
Response of Soil Microbial Community Composition and Diversity at Different Gradients of Grassland Degradation in Central Mongolia
Abstract
:1. Introduction
2. Materials and Methods
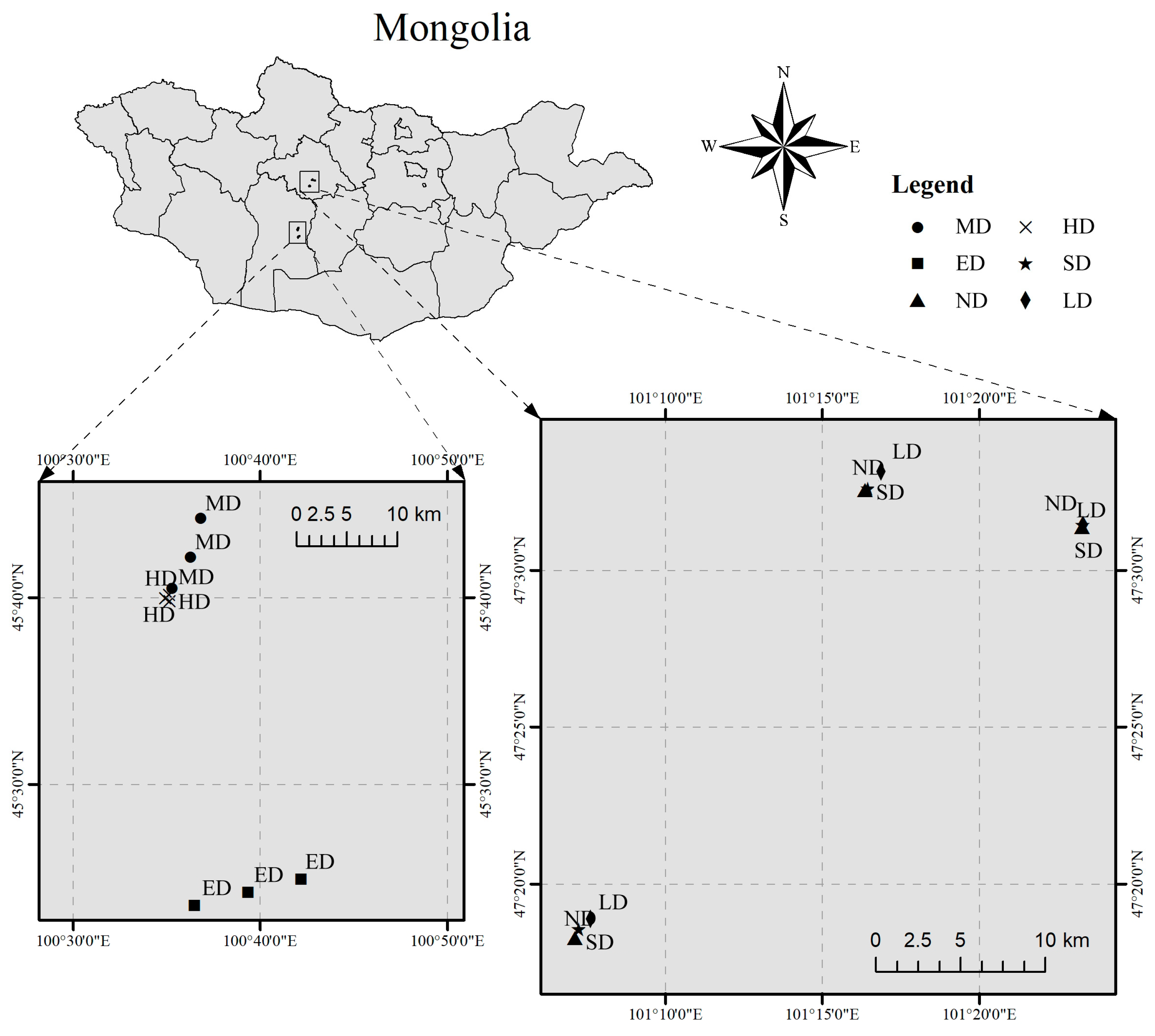
2.1. Soil Sampling and Preparation
2.2. Soil Chemical/Physical Properties and Enzyme Activities
2.3. DNA Extraction, Amplification and Sequencing
2.4. Bioinformatics and Statistical Analysis
3. Results
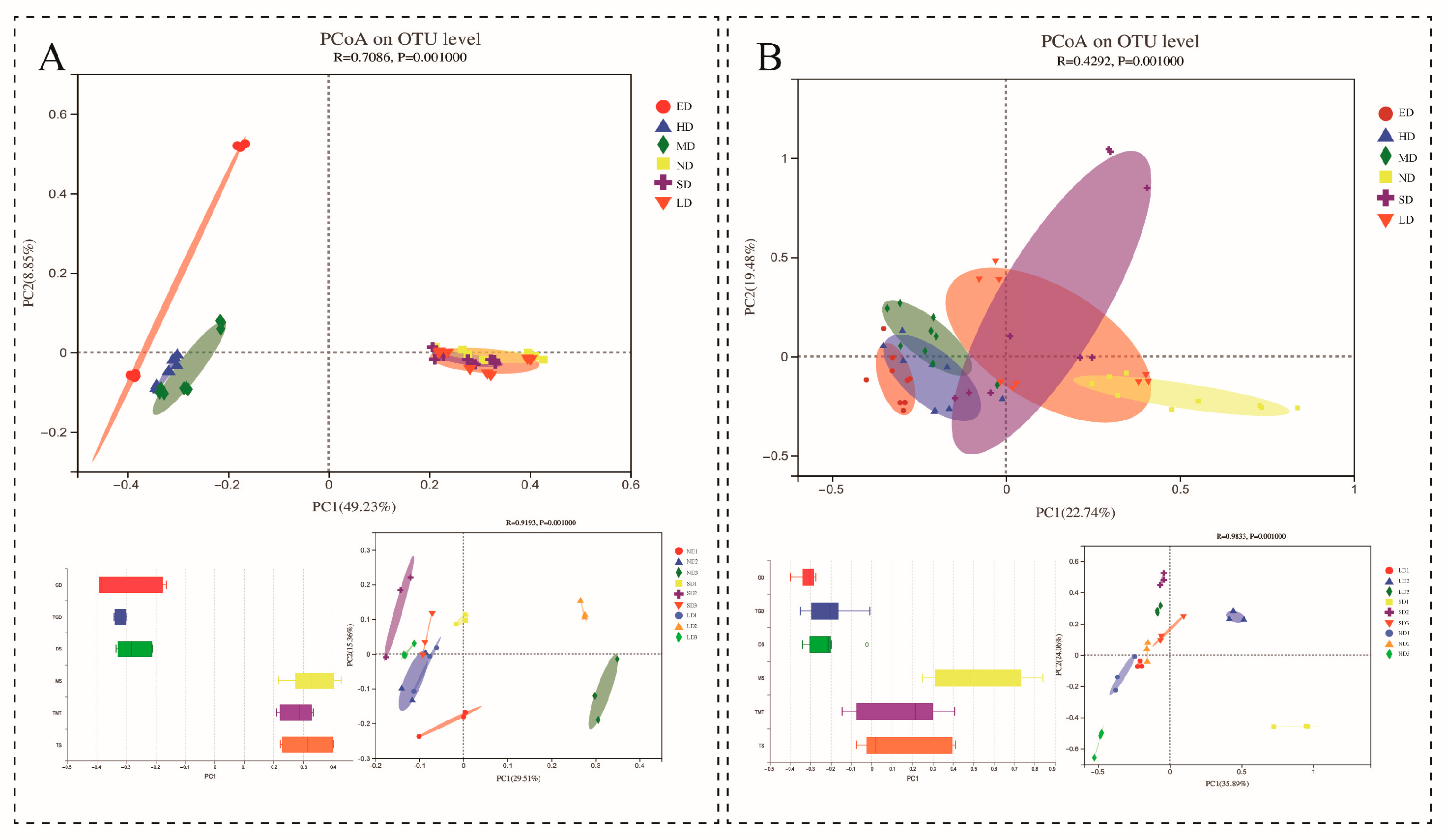
3.1. Microbial Community Composition and Diversity
3.2. Soil Microbial Difference Analysis between Different Grassland Degradation Gradients
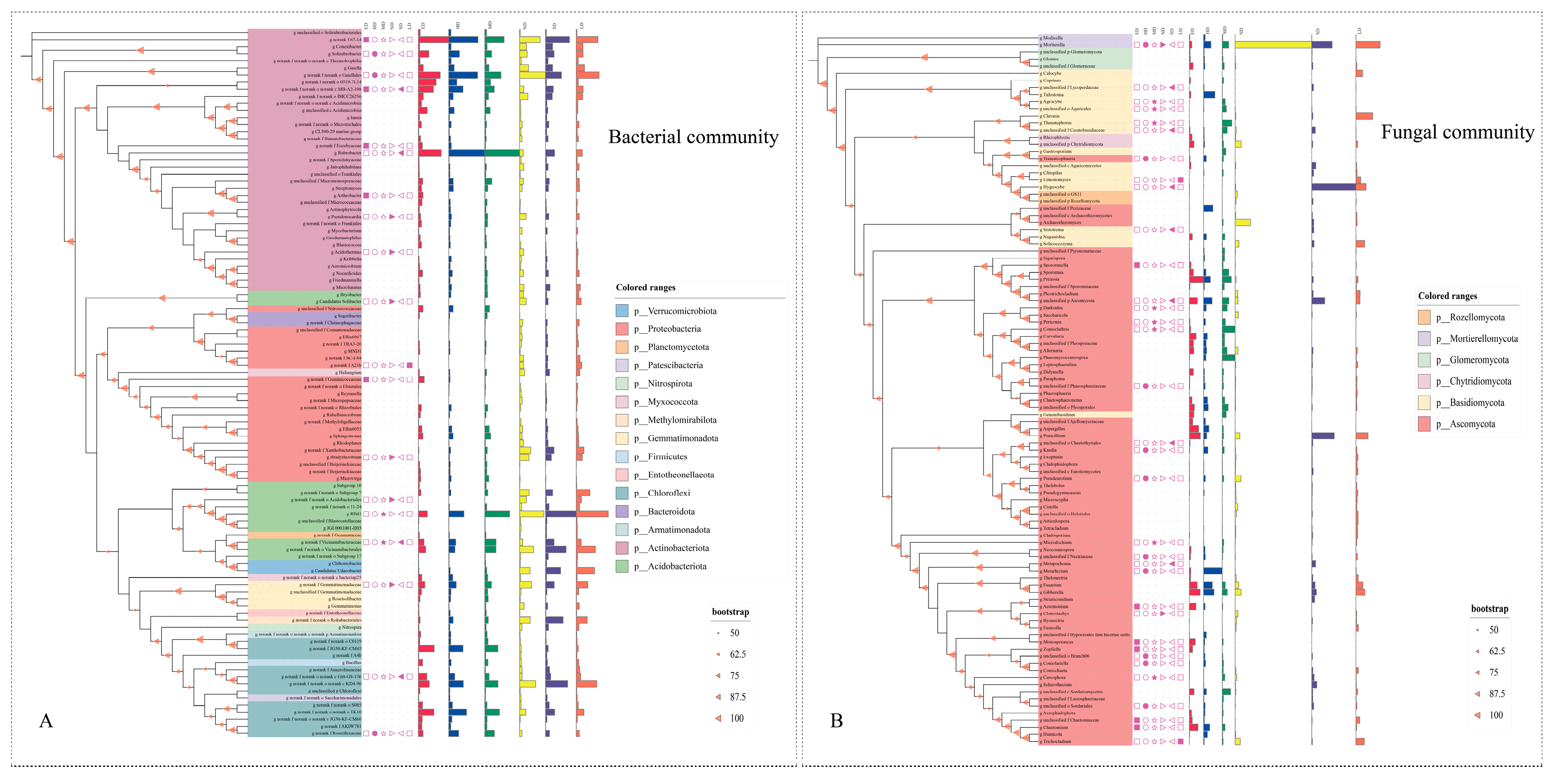
3.3. Phylogenetic Analysis
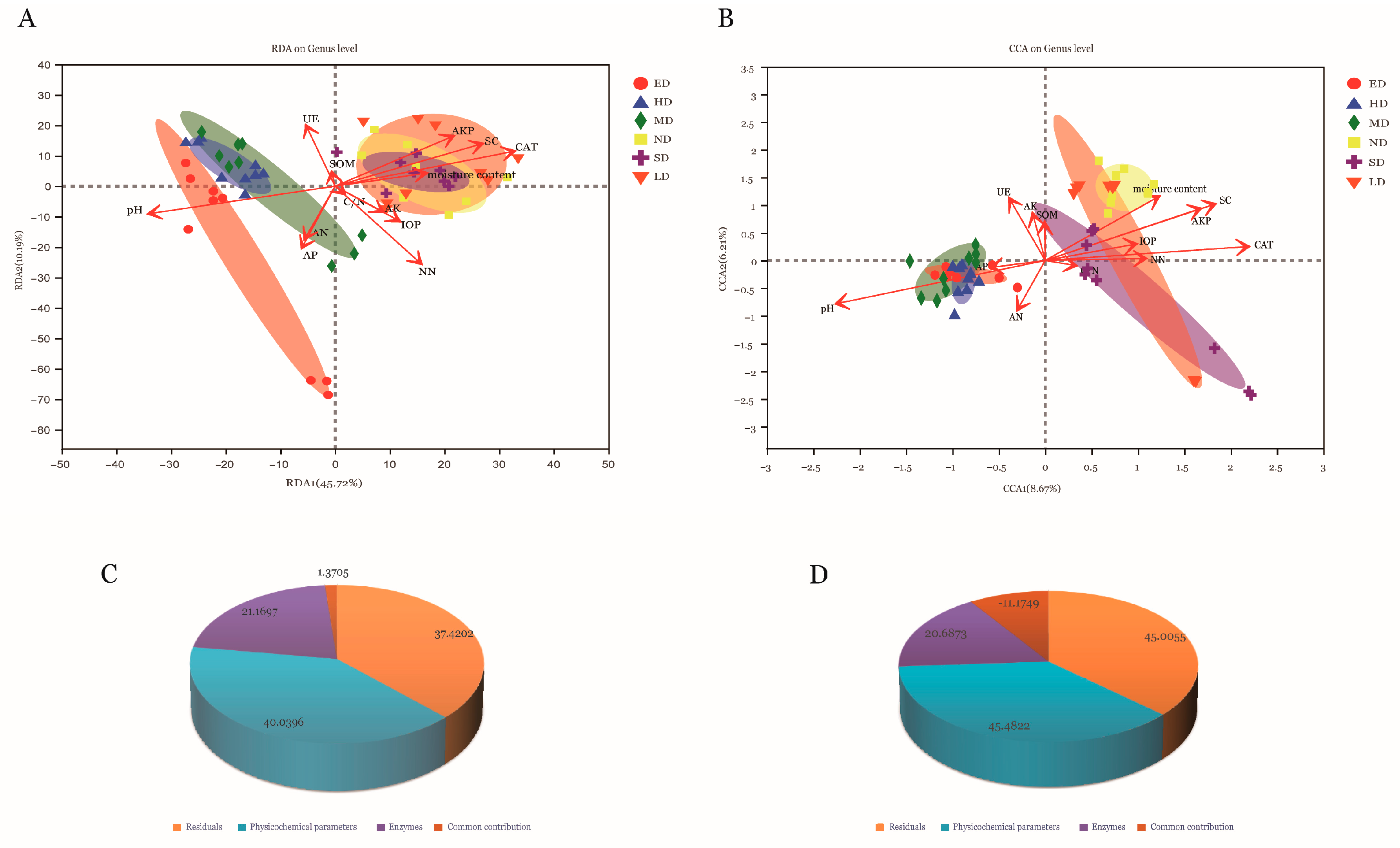
3.4. Relationships between Microbial Community Structures and Environmental Factors
4. Discussion
4.1. Changes in the Soil Microbial Communities at Different Grassland Degradation Gradients
4.2. Comparison of Differences and Phylogenetic Trees of Soil Microorganisms at Different Grassland Degradation Gradients
4.3. Relationships between Soil Microbial Community Structure and Environmental Factors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Tumenjargal, S.; Fassnacht, S.R.; Venable, N.B.H.; Kingston, A.P.; Fernández-Giménez, M.E.; Batbuyan, B.; Laituri, M.J.; Kappas, M.; Adyabadam, G. Variability and change of climate extremes from indigenous herder knowledge and at meteorological stations across central Mongolia. Front. Earth Sci. 2020, 14, 286–297. [Google Scholar] [CrossRef]
