
Hybrid Deep Learning Approaches for Improved Genomic Prediction in Crop Breeding

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets Used for Genomic Prediction
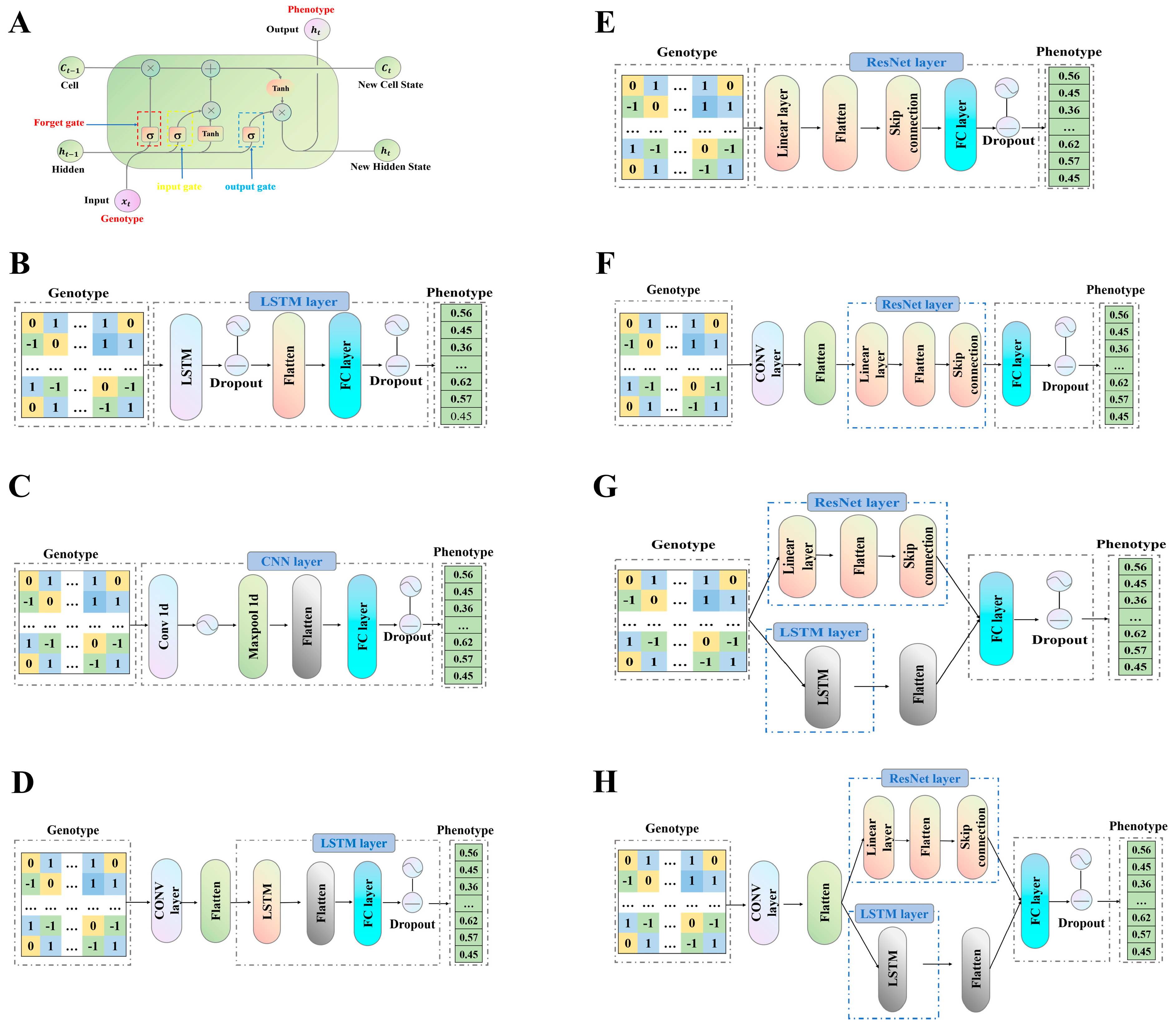
2.2. Design and Aichitecture of Hybrid Models
2.3. Model Training Parameters
2.4. Model Evaluation
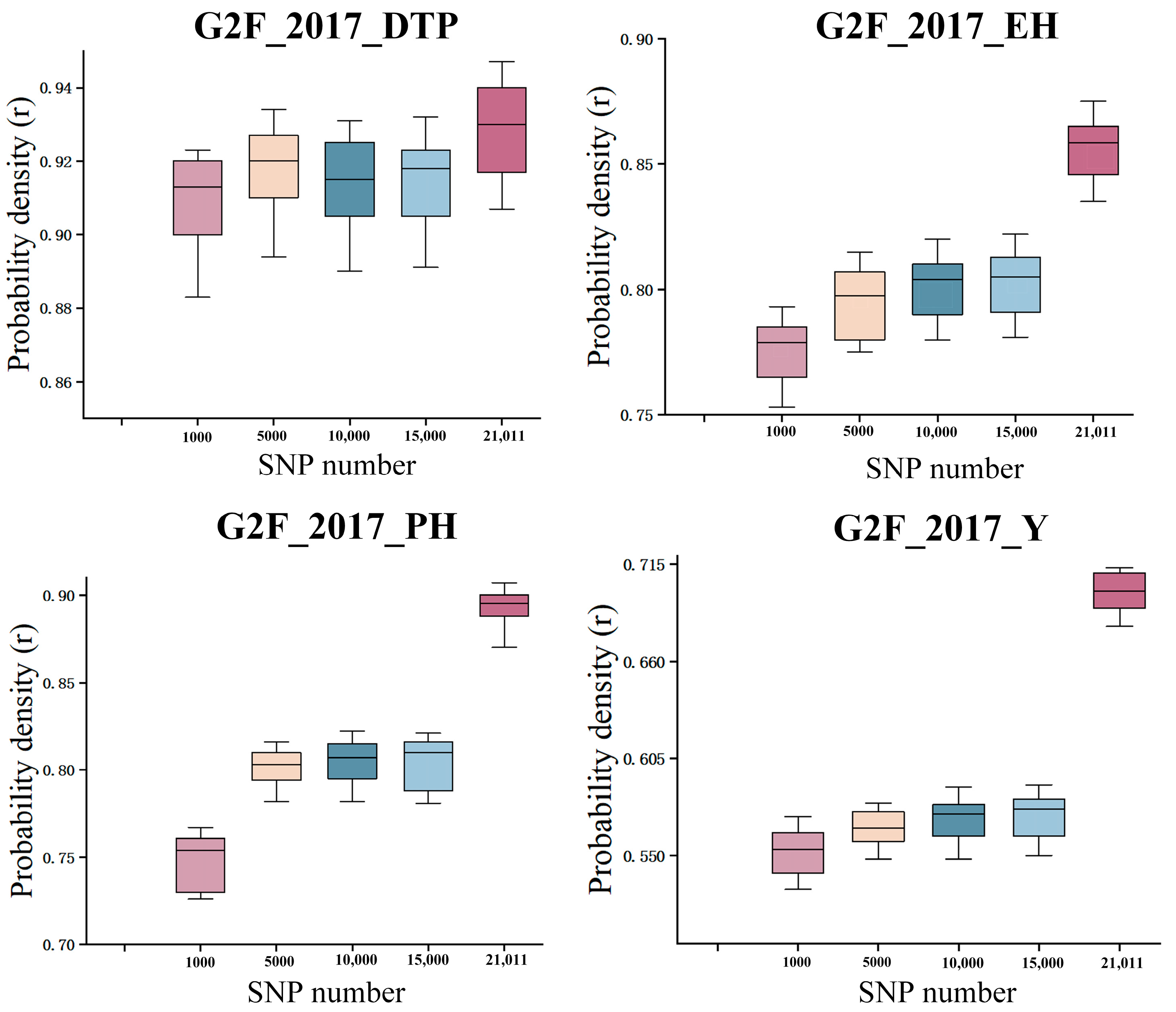
2.5. The Impact of the Number of SNPs on Prediction Methods
2.6. A Comparison of the Application Effects of GS Based on LSTM-ResNet
3. Results
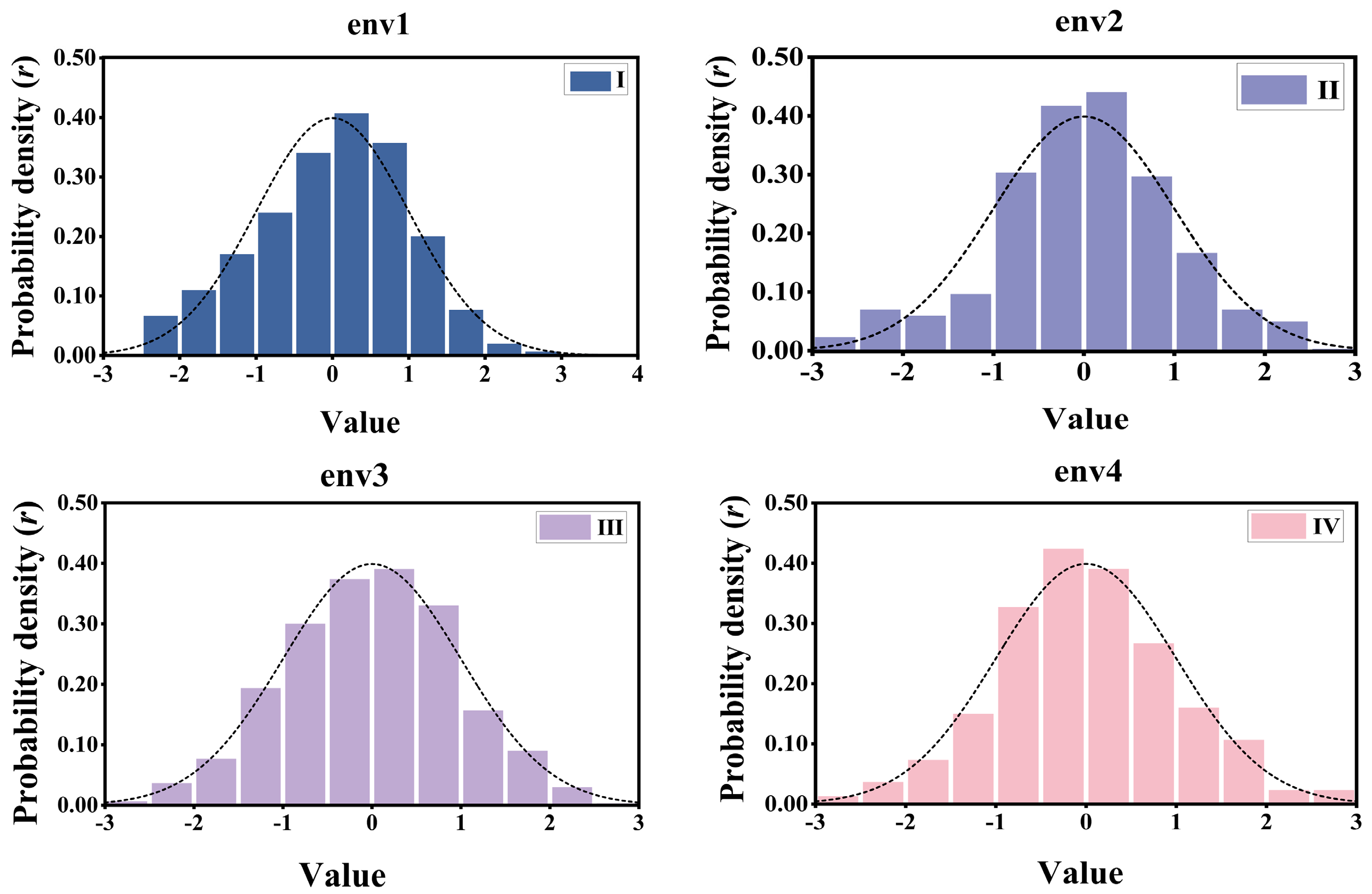
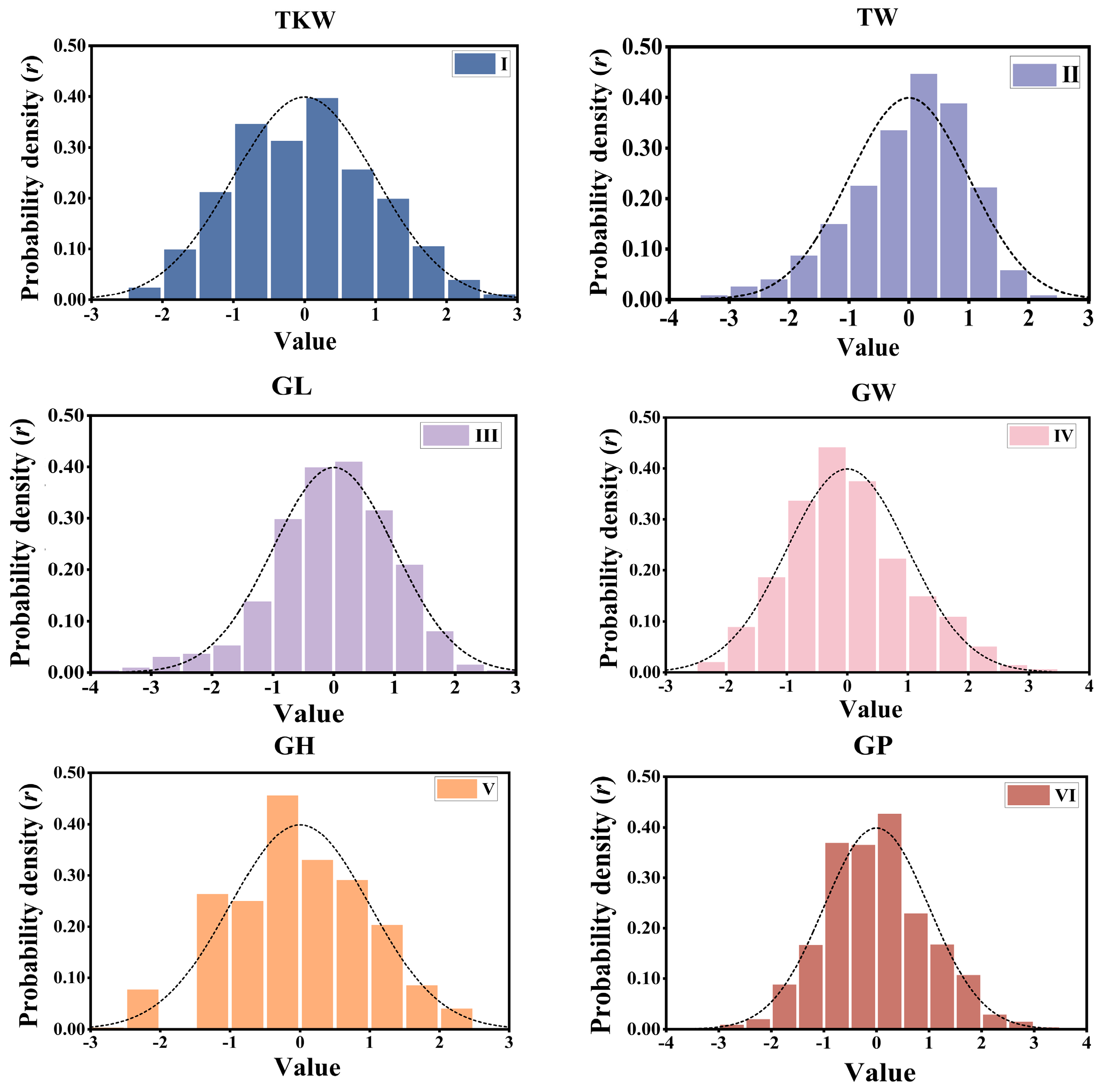
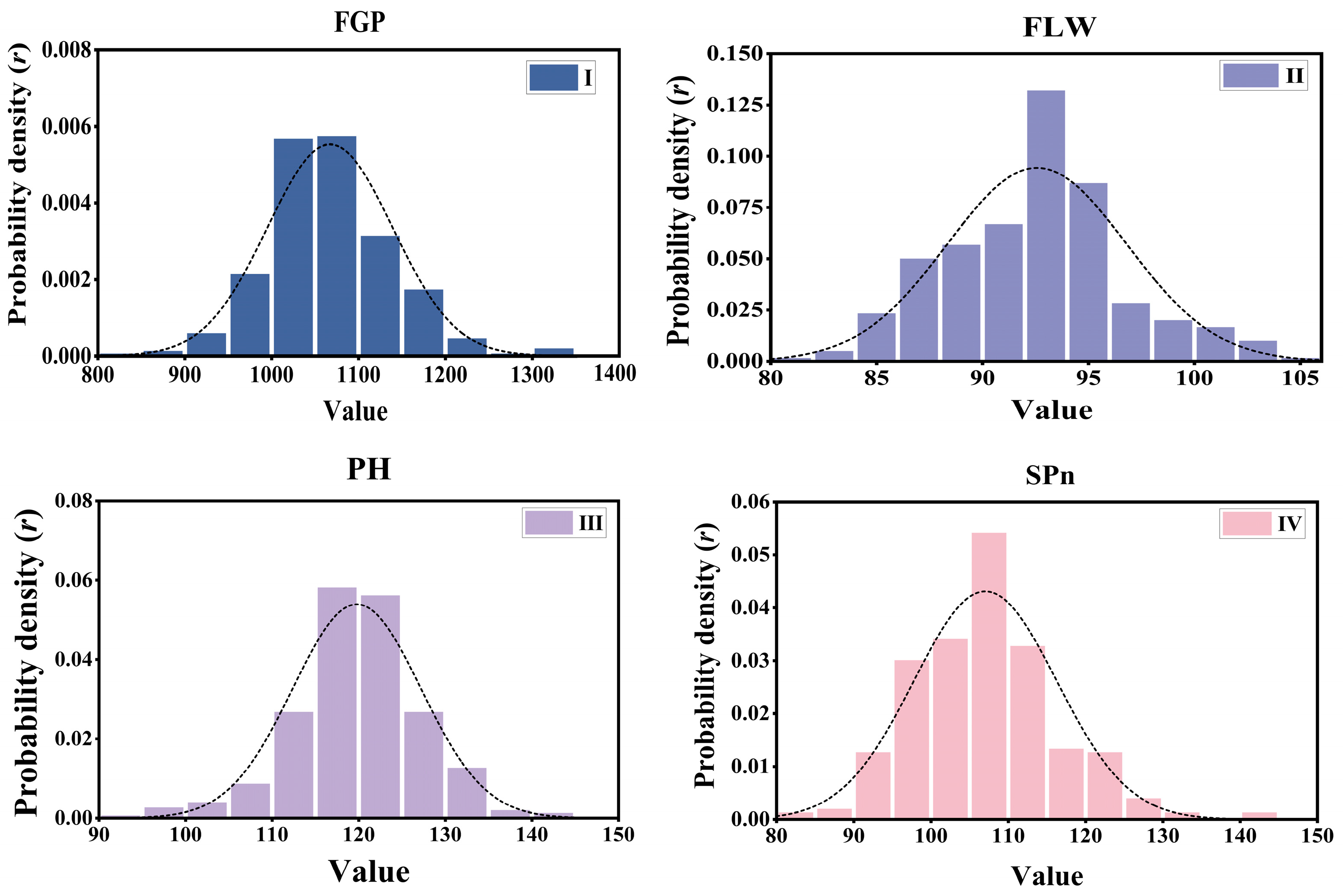
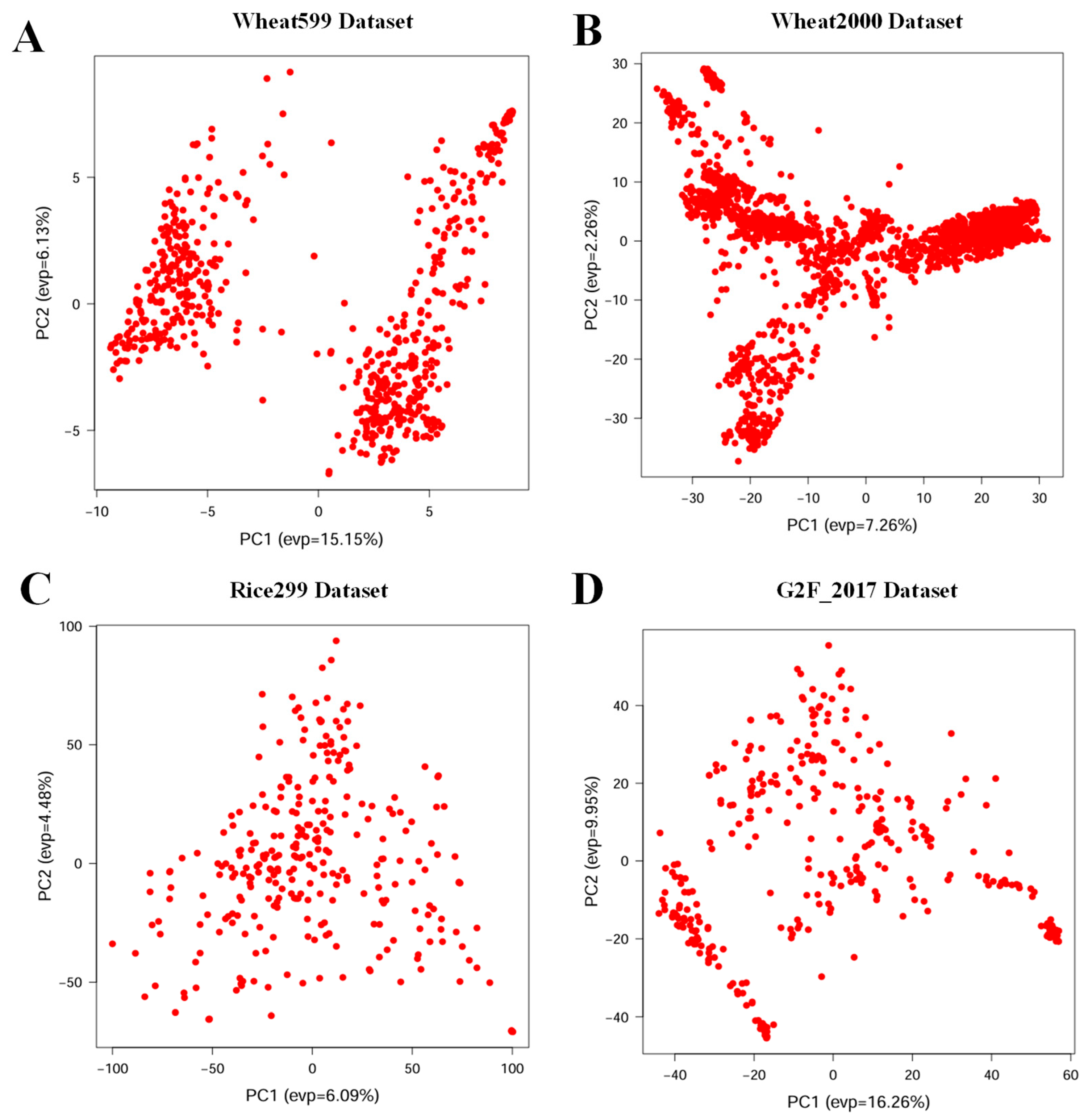
3.1. Phenotypic Distribution and Genetic Diversity
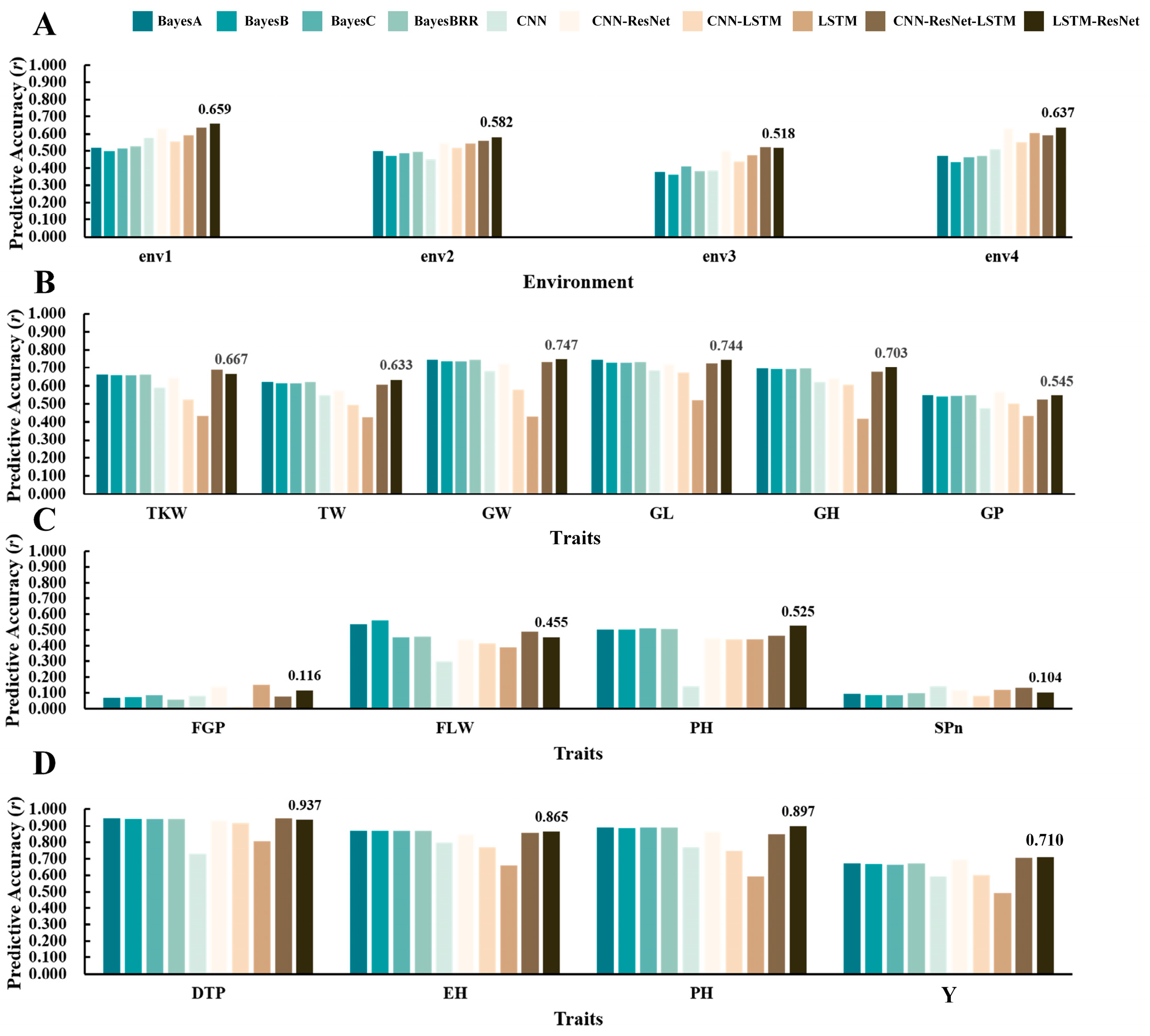
3.2. Evaluation of Prediction Accuracy for Hybrid Models and Other Methods
3.3. Comparison of Hybrid Deep Learning Model Architectures and Their Predictive Performance
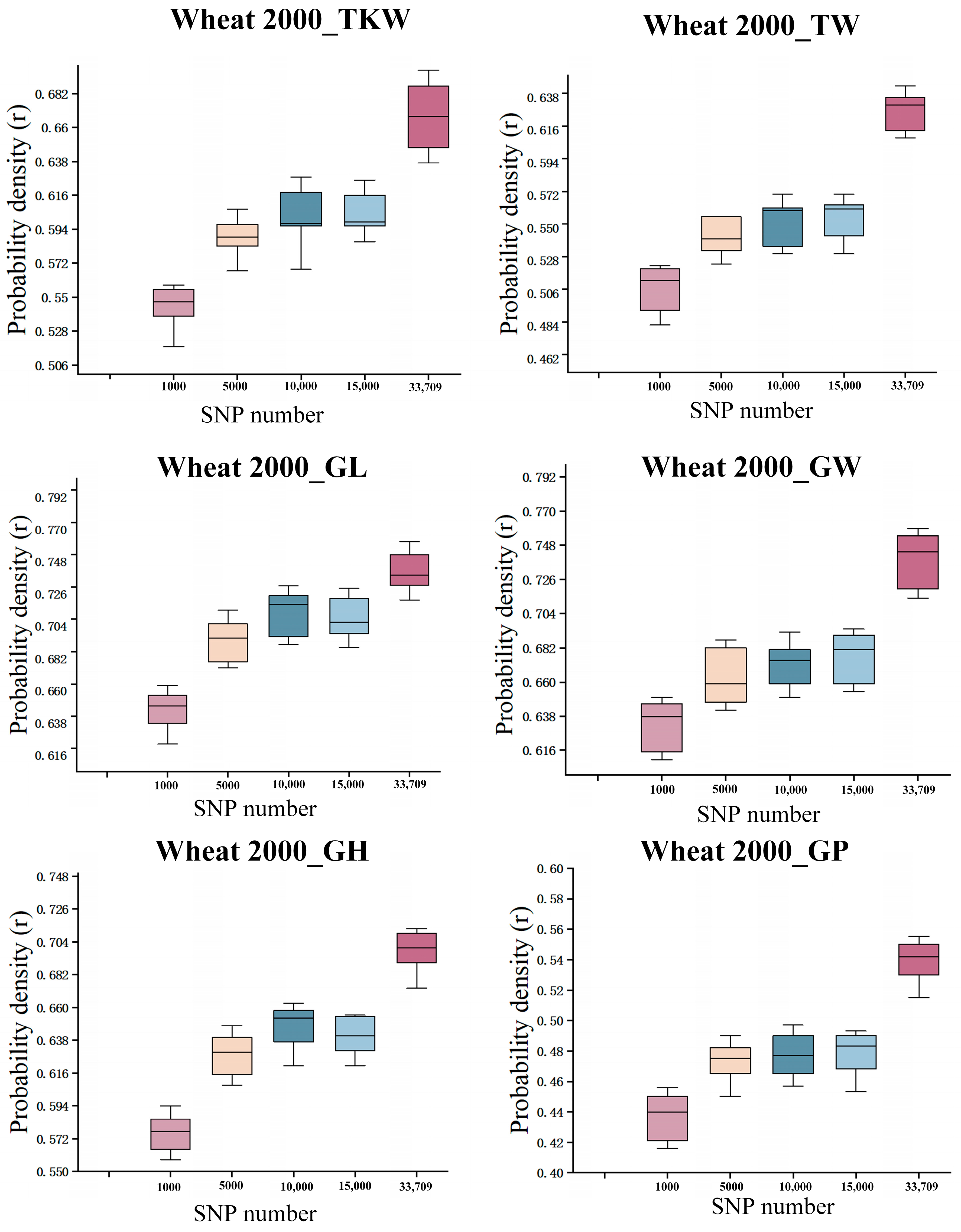
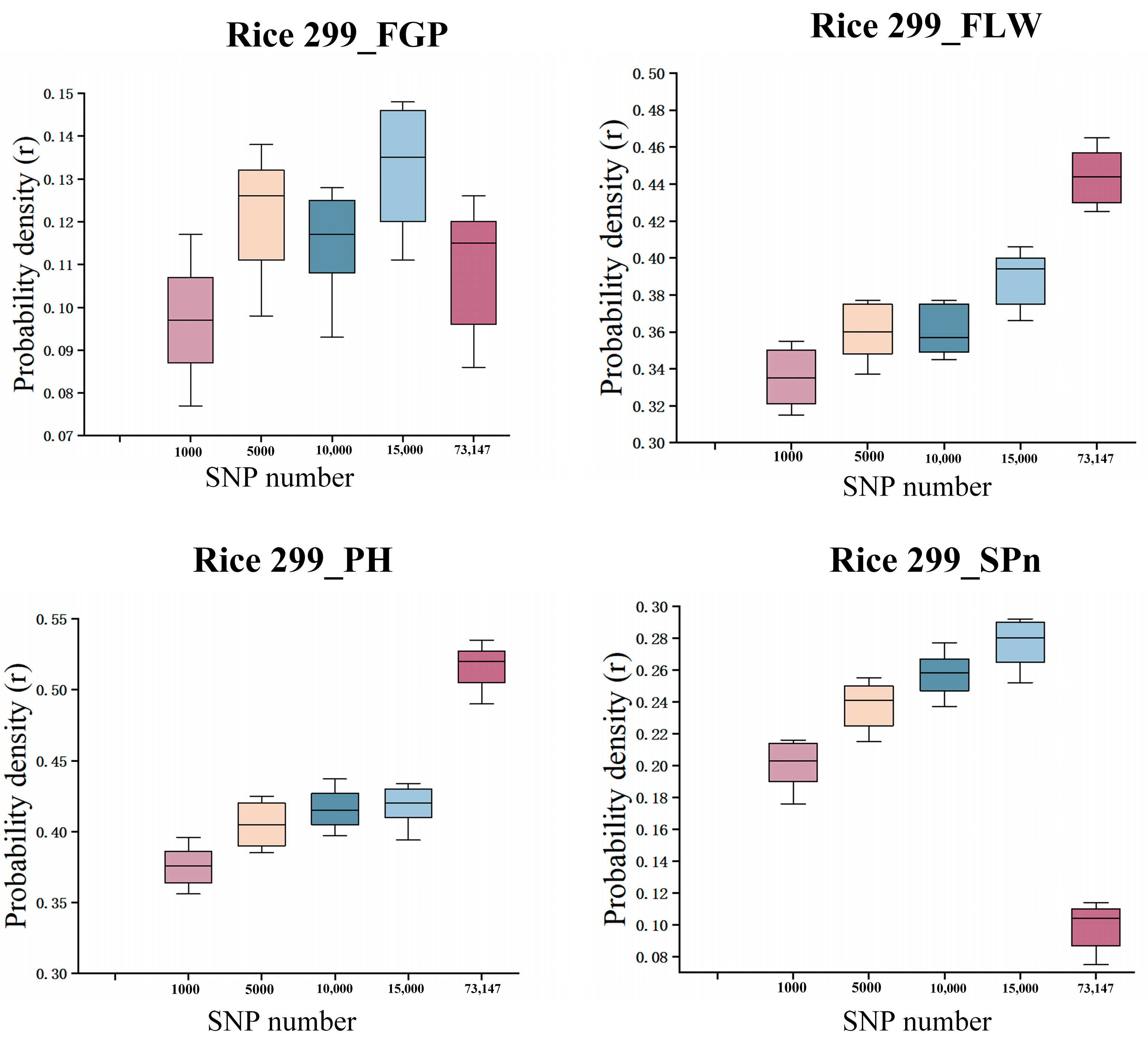
3.4. The Impact of Randomly Selecting the Number of SNPs on Prediction Methods
3.5. The Application Outcomes of GS Based on LSTM-ResNet
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ma, K.; Zhao, Y.; Wang, X.; Zhou, K.; Yu, G.; Li, C.; Li, P.; Yang, Z.; Xu, C. Genomic selection: A breakthrough technology in rice breeding. Crop J. 2021, 9, 669–677. [Google Scholar] [CrossRef]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; De Los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y. Genomic selection in plant breeding: Methods, models, and perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef]
- Tong, H.; Nikoloski, Z. Machine learning approaches for crop improvement: Leveraging phenotypic and genotypic big data. J. Plant Physiol. 2021, 257, 153354. [Google Scholar] [CrossRef]
- Gao, H.; Christensen, O.F.; Madsen, P.; Nielsen, U.S.; Zhang, Y.; Lund, M.S.; Su, G. Comparison on genomic predictions using three GBLUP methods and two single-step blending methods in the Nordic Holstein population. Genet. Sel. Evol. 2012, 44, 8. [Google Scholar] [CrossRef]
