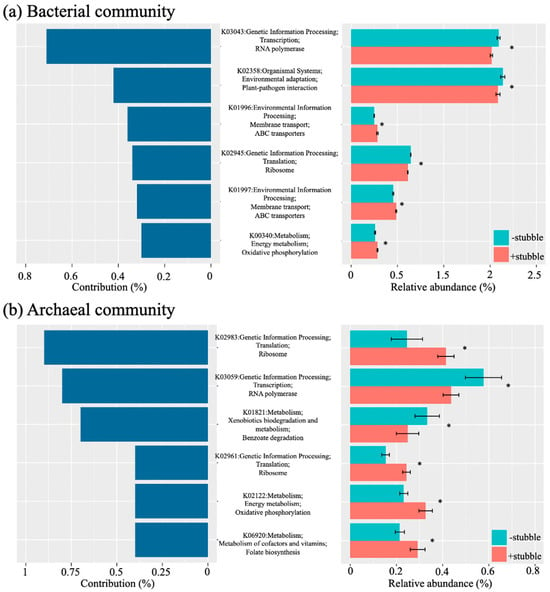
Figure 4.
Key functions contributing to the differentiation of soil bacterial (a) and archaeal (b) microbiome composition by stubble management and the relative abundance of the key functions under −stubble and +stubble treatments. Similarity percentages breakdown analysis was performed to identify the contribution to the differentiation of microbiome composition. The identified functions were annotated for KEGG orthologous groups in three levels. The symbol * indicates significance at p < 0.05 in response to stubble treatment based on the p-value corrected by Benjamini–Hochberg FDR.
The function “Ribosome” (K02983) accounted for the most, nearly 0.9% of the total contribution to the soil archaeal community (Figure 4b), followed by the functions “RNA polymerase” (K03059) and “Benzoate degradation” (K01821). Although the relative abundance of “Ribosome” (K02983) was naturally much lower than that of “RNA polymerase” (K03059), it was increased to a level at which the reduced “RNA polymerase” (K03059) fell into due to their opposite responses to stubble retention treatment (Figure 4b). The functions “Ribosome” (K02961), “Oxidative phosphorylation” (K02122), and “Folate biosynthesis” (K06920) were also favored by stubble retention treatment; however, each contributed less than half of “Ribosome” (K02983).
We conducted a correlation analysis between the taxonomic and functional diversity of different microbial kingdoms. The taxonomic alpha diversity was correlated with the functional alpha diversity for the archaeal community (r < 0) and the fungal community (r > 0, Figure 5a). For the beta diversity, the bacterial taxonomic composition was correlated with its functional composition (Figure 5b). Furthermore, we examined the correlation of the soil physical and chemical properties with the species and functional diversity across various communities. Only the soil properties significantly affected by stubble and tillage management were used for the analysis. SOC had no relationship with microbiome diversity (data not shown). The fungal community was not influenced by the soil properties. Bacterial taxonomic and functional diversity exhibited positive correlations with soil total and Colwell P, while archaeal diversity displayed negative correlations with total N and Colwell P. Total N and Colwell P also significantly influenced the taxonomic and functional structure of the bacterial microbiome, but only the functional composition of the archaeal community.
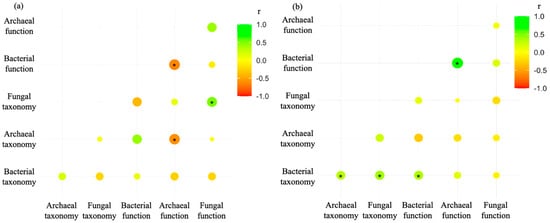
Figure 5.
Correlation coefficient (r) between the taxonomic and functional diversity of different microbial kingdoms. (a) Pearson correlation of the Shannon index and (b) mantel analysis among the taxonomic and functional composition of different microbial kingdoms. The symbol * indicates significance at p < 0.05.