- Kusakabe, R.; Taniguchi, T.; Goomaral, A.; Undarmaa, J.; Yamanaka, N.; Yamato, M. Arbuscular mycorrhizal fungal communities under gradients of grazing in Mongolian grasslands of different aridity. Mycorrhiza 2018, 28, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Jamsranjav, C.; Fernandez-Gimenez, M.; Reid, R.S.; Adya, B. Opportunities to integrate herders’ indicators into formal rangeland monitoring: An example from Mongolia. Ecol. Appl. 2019, 29, e01899. [Google Scholar] [CrossRef] [PubMed]
- Su, N.; Jarvie, S.; Yan, Y.; Gong, X.; Li, F.; Han, P.; Zhang, Q. Landscape context determines soil fungal diversity in a fragmented habitat. CATENA 2022, 213, 106163. [Google Scholar] [CrossRef]
- Lladó, S.; López-Mondéjar, R.; Baldrian, P. Forest Soil Bacteria: Diversity, Involvement in Ecosystem Processes, and Response to Global Change. Microbiol. Mol. Biol. Rev. 2017, 81, e00063-16. [Google Scholar] [CrossRef]
- Mącik, M.; Gryta, A.; Sas-Paszt, L.; Frąc, M. The Status of Soil Microbiome as Affected by the Application of Phosphorus Biofertilizer: Fertilizer Enriched with Beneficial Bacterial Strains. Int. J. Mol. Sci. 2020, 21, 8003. [Google Scholar] [CrossRef] [PubMed]
- Jansson, J.K.; Hofmockel, K.S. Soil microbiomes and climate change. Nat. Rev. Microbiol. 2020, 18, 35–46. [Google Scholar] [CrossRef]
- Wei, H.; Wang, L.; Hassan, M.; Xie, B. Succession of the functional microbial communities and the metabolic functions in maize straw composting process. Bioresour. Technol. 2018, 256, 333–341. [Google Scholar] [CrossRef]
- Cui, Y.; Fang, L.; Guo, X.; Wang, X.; Wang, Y.; Li, P.; Zhang, Y.; Zhang, X. Responses of soil microbial communities to nutrient limitation in the desert-grassland ecological transition zone. Sci. Total Environ. 2018, 642, 45–55. [Google Scholar] [CrossRef]