- Endelman, J.B. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- Pérez, P.; de Los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the Bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef]
- Munoz, P.; Resende, M.; Peter, G.; Huber, D.; Kirst, M.; Quesada, T. Effect of BLUP prediction on genomic selection: Practical considerations to achieve greater accuracy in genomic selection. BMC Proc. 2011, 5, P49. [Google Scholar] [CrossRef]
- Jiang, S.; Cheng, Q.; Yan, J.; Fu, R.; Wang, X. Genome optimization for improvement of maize breeding. Theor. Appl. Genet. 2020, 133, 1491–1502. [Google Scholar] [CrossRef]
- Ogutu, J.O.; Piepho, H.-P.; Schulz-Streeck, T. A comparison of random forests, boosting and support vector machines for genomic selection. BMC Proc. 2011, 5, S11. [Google Scholar] [CrossRef] [PubMed]
- Montesinos-López, A.; Montesinos-López, O.A.; Montesinos-López, J.C.; Flores-Cortes, C.A.; de la Rosa, R.; Crossa, J. A guide for kernel generalized regression methods for genomic-enabled prediction. Heredity 2021, 126, 577–596. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Basak, S.; Saikia, P.; Paul, S.; Tsalavoutis, V.; Atiah, F.; Ravi, V.; Peters, A. A review of deep learning with special emphasis on architectures, applications and recent trends. Knowl.-Based Syst. 2020, 194, 105596. [Google Scholar] [CrossRef]
- Chan, K.Y.; Abu-Salih, B.; Qaddoura, R.; Ala’M, A.-Z.; Palade, V.; Pham, D.-S.; Del Ser, J.; Muhammad, K. Deep neural networks in the cloud: Review, applications, challenges and research directions. Neurocomputing 2023, 545, 126327. [Google Scholar] [CrossRef]
- Chen, J.-F.; Do, Q.H.; Hsieh, H.-N. Training artificial neural networks by a hybrid PSO-CS algorithm. Algorithms 2015, 8, 292–308. [Google Scholar] [CrossRef]
- LeCun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 1998, 86, 2278–2324. [Google Scholar] [CrossRef]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Montesinos-López, A.; Montesinos-López, O.A.; Gianola, D.; Crossa, J.; Hernández-Suárez, C.M. Multi-environment genomic prediction of plant traits using deep learners with dense architecture. G3 Genes Genomes Genet. 2018, 8, 3813–3828. [Google Scholar] [CrossRef]