4. Discussion
This study assembled a gene catalog of the soil metagenome, then identified three groups of bacterial, archaeal, and fungal communities for taxonomic and functional analysis. We demonstrate that tillage and stubble management had distinct impacts on the soil microbiome in different microbial kingdoms. There was no interaction between tillage and stubble management on the species or functional composition of any of the three communities; the individual effect of stubble management was stronger than that of the tillage practice in influencing the composition and diversity of the prokaryotic community, as the response of the fungal community was more stable to different cropping management techniques. In addition, coupling of the taxonomic and functional structure was only found in the bacterial community. The bacterial and archaeal microbiome were highly correlated in the taxonomic and functional composition under tillage and stubble management, but differed from the fungal community. Stubble management modulated the soil abiotic properties, such as SOC and available P, which likely accounted for its influence on the soil microbiome.
4.1. Effects of Stubble and Tillage Management on the Structure and Diversity of Microbial Community
In our study, there were no effects of tillage on the structure and diversity of the soil prokaryotic community (i.e., bacteria and archaea), but the effects of stubble on the taxonomic and functional composition were significant. Given that stubble serves as an additional carbon and nutrient source for soil prokaryotic organisms in the decomposition process of plant residues [54], retaining stubble enhances the amount of organic matter in the soil, fueling microbial activity with more energy and nutrients [55]. A strong effect of stubble management on the taxonomic structure of soil prokaryotic communities was also found in previous studies [28,56,57]. According to our results, changes in phylum composition altered the taxonomic structure of prokaryotic communities. We found that only two phyla, Proteobacteria and Chloroflexi, were sensitive to stubble management, but with opposite responses, where a higher abundance of Proteobacteria occurred when stubble was retained and, regarding a higher abundance of Chloroflexi, when stubble was removed (Figure 2a). Interestingly, the same two phyla were also identified as the most responsive bacterial phyla to stubble retention in another study [58]. The stubble-driven increment in SOM may have been key to the increase in Proteobacteria, because the fast-growing copiotrophic members of the Proteobacteria were favored by the high-C micro-environment [59]. In contrast, due to its sensitivity to the oligotrophic habitat, Chloroflexi was less competitive in the niche with a high nutrient content [55].
In addition, this study identified the impacts of stubble retention on the functional structure of soil prokaryotic communities. An increment in SOM decomposition and nutrient release in stubble retention treatment can enhance the microbial functions associated with carbon and nutrient (i.e., nitrogen and phosphorus) cycling to facilitate the adaption to diverse soil conditions [35,36], such as N-fixing, nitrifying, and denitrifying functions in the nitrogen cycle and the oxidizing functions in SOM decomposition [11,60,61]. Our findings demonstrate that the status of soil nutrients, rather than organic carbon, affected the functional structure of soil prokaryotic communities, although the stubble retention increased both SOC and soil nutrients of N, P, and K (Table 1). A possible reason is that the degradation of SOC is a prolonged and intricate process that gradually modulates the soil microbiome over the long term [62], whereas soil nutrients can directly provide short-term nutritional resources [63], thus exerting a more immediate impact on microbiome function and structure.
Differently from the prokaryotic communities, the diversity and structure of fungal community were more stable, and modulated by the stubble and tillage management to a lesser extent; only alpha diversity was influenced by the tillage management. Similar to our observation, higher alpha diversity was also detected when NT or minimal tillage was applied in diverse cropping systems [64].
4.2. Key Species and Functions Determining Prokaryotic Community
The key bacterial species driving the dissimilarity in the bacterial community between stubble retention and stubble removal treatments were the members of the Proteobacteria, including Rhodospirillum sp., S. aerolata, and Methyloceanibacter sp. Given that Rhodospirillum sp. is associated with crop disease occurrence [65], our result regarding its great abundance in the stubble-retention treatment appears to align with the previous finding of higher disease incidence when stubble was retained [66]; however, Rhodospirillum is known to be facultative anaerobic and is not known as a causal agent [67]. Rhodospirillum sp. is known for its ability to fix nitrogen; therefore, it might have contributed to the boost in nitrogen fixation-related genes [68]. S. aerolata is a denitrifying bacterium carrying the nirS gene [69]. The abundance of nirS was higher in soils with organic fertilizer than in soils with mineral fertilizer, suggesting that denitrifying micro-organisms are enriched with organic matter addition into the soil [69], which is in line with our finding that, when stubble was retained, the relative abundance of S. aerolata was significantly higher compared with that under stubble removal. Methyloceanibacter sp. is recognized for its ability to perform carbon fixation [70] and could potentially play a significant role in the carbon metabolism of the soil microbiome.