- Metcalf, J.L.; Xu, Z.Z.; Weiss, S.; Lax, S.; Van Treuren, W.; Hyde, E.R.; Song, S.J.; Amir, A.; Larsen, P.; Sangwan, N.; et al. Microbial community assembly and metabolic function during mammalian corpse decomposition. Science 2016, 351, 158–162. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Kang, H.; Cui, Y.; Lu, S.; Yang, H.; Zhu, J.; Fu, Z.; Yan, C.; Wang, D. Consistent Plant and Microbe Nutrient Limitation Patterns During Natural Vegetation Restoration. Front. Plant Sci. 2022, 13, 885984. [Google Scholar] [CrossRef]
- Cui, H.; Sun, W.; Delgado-Baquerizo, M.; Song, W.; Ma, J.-Y.; Wang, K.; Ling, X. The effects of mowing and multi-level N fertilization on soil bacterial and fungal communities in a semiarid grassland are year-dependent. Soil Biol. Biochem. 2020, 151, 108040. [Google Scholar] [CrossRef]
- Chen, L.; Baoyin, T.; Minggagud, H. Effects of mowing regimes on above- and belowground biota in semi-arid grassland of northern China. J. Environ. Manag. 2020, 277, 111441. [Google Scholar] [CrossRef]
- Yang, X.; Zhu, K.; Loik, M.E.; Sun, W. Differential responses of soil bacteria and fungi to altered precipitation in a meadow steppe. Geoderma 2020, 384, 114812. [Google Scholar] [CrossRef]
- Erlandson, S.; Wei, X.; Savage, J.; Cavender-Bares, J.; Peay, K. Soil abiotic variables are more important than Salicaceae phylogeny or habitat specialization in determining soil microbial community structure. Mol. Ecol. 2018, 27, 2007–2024. [Google Scholar] [CrossRef]
- Nan, J.; Chao, L.; Ma, X.; Xu, D.; Mo, L.; Zhang, X.; Zhao, X.; Bao, Y. Microbial diversity in the rhizosphere soils of three Stipa species from the eastern Inner Mongolian grasslands. Glob. Ecol. Conserv. 2020, 22, e00992. [Google Scholar] [CrossRef]