- Ma, W.; Qiu, Z.; Song, J.; Li, J.; Cheng, Q.; Zhai, J.; Ma, C. A deep convolutional neural network approach for predicting phenotypes from genotypes. Planta 2018, 248, 1307–1318. [Google Scholar] [CrossRef]
- Wang, K.; Abid, M.A.; Rasheed, A.; Crossa, J.; Hearne, S.; Li, H. DNNGP, a deep neural network-based method for genomic prediction using multi-omics data in plants. Mol. Plant 2023, 16, 279–293. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, D.; He, F.; Wang, J.; Joshi, T.; Xu, D. Phenotype prediction and genome-wide association study using deep convolutional neural network of soybean. Front. Genet. 2019, 10, 1091. [Google Scholar] [CrossRef] [PubMed]
- Zingaretti, L.M.; Gezan, S.A.; Ferrão, L.F.V.; Osorio, L.F.; Monfort, A.; Muñoz, P.R.; Whitaker, V.M.; Pérez-Enciso, M. Exploring deep learning for complex trait genomic prediction in polyploid outcrossing species. Front. Plant Sci. 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, F.; Yang, W.; Peng, S.; Zhou, J. A survey of convolutional neural networks: Analysis, applications, and prospects. IEEE Trans. Neural Netw. Learn. Syst. 2021, 33, 6999–7019. [Google Scholar] [CrossRef]
- Lindemann, B.; Müller, T.; Vietz, H.; Jazdi, N.; Weyrich, M. A survey on long short-term memory networks for time series prediction. Procedia Cirp 2021, 99, 650–655. [Google Scholar] [CrossRef]
- Wu, D.; Wang, Y.; Xia, S.-T.; Bailey, J.; Ma, X. Skip connections matter: On the transferability of adversarial examples generated with resnets. arXiv 2020, arXiv:2002.05990. [Google Scholar]
- Zhang, D.; Yang, F.; Li, J.; Liu, Z.; Han, Y.; Zhang, Q.; Pan, S.; Zhao, X.; Wang, K. Progress and perspectives on genomic selection models for crop breeding. Technol. Agron. 2025, 5, e006. [Google Scholar] [CrossRef]
- Mclaren, C.G.; Bruskiewich, R.M.; Portugal, A.M.; Cosico, A.B. The International Rice Information System. A Platform for Meta-Analysis of Rice Crop Data. Plant Physiol. 2005, 139, 637–642. [Google Scholar] [CrossRef]
- Crossa, J.; Jarquín, D.; Franco, J.; Pérez-Rodríguez, P.; Burgueño, J.; Saint-Pierre, C.; Vikram, P.; Sansaloni, C.; Petroli, C.; Akdemir, D. Genomic prediction of gene bank wheat landraces. G3 Genes Genomes Genet. 2016, 6, 1819–1834. [Google Scholar] [CrossRef]