The key archaeal species driving the dissimilarity of archaeal community between stubble retention and stubble removal treatments were the members of Thaumarchaeota including Nitrosopumilus and Nitrososphaera. These two genera are classified as ammonia-oxidizing archaea (AOA) [71]. AOA are able to oxidize ammonia, a key step in the conversion of atmospheric nitrogen into a biologically available form [71]. The introduction of biochar into soil has been found to promote AOA growth, enhancing soil organic carbon, total nitrogen, and ammonium nitrogen levels, ultimately increasing energy availability [72]. The stubble retention treatment in our study created a C- and N-enriched condition, resulting in the rise in AOA abundance and diversity, which might contribute to the nitrogen cycle and improved soil quality.
The function of genetic information processing, specifically in the transcription of RNA polymerase, played a significant role in driving the dissimilarity in the bacterial and archaeal function structure in response to stubble treatments. In particular, the relative abundance of these two key KOs was lower in soil samples from the stubble retention treatment compared with stubble removal. Stubble retention increases the availability of carbon resources in the soil, resulting in a higher proportion of genes functional for metabolic activity [59], and might lower the relative abundance of genetic information processing functions.
In addition to the genetic information processing functions mentioned above, “plant–pathogen interaction” is another significant functional unit causing alterations in bacterial functional composition. The relative abundance of this functional unit was higher in the stubble-removal treatment than that in the stubble-retention treatment, indicating the complex interactions between pathogens, disease-suppressive microbes, and crop residues in the soil [13].
5. Conclusions
This study reveals the complex interplay between tillage and stubble management on the soil microbiome, including the taxonomic and functional diversity of bacterial, archaeal, and fungal communities. It emphasizes that tillage practices did not significantly alter the prokaryotic community in the soil, while stubble management notably influenced both the taxonomic and functional composition of these microbial communities. Interestingly, the fungal community responded differently, showing that changes in alpha diversity were linked to tillage methods. Future research will aim to isolate the beneficial microbial species identified through metagenomic analysis in this study, screen for plant growth-promoting microbes, and develop eco-friendly biofertilizers and biopesticides to promote sustainable agricultural practices.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/agriculture15020143/s1, Table S1: The number of sequenced reads and mapped genes of the 16 samples in present study.
Author Contributions
Conceptualization, C.X., Y.Z. and M.D.D.; methodology, C.X., Y.Z. and J.L.; software, C.X., Y.Z. and R.T.; validation, C.X., Y.Z. and R.T.; formal analysis, C.X., Y.Z. and R.T.; investigation, C.X., Y.Z., R.T., J.L. and M.D.D.; resources, J.L. and M.D.D.; data curation, C.X. and Y.Z.; writing—original draft preparation, C.X., Y.Z., R.T., J.L. and M.D.D.; writing—review and editing, C.X., Y.Z., R.T., J.L. and M.D.D.; visualization, C.X., Y.Z. and R.T.; supervision, Y.Z., R.T., J.L. and M.D.D.; project administration, J.L. and M.D.D.; funding acquisition, J.L. and M.D.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Australian Research Council (project ID: LP200200813); and the field trial was funded by the Shandong Provincial Natural Science Foundation of China (project ID: ZR2022MC215 and ZR2023MC045).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Acknowledgments
We acknowledge staff from Ecology Institute of Qilu University of Technology (Shandong Academy of Sciences) for soil sampling.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Shia, A.; Cavagnarob, T.R.; Sumbyb, K.M.; McDonaldb, G.; Dentonb, M.D.; Royb, S.J.; Schillinga, R.K. Impact of agronomic management on the soil microbiome: A southern Australian dryland broadacre perspective. Adv. Agron. 2024, 186, 113. [Google Scholar] [CrossRef]