- Toju, H.; Peay, K.G.; Yamamichi, M.; Narisawa, K.; Hiruma, K.; Naito, K.; Fukuda, S.; Ushio, M.; Nakaoka, S.; Onoda, Y.; et al. Core microbiomes for sustainable agroecosystems. Nat. Plants 2018, 4, 247–257. [Google Scholar] [CrossRef]
- Wan, W.; Tan, J.; Wang, Y.; Qin, Y.; He, H.; Wu, H.; Zuo, W.; He, D. Responses of the rhizosphere bacterial community in acidic crop soil to pH: Changes in diversity, composition, interaction, and function. Sci. Total Environ. 2019, 700, 134418. [Google Scholar] [CrossRef]
- Ramírez, P.B.; Fuentes-Alburquenque, S.; Díez, B.; Vargas, I.; Bonilla, C.A. Soil microbial community responses to labile organic carbon fractions in relation to soil type and land use along a climate gradient. Soil Biol. Biochem. 2019, 141, 107692. [Google Scholar] [CrossRef]
- Zhao, X.; Huang, J.; Lu, J.; Sun, Y. Study on the influence of soil microbial community on the long-term heavy metal pollution of different land use types and depth layers in mine. Ecotoxicol. Environ. Saf. 2018, 170, 218–226. [Google Scholar] [CrossRef]
- Zhou, C.; Heal, K.; Tigabu, M.; Xia, L.; Hu, H.; Yin, D.; Ma, X. Corrigendum to “Biochar addition to forest plantation soil enhances phosphorus availability and soil bacterial community diversity”. For. Ecol. Manag. 2020, 461, 117857. [Google Scholar] [CrossRef]