- Spindel, J.; Begum, H.; Akdemir, D.; Virk, P.; Collard, B.; Redona, E.; Atlin, G.; Jannink, J.-L.; McCouch, S.R. Genomic selection and association mapping in rice (Oryza sativa): Effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet. 2015, 11, e1004982. [Google Scholar] [CrossRef]
- McFarland, B.A.; AlKhalifah, N.; Bohn, M.; Bubert, J.; Buckler, E.S.; Ciampitti, I.; Edwards, J.; Ertl, D.; Gage, J.L.; Falcon, C.M. Maize genomes to fields (G2F): 2014–2017 field seasons: Genotype, phenotype, climatic, soil, and inbred ear image datasets. BMC Res. Notes 2020, 13, 71. [Google Scholar] [CrossRef]
- Paszke, A.; Gross, S.; Chintala, S.; Chanan, G.; Yang, E.; DeVito, Z.; Lin, Z.; Desmaison, A.; Antiga, L.; Lerer, A. Automatic differentiation in pytorch. In Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Meher, P.K.; Rustgi, S.; Kumar, A. Performance of Bayesian and BLUP alphabets for genomic prediction: Analysis, comparison and results. Heredity 2022, 128, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Bengio, Y.; Grandvalet, Y. No unbiased estimator of the variance of k-fold cross-validation. J. Mach. Learn. Res. 2004, 5, 1089–1105. [Google Scholar] [CrossRef]
- Hayes, B. Overview of statistical methods for genome-wide association studies (GWAS). In Genome-Wide Association Studies and Genomic Prediction; Humana Press: Totowa, NJ, USA, 2013; pp. 149–169. [Google Scholar] [CrossRef]
- Stroup, W.W.; Ptukhina, M.; Garai, J. Generalized Linear Mixed Models: Modern Concepts, Methods and Applications; Chapman and Hall/CRC: Boca Raton, FL, USA, 2024. [Google Scholar]
- Wang, J.; Zhang, Z. GAPIT version 3: Boosting power and accuracy for genomic association and prediction. Genom. Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Greenacre, M.; Groenen, P.J.; Hastie, T.; d’Enza, A.I.; Markos, A.; Tuzhilina, E. Principal component analysis. Nat. Rev. Methods Primers 2022, 2, 100. [Google Scholar] [CrossRef]
- Boopathi, N.M.; Boopathi, N.M. Marker-assisted selection (MAS). In Genetic Mapping and Marker Assisted Selection: Basics, Practice and Benefits; Springer: New Delhi, India, 2020; pp. 343–388. [Google Scholar]