- Lal, R.; Eckert, D.; Fausey, N.; Edwards, W. Conservation tillage in sustainable agriculture. Sustain. Agric. Syst. 1990, 203–225. [Google Scholar]
- Alam, M.K.; Islam, M.M.; Salahin, N.; Hasanuzzaman, M. Effect of tillage practices on soil properties and crop productivity in wheat-mungbean-rice cropping system under subtropical climatic conditions. Sci. World J. 2014, 2014, 437283. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Canqui, H.; Ruis, S.J. No-tillage and soil physical environment. Geoderma 2018, 326, 164–200. [Google Scholar] [CrossRef]
- Sarkar, R.; Kar, S. Temporal changes in fertility and physical properties of soil under contrasting tillage-crop residue management for sustainable rice-wheat system on sandy-loam soil. J. Crop Improv. 2011, 25, 262–290. [Google Scholar] [CrossRef]
- Roper, M.M.; Ward, P.R.; Keulen, A.F.; Hill, J.R. Under no-tillage and stubble retention, soil water content and crop growth are poorly related to soil water repellency. Soil Tillage Res. 2013, 126, 143–150. [Google Scholar] [CrossRef]
- Busari, M.A.; Kukal, S.S.; Kaur, A.; Bhatt, R.; Dulazi, A.A. Conservation tillage impacts on soil, crop and the environment. Int. Soil Water Conserv. Res. 2015, 3, 119–129. [Google Scholar] [CrossRef]
- Allen, D.E.; Singh, B.P.; Dalal, R.C. Soil health indicators under climate change: A review of current knowledge. In Soil Health and Climate Change; Springer: Berlin/Heidelberg, Germany, 2011; Volume 29. [Google Scholar] [CrossRef]
- Chen, H.Q.; Hou, R.X.; Gong, Y.S.; Li, H.W.; Fan, M.S.; Kuzyakov, Y. Effects of 11 years of conservation tillage on soil organic matter fractions in wheat monoculture in Loess Plateau of China. Soil Tillage Res. 2009, 106, 85–94. [Google Scholar] [CrossRef]
- Lv, L.; Gao, Z.; Liao, K.; Zhu, Q.; Zhu, J. Impact of conservation tillage on the distribution of soil nutrients with depth. Soil Tillage Res. 2023, 225, 105527. [Google Scholar] [CrossRef]
- Zhang, L.; Su, X.; Meng, H.; Men, Y.; Liu, C.; Yan, X.; Mao, L. Cotton stubble return and subsoiling alter soil microbial community, carbon and nitrogen in coastal saline cotton fields. Soil Tillage Res. 2023, 226, 105585. [Google Scholar] [CrossRef]
- Obayomi, O.; Seyoum, M.M.; Ghazaryan, L.; Tebbe, C.C.; Murase, J.; Bernstein, N.; Gillor, O. Soil texture and properties rather than irrigation water type shape the diversity and composition of soil microbial communities. Appl. Soil Ecol. 2021, 161, 103834. [Google Scholar] [CrossRef]
- Pankhurst, C.E.; McDonald, H.J.; Hawke, B.G.; Kirkby, C.A. Effect of tillage and stubble management on chemical and microbiological properties and the development of suppression towards cereal root disease in soils from two sites in NSW, Australia. Soil Biol. Biochem. 2002, 34, 833–840. [Google Scholar] [CrossRef]
- Wang, X.B.; Yao, J.; Zhang, H.Y.; Wang, X.G.; Li, K.H.; Lu, X.T.; Wang, Z.W.; Zhou, J.Z.; Han, X.G. Environmental and spatial variables determine the taxonomic but not functional structure patterns of microbial communities in alpine grasslands. Sci. Total Environ. 2019, 654, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Alemneh, A.A.; Zhou, Y.; Ryder, M.H.; Denton, M.D. Is phosphate solubilizing ability in plant growth-promoting rhizobacteria isolated from chickpea linked to their ability to produce ACC deaminase? J. Appl. Microbiol. 2021, 131, 2416–2432. [Google Scholar] [CrossRef]
- Zhou, Y.; Lambrides, C.J.; Li, J.; Xu, Q.; Toh, R.; Tian, S.; Yang, P.; Yang, H.; Ryder, M.; Denton, M.D. Nitrifying microbes in the rhizosphere of perennial grasses are modified by biological nitrification inhibition. Microorganisms 2020, 8, 1687. [Google Scholar] [CrossRef]
- Sui, L.; Li, J.; Philp, J.; Yang, K.; Wei, Y.; Li, H.; Li, J.; Li, L.; Ryder, M.; Toh, R.; et al. Trichoderma atroviride seed dressing influenced the fungal community and pathogenic fungi in the wheat rhizosphere. Sci. Rep. 2022, 12, 9677. [Google Scholar] [CrossRef]
- Anderson, G. The Impact of Tillage Practices and Crop Residue (Stubble) Retention in the Cropping System of Western Australia; Department of Agriculture and Food: Perth, WA, Australia, 2009.