- Zhang, P.; Li, B.; Wu, J.; Hu, S. Invasive plants differentially affect soil biota through litter and rhizosphere pathways: A meta-analysis. Ecol. Lett. 2018, 22, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.F. Conservation evaluation and phylogenetic diversity. Biol Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Chen, J.; Xu, D.; Chao, L.; Liu, H.; Bao, Y. Microbial assemblages associated with the rhizosphere and endosphere of an herbage, Leymus chinensis. Microb. Biotechnol. 2020, 13, 1390–1402. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, Q.; Jarvie, S.; Yan, Y.; Han, P.; Liu, T.; Guo, K.; Ren, L.; Yue, K.; Wu, H.; et al. Ecosystem restoration through aerial seeding: Interacting plant–soil microbiome effects on soil multifunctionality. Land Degrad. Dev. 2021, 32, 5334–5347. [Google Scholar] [CrossRef]
- Marano, A.V.; Pires-Zottarelli, C.L.A.; Barrera, M.D.; Steciow, M.M.; Gleason, F.H. Diversity, role in decomposition, and succession of zoosporic fungi and straminipiles on submerged decaying leaves in a woodland stream. Hydrobiologia 2010, 659, 93–109. [Google Scholar] [CrossRef]
- Lee, E.-H.; Sohn, B.-J. Recent increasing trend in dust frequency over Mongolia and Inner Mongolia regions and its association with climate and surface condition change. Atmos. Environ. 2011, 45, 4611–4616. [Google Scholar] [CrossRef]
- Yuan, J.; Wen, T.; Zhang, H.; Zhao, M.; Penton, C.R.; Thomashow, L.S.; Shen, Q. Predicting disease occurrence with high accuracy based on soil macroecological patterns of Fusarium wilt. ISME J. 2020, 14, 2936–2950. [Google Scholar] [CrossRef]
- Wu, S.-H.; Huang, B.-H.; Huang, C.-L.; Li, G.; Liao, P.-C. The Aboveground Vegetation Type and Underground Soil Property Mediate the Divergence of Soil Microbiomes and the Biological Interactions. Microb. Ecol. 2017, 75, 434–446. [Google Scholar] [CrossRef]