- Heffner, E.L.; Jannink, J.L.; Sorrells, M.E. Genomic selection accuracy using multifamily prediction models in a wheat breeding program. Plant Genome 2011, 4, 65–75. [Google Scholar] [CrossRef]
- Wang, X.; Yang, Z.; Xu, C. A comparison of genomic selection methods for breeding value prediction. Sci. Bull. 2015, 60, 925–935. [Google Scholar] [CrossRef]
- Ma, X.; Wang, H.; Wu, S.; Han, B.; Cui, D.; Liu, J.; Zhang, Q.; Xia, X.; Song, P.; Tang, C.; et al. DeepCCR: Large-scale genomics-based deep learning method for improving rice breeding. Plant Biotechnol. J. 2024, 22, 2691–2693. [Google Scholar] [CrossRef]
- Gao, P.; Zhao, H.; Luo, Z.; Lin, Y.; Feng, W.; Li, Y.; Kong, F.; Li, X.; Fang, C.; Wang, X. SoyDNGP: A web-accessible deep learning framework for genomic prediction in soybean breeding. Brief. Bioinform. 2023, 24, bbad349. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, Y.; Ying, Z.; Li, L.; Wang, J.; Yu, H.; Zhang, M.; Feng, X.; Wei, X.; Xu, X. A transformer-based genomic prediction method fused with knowledge-guided module. Brief. Bioinform. 2024, 25, bbad438. [Google Scholar] [CrossRef]
- Wang, H.; Yan, S.; Wang, W.; Chen, Y.; Hong, J.; He, Q.; Diao, X.; Lin, Y.; Chen, Y.; Cao, Y.; et al. Cropformer: An interpretable deep learning framework for crop genomic prediction. Plant Commun. 2025, 6, 101223. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Alam, M.S.B.; Hassan, M.; Rozbu, M.R.; Ishtiak, T.; Rafa, N.; Mofijur, M.; Shawkat Ali, A.; Gandomi, A.H. Deep learning modelling techniques: Current progress, applications, advantages, and challenges. Artif. Intell. Rev. 2023, 56, 13521–13617. [Google Scholar] [CrossRef]
- Abdollahi-Arpanahi, R.; Gianola, D.; Peñagaricano, F. Deep learning versus parametric and ensemble methods for genomic prediction of complex phenotypes. Genet. Sel. Evol. 2020, 52, 12. [Google Scholar] [CrossRef] [PubMed]
- Pook, T.; Freudenthal, J.; Korte, A.; Simianer, H. Using Local Convolutional Neural Networks for Genomic Prediction. Front. Genet. 2020, 11, 561497. [Google Scholar] [CrossRef]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. Lightgbm: A highly efficient gradient boosting decision tree. Adv. Neural Inf. Process. Syst. 2017, 30, 3149–3157. [Google Scholar]