- Ayilara, M.S.; Olanrewaju, O.S.; Babalola, O.O.; Odeyemi, O. Waste management through composting: Challenges and potentials. Sustainability 2020, 12, 4456. [Google Scholar] [CrossRef]
- Gupta, A.; Singh, U.B.; Sahu, P.K.; Paul, S.; Kumar, A.; Malviya, D.; Singh, S.; Kuppusamy, P.; Singh, P.; Paul, D.; et al. Linking soil microbial diversity to modern agriculture practices: Review. Int. J. Environ. Res. Public Health 2022, 19, 3141. [Google Scholar] [CrossRef]
- Hubbe, M.; Nazhad, M.; Sanchez, C. Composting as a way to convert cellulosic biomass and organic waste into high-value soil amendments: A Review. Bioresource 2010, 5, 2808–2854. [Google Scholar] [CrossRef]
- Zuber, S.M.; Villamil, M.B. Meta-analysis approach to assess effect of tillage on microbial biomass and enzyme activities. Soil Biol. Biochem. 2016, 97, 176–187. [Google Scholar] [CrossRef]
- Chen, X.; Henriksen, T.M.; Svensson, K.; Korsaeth, A. Long-term effects of agricultural production systems on structure and function of the soil microbial community. Appl. Soil Ecol. 2020, 147, 103387. [Google Scholar] [CrossRef]
- Jackson, L.E.; Calderon, F.J.; Steenwerth, K.L.; Scow, K.M.; Rolston, D.E. Responses of soil microbial processes and community structure to tillage events and implications for soil quality. Geoderma 2003, 114, 305–317. [Google Scholar] [CrossRef]
- Wang, Y.; Li, C.; Tu, C.; Hoyt, G.D.; DeForest, J.L.; Hu, S. Long-term no-tillage and organic input management enhanced the diversity and stability of Soil Microbial Community. Sci. Total Environ. 2017, 609, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.L.; Challacombe, J.F.; Janssen, P.H.; Henrissat, B.; Coutinho, P.M.; Wu, M.; Kuske, C.R. Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl. Environ. Microbiol. 2009, 75, 2046–2056. [Google Scholar] [CrossRef]
- Fang, Y.Y.; Van Zwieten, L.; Rose, M.T.; Vasileiadis, S.; Donner, E.; Vancov, T.; Rigg, J.L.; Weng, Z.; Lombi, E.; Drigo, B.; et al. Unraveling microbiomes and functions associated with strategic tillage, stubble, and fertilizer management. Agric. Ecosyst. Environ. 2022, 323, 107686. [Google Scholar] [CrossRef]
- Wakelin, S.A.; Colloff, M.J.; Harvey, P.R.; Marschner, P.; Gregg, A.L.; Rogers, S.L. The effects of stubble retention and nitrogen application on soil microbial community structure and functional gene abundance under irrigated maize. FEMS Microbiol. Ecol. 2007, 59, 661–670. [Google Scholar] [CrossRef]
- Hobbie, E.A.; Horton, T.R. Evidence that saprotrophic fungi mobilise carbon and mycorrhizal fungi mobilise nitrogen during litter decomposition. New Phytol. 2007, 173, 447–449. [Google Scholar] [CrossRef]
- Ma, A.; Zhuang, X.; Wu, J.; Cui, M.; Lv, D.; Liu, C.; Zhuang, G. Ascomycota members dominate fungal communities during straw residue decomposition in arable soil. PLoS ONE 2013, 8, e66146. [Google Scholar] [CrossRef]
- Hoyle, F.C.; Murphy, D.V. Seasonal changes in microbial function and diversity associated with stubble retention versus burning. Soil Res. 2006, 44, 407. [Google Scholar] [CrossRef]
- Sarker, J.R.; Singh, B.P.; Cowie, A.L.; Fang, Y.; Collins, D.; Badgery, W.; Dalal, R.C. Agricultural management practices impacted carbon and nutrient concentrations in soil aggregates, with minimal influence on aggregate stability and total carbon and nutrient stocks in contrasting soils. Soil Tillage Res. 2018, 178, 209–223. [Google Scholar] [CrossRef]