- Liang, Y.; Jiang, Y.; Wang, F.; Wen, C.; Deng, Y.; Xue, K.; Qin, Y.; Yang, Y.; Wu, L.; Zhou, J.; et al. Long-term soil transplant simulating climate change with latitude significantly alters microbial temporal turnover. ISME J. 2015, 9, 2561–2572. [Google Scholar] [CrossRef]
- Chen, Y.; Ma, S.; Jiang, H.; Hu, Y.; Lu, X. Influences of litter diversity and soil moisture on soil microbial communities in decomposing mixed litter of alpine steppe species. Geoderma 2020, 377, 114577. [Google Scholar] [CrossRef]
- Fan, M.; Li, J.; Tang, Z.; Shangguan, Z. Soil bacterial community succession during desertification in a desert steppe ecosystem. Land Degrad. Dev. 2020, 31, 1662–1674. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, S.; Niu, B.; Chen, Q.; Wang, J.; Zhao, J.; Luo, T.; Zhang, G. Effect of increasing precipitation and warming on microbial community in Tibetan alpine steppe. Environ. Res. 2020, 189, 109917. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Na, R.; Koziol, L.; Schellenberg, M.P.; Li, X.; Ta, N.; Jin, K.; Wang, H. Response of bacterial communities and plant-mediated soil processes to nitrogen deposition and precipitation in a desert steppe. Plant Soil 2020, 448, 277–297. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Y.; Zhang, P.; Song, G.; Hui, R.; Wang, Z.; Wang, J. Development of bacterial communities in biological soil crusts along a revegetation chronosequence in the Tengger Desert, northwest China. Biogeosciences 2017, 14, 3801–3814. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, X.; Tai, X.; Sun, L.; Wu, M.; Zhang, W.; Chen, X.; Zhang, G.; Chen, T.; Liu, G.; et al. Variation in Actinobacterial Community Composition and Potential Function in Different Soil Ecosystems Belonging to the Arid Heihe River Basin of Northwest China. Front. Microbiol. 2019, 10, 2209. [Google Scholar] [CrossRef]