- He, X.; Wang, K.; Zhang, L.; Zhang, D.; Yang, F.; Zhang, Q.; Pan, S.; Li, J.; Bai, L.; Sun, J.; et al. HGATGS: Hypergraph Attention Network for Crop Genomic Selection. Agriculture 2025, 15, 409. [Google Scholar] [CrossRef]
- Helyar, S.J.; Hemmer-Hansen, J.; Bekkevold, D.; Taylor, M.I.; Ogden, R.; Limborg, M.T.; Cariani, A.; Maes, G.E.; Diopere, E.; Carvalho, G. Application of SNPs for population genetics of nonmodel organisms: New opportunities and challenges. Mol. Ecol. Resour. 2011, 11, 123–136. [Google Scholar] [CrossRef]
- Chapman, D.; Pescott, O.L.; Roy, H.E.; Tanner, R. Improving species distribution models for invasive non-native species with biologically informed pseudo-absence selection. J. Biogeogr. 2019, 46, 1029–1040. [Google Scholar] [CrossRef]
- Uppu, S.; Krishna, A.; Gopalan, R.P. A review on methods for detecting SNP interactions in high-dimensional genomic data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 15, 599–612. [Google Scholar] [CrossRef]
- e Sousa, M.B.; Galli, G.; Lyra, D.H.; Granato, Í.S.C.; Matias, F.I.; Alves, F.C.; Fritsche-Neto, R. Increasing accuracy and reducing costs of genomic prediction by marker selection. Euphytica 2019, 215, 18. [Google Scholar] [CrossRef]
- Kuppuraj, S.A.M. Genomic Selection for Phenotype. In Climate-Smart Rice Breeding; Springer: Singapore, 2024; p. 167. [Google Scholar]
- Xu, Y.; Lu, Y.; Xie, C.; Gao, S.; Wan, J.; Prasanna, B.M. Whole-genome strategies for marker-assisted plant breeding. Mol. Breed. 2012, 29, 833–854. [Google Scholar] [CrossRef]
- Voss-Fels, K.P.; Cooper, M.; Hayes, B.J. Accelerating crop genetic gains with genomic selection. Theor. Appl. Genet. 2019, 132, 669–686. [Google Scholar] [CrossRef] [PubMed]
- Alahmad, S.; Rambla, C.; Voss-Fels, K.P.; Hickey, L.T. Accelerating breeding cycles. In Wheat Improvement: Food Security in a Changing Climate; Springer International Publishing: Cham, Switzerland, 2022; pp. 557–571. [Google Scholar]
- Montesinos-López, O.A.; Montesinos-López, A.; Hernandez-Suarez, C.M.; Barrón-López, J.A.; Crossa, J. Deep-learning power and perspectives for genomic selection. Plant Genome 2021, 14, e20122. [Google Scholar] [CrossRef] [PubMed]