- Nazaries, L.; Singh, B.P.; Sarker, J.R.; Fang, Y.; Klein, M.; Singh, B.K. The response of soil multi-functionality to agricultural management practices can be predicted by key soil abiotic and biotic properties. Agric. Ecosyst. Environ. 2021, 307, 107206. [Google Scholar] [CrossRef]
- Yang, T.; Lupwayi, N.; Marc, S.A.; Siddique, K.H.; Bainard, L.D. Anthropogenic drivers of soil microbial communities and impacts on soil biological functions in agroecosystems. Glob. Ecol. Conserv. 2021, 27, e01521. [Google Scholar] [CrossRef]
- Hayatsu, M.; Tago, K.; Saito, M. Various players in the nitrogen cycle: Diversity and functions of the microorganisms involved in nitrification and denitrification. J. Soil Sci. Plant Nutr. 2008, 54, 33–45. [Google Scholar] [CrossRef]
- Kuypers, M.M.M.; Marchant, H.K.; Kartal, B. The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 2018, 16, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Paul, E.; Frey, S. (Eds.) Soil Microbiology, Ecology and Biochemistry; Elsevier: Amsterdam, The Netherlands, 2023. [Google Scholar]
- Mayer, M.; Rewald, B.; Matthews, B.; Sanden, H.; Rosinger, C.; Katzensteiner, K.; Godbold, D.L. Soil fertility relates to fungal-mediated decomposition and organic matter turnover in a temperate mountain forest. New Phytol. 2021, 231, 777–790. [Google Scholar] [CrossRef]
- Deckers, J.A.; Nachtergaele, F.; Spaargaren, O.C. (Eds.) World Reference Base for Soil Resources Introduction; Acco: Louven, Belgium, 1998. [Google Scholar]
- Rayment, G.E.; Lyons, D.J. Soil Chemical Methods Australasia; CSIRO: Canberra, Australia, 2011; Volume 3.
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, J.; Hu, G.; Zhu, H. Gene prediction in metagenomic fragments based on the SVM algorithm. BMC Bioinform. 2013, 14, S12. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef]
- Bahram, M.; Hildebrand, F.; Forslund, S.K.; Anderson, J.L.; Soudzilovskaia, N.A.; Bodegom, P.M.; Bengtsson-Palme, J.; Anslan, S.; Coelho, L.P.; Harend, H.; et al. Structure and function of the global topsoil microbiome. Nature 2018, 560, 233–237. [Google Scholar] [CrossRef]
- Wang, L.; Lu, P.; Feng, S.; Hamel, C.; Sun, D.; Siddique, K.H.M.; Gan, G.Y. Strategies to improve soil health by optimizing the plant–soil–microbe–anthropogenic activity nexus. Agric. Ecosyst. Environ. 2024, 359, 108750. [Google Scholar] [CrossRef]
- Tian, W.; Wang, L.; Li, Y.; Zhuang, K.; Li, G.; Zhang, J.; Xiao, X.; Xi, Y. Responses of microbial activity, abundance, and community in wheat soil after three years of heavy fertilization with manure-based compost and inorganic nitrogen. Agric. Ecosyst. Environ. 2015, 213, 219–227. [Google Scholar] [CrossRef]
- Arunrat, N.; Sereenonchai, S.; Sansupa, C.; Kongsurakan, P.; Hatano, R. Effect of rice straw and stubble burning on soil physicochemical properties and bacterial communities in central thailand. Biology 2023, 12, 501. [Google Scholar] [CrossRef] [PubMed]
- Marsch, R.; Verhulst, N.; Govaerts, B.; Dendooven, L. Bacterial indicator taxa in soils under different long-term agricultural management. J. Appl. Microbiol. 2016, 120, 921–933. [Google Scholar] [CrossRef]
- Yu, D.; Wen, Z.; Li, X.; Song, X.; Wu, H.; Yang, P. Effects of straw return on bacterial communities in a wheat-maize rotation system in the North China Plain. PLoS ONE 2018, 13, e0198087. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xu, W.; Li, J.; Yu, Z.; Zeng, Q.; Tan, W.; Mi, W. Short-term effect of manure and straw application on bacterial and fungal community compositions and abundances in an acidic paddy soil. J. Soils Sediments 2021, 21, 3057–3071. [Google Scholar] [CrossRef]