- Tang, K.; Yuan, B.; Jia, L.; Pan, X.; Feng, F.; Jin, K. Spatial and temporal distribution of aerobic anoxygenic phototrophic bacteria: Key functional groups in biological soil crusts. Environ. Microbiol. 2021, 23, 3554–3567. [Google Scholar] [CrossRef]
- Kristensen, J.M.; Singleton, C.; Clegg, L.-A.; Petriglieri, F.; Nielsen, P.H. High Diversity and Functional Potential of Undescribed “Acidobacteriota” in Danish Wastewater Treatment Plants. Front. Microbiol. 2021, 12, 643950. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Z.; Zhao, J.; Ma, J.; Yu, Q.; Zou, P.; Lin, H.; Ma, J. Fate of heavy metals and bacterial community composition following biogas slurry application in a single rice cropping system. J. Soils Sediments 2022, 22, 968–981. [Google Scholar] [CrossRef]
- Zhu, X.; Zhao, X.; Lin, Q.; Li, G. Distribution Characteristics of phoD-Harbouring Bacterial Community Structure and Its Roles in Phosphorus Transformation in Steppe Soils in Northern China. J. Soil Sci. Plant Nutr. 2021, 21, 1531–1541. [Google Scholar] [CrossRef]
- Wei, P.; An, S.; Dong, Y.; Sun, Z.; Hou, Y.; Bieerdawulieti, X. Diversity and Community Structure of Soil Fungi in Three Typical Desert Plant Formations in the Junggar Basin, Northwest China. Eurasian Soil Sci. 2021, 54, 1945–1956. [Google Scholar] [CrossRef]
- Uke, A.; Nakazono-Nagaoka, E.; Chuah, J.-A.; Zain, N.-A.A.; Amir, H.-G.; Sudesh, K.; Abidin, N.Z.H.A.Z.