- Nagahisarchoghaei, M.; Karimi, M.M.; Rahimi, S.; Cummins, L.; Ghanbari, G. Generative local interpretable model-agnostic explanations. In Proceedings of the The International FLAIRS Conference, Clearwater, FL, USA, 14–17 May 2023. [Google Scholar]
- Van den Broeck, G.; Lykov, A.; Schleich, M.; Suciu, D. On the tractability of SHAP explanations. J. Artif. Intell. Res. 2022, 74, 851–886. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Dataset | Population Size | Marker Number | Environment | Trait | Abbr. |
|---|---|---|---|---|---|---|
| Wheat | Wheat599 | 599 | 1279 | env1 | grain yield | GY |
| Wheat | Wheat599 | 599 | 1279 | env2 | grain yield | GY |
| Wheat | Wheat599 | 599 | 1279 | env3 | grain yield | GY |
| Wheat | Wheat599 | 599 | 1279 | env4 | grain yield | GY |
| Wheat | Wheat2000 | 2000 | 33,709 | 1 | thousand kernel weight | TKW |
| Wheat | Wheat2000 | 2000 | 33,709 | 1 | test weight | TW |
| Wheat | Wheat2000 | 2000 | 33,709 | 1 | grain length | GL |
| Wheat | Wheat2000 | 2000 | 33,709 | 1 | grain width | GW |
| Wheat | Wheat2000 | 2000 | 33,709 | 1 | grain hardness | GH |
| Wheat | Wheat2000 | 2000 | 33,709 | 1 | grain protein | GP |
| Rice | Rice299 | 299 | 73,147 | 1 | filled grain percentage | FGP |
| Rice | Rice299 | 299 | 73,147 | 1 | flag leaf width | FLW |
| Rice | Rice299 | 299 | 73,147 | 1 | plant height | PH |
| Rice | Rice299 | 299 | 73,147 | 1 | spikelet number per Panicle | SPN |
| Maize | G2F_2017 | 356 | 21,011 | 1 | days to pollen | DTP |
| Maize | G2F_2017 | 356 | 21,011 | 1 | ear height | EH |
| Maize | G2F_2017 | 356 | 21,011 | 1 | plant height | PH |
| Maize | G2F_2017 | 356 | 21,011 | 1 | yield | Y |
| Model/Layer | CNN Layer | Flatten Layer | ResNet Layer | Fully Connected (FC) Layer | Dropout Layer | ReLU Layer | The Quantity of Highest Scores | Traits with Highest Accuracy |
|---|---|---|---|---|---|---|---|---|
| CNN-ResNet | Conv1d, ReLU, Maxpool1d, Flatten | √ | Linear, Flatten, Skip Connected | √ | √ | √ | 1 | Wheat 2000_GP |
| CNN-LSTM | Conv1d, ReLU, Maxpool1d, Flatten | √ | × | √ | √ | √ | × | × |