- Lai, H.L.; Gao, F.Y.; Su, H.; Zheng, P.; Li, Y.Y.; Yao, H.Y. Nitrogen distribution and soil microbial community characteristics in a legume-cereal intercropping system: A review. Agronomy 2022, 12, 1900. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Shi, Y.C.; Dong, Y.X.; Lapen, D.R.; Liu, J.H.; Chen, W. Subsoiling and conversion to conservation tillage enriched nitrogen cycling bacterial communities in sandy soils under long-term maize monoculture. Soil Tillage Res. 2022, 215, 105197. [Google Scholar] [CrossRef]
- Liu, C.; Lu, M.; Cui, J.; Li, B.; Fang, C. Effects of straw carbon input on carbon dynamics in agricultural soils: A meta-analysis. Glob. Change Biol. 2014, 20, 1366–1381. [Google Scholar] [CrossRef]
- Huang, T.; Yang, N.; Lu, C.; Qin, X.; Siddique, K.H.M. Soil Organic Carbon, total nitrogen, available nutrients, and yield under different straw returning methods. Soil Tillage Res. 2021, 214, 105171. [Google Scholar] [CrossRef]
- Finn, D.R.; Lee, S.; Lanzén, A.; Bertrand, M.; Nicol, G.W.; Hazard, C. Cropping systems impact changes in soil fungal, but not prokaryote, alpha-diversity and community composition stability over a growing season in a long-term field trial. FEMS Microbiol. Ecol. 2021, 97, fiab136. [Google Scholar] [CrossRef]
- Donn, S.; Almario, J.; Muller, D.; Moënne-Loccoz, Y.; Gupta, V.V.; Kirkegaard, J.A.; Richardson, A.E. Rhizosphere microbial communities associated with Rhizoctonia damage at the field and disease patch scale. Appl. Soil Ecol. 2014, 78, 37–47. [Google Scholar] [CrossRef]
- De Boer, R.F.; Steed, G.R.; Kollmorgen, J.F.; Macauley, B.J. Effects of rotation, stubble retention and cultivation on take-all and eyespot of wheat in northeastern Victoria, Australia. Soil Tillage Res. 1993, 25, 263–280. [Google Scholar] [CrossRef]
- Campbell, A.R.; Titus, B.R.; Kuenzi, M.R.; Rodriguez-Perez, F.; Brunsch, A.D.; Schroll, M.M.; Shepherd, J.N. Investigation of candidate genes involved in the rhodoquinone biosynthetic pathway in Rhodospirillum rubrum. PLoS ONE 2019, 14, e0217281. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Yang, J.; Yu, N.; Luo, L.; Wang, E. Biological nitrogen fixation in cereal crops: Progress, strategies, and Perspectives. Plant Commun. 2023, 4, 100499. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Chen, C.; Bai, M.; Xu, T.; Yang, H.; Shi, A.; Li, J. Abundance and diversity of denitrifying bacterial communities associated with N2O emission under long-term organic farming. Eur. J. Soil Biol. 2020, 97, 103153. [Google Scholar] [CrossRef]
- Garritano, A.N.; Song, W.; Thomas, T. Carbon fixation pathways across the bacterial and archaeal tree of life. PNAS Nexus 2022, 1, pgac226. [Google Scholar] [CrossRef]
- Klein, T.; Poghosyan, L.; Barclay, J.E.; Murrell, J.C.; Hutchings, M.I.; Lehtovirta-Morley, L.E. Cultivation of ammonia-oxidising archaea on solid medium. FEMS Microbiol. Lett. 2022, 369, fnac029. [Google Scholar] [CrossRef]
- Zheng, J.; Luan, L.; Luo, Y.; Fan, J.B.; Xu, Q.S.; Sun, B.; Jiang, Y.J. Biochar and lime amendments promote soil nitrification and nitrogen use efficiency by differentially mediating ammonia-oxidizer community in an acidic soil. Appl. Soil Ecol. 2022, 180, 104619. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).