| CNN-ResNet-LSTM | Conv1d, ReLU, Maxpool1d, Flatten | √ | Linear, Flatten, Skip Connected | √ | √ | √ | 4 | Wheat 599_env3; Wheat 2000_TKW; Rice 299_SPn; G2F_2017_DTP |
| LSTM-ResNet | × | × | Linear, Flatten, Skip Connected | √ | √ | √ | 10 | Wheat 599_env1/env2/env4; Wheat 2000_TW/GL/GW/GH; Rice 299_PH; G2F_2017_PH/Yield |
| LSTM | × | × | × | √ | √ | √ | 1 | Wheat 599_FGP |
| BayesA | × | × | × | × | × | × | 1 | G2F_2017_EH |
| BayesB | × | × | × | × | × | × | 1 | Rice 299_FLW |
| Datasets | GS | MAS TOP 100 SNP | MAS TOP 10 SNP | Random |
|---|---|---|---|---|
| Wheat599_env1 | 0.539 | 0.569 | 0.281 | 0.021 |
| Wheat599_env2 | 0.447 | 0.449 | 0.233 | −0.113 |
| Wheat599_env3 | 0.349 | 0.593 | 0.084 | −0.168 |
| Wheat599_env4 | 0.444 | 0.491 | 0.331 | −0.034 |
| Wheat2000_TKW | 0.540 | 0.457 | 0.231 | −0.078 |
| Wheat2000_TW | 0.463 | 0.104 | 0.097 | 0.022 |
| Wheat2000_GL | 0.552 | 0.572 | 0.294 | −0.122 |
| Wheat2000_GW | 0.586 | 0.466 | 0.238 | −0.006 |
| Wheat2000_GH | 0.591 | 0.538 | 0.235 | −0.002 |
| Wheat2000_GP | 0.458 | 0.572 | 0.292 | −0.052 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Zhang, D.; Han, Y.; Liu, Z.; Zhang, Q.; Zhang, Q.; Wang, X.; Pan, S.; Sun, J.; Wang, K. Hybrid Deep Learning Approaches for Improved Genomic Prediction in Crop Breeding. Agriculture 2025, 15, 1171. https://doi.org/10.3390/agriculture15111171
Li R, Zhang D, Han Y, Liu Z, Zhang Q, Zhang Q, Wang X, Pan S, Sun J, Wang K. Hybrid Deep Learning Approaches for Improved Genomic Prediction in Crop Breeding. Agriculture. 2025; 15(11):1171. https://doi.org/10.3390/agriculture15111171
Chicago/Turabian StyleLi, Ran, Dongfeng Zhang, Yanyun Han, Zhongqiang Liu, Qiusi Zhang, Qi Zhang, Xiaofeng Wang, Shouhui Pan, Jiahao Sun, and Kaiyi Wang. 2025. "Hybrid Deep Learning Approaches for Improved Genomic Prediction in Crop Breeding" Agriculture 15, no. 11: 1171. https://doi.org/10.3390/agriculture15111171
APA StyleLi, R., Zhang, D., Han, Y., Liu, Z., Zhang, Q., Zhang, Q., Wang, X., Pan, S., Sun, J., & Wang, K. (2025). Hybrid Deep Learning Approaches for Improved Genomic Prediction in Crop Breeding. Agriculture, 15(11), 1171. https://doi.org/10.3390/agriculture15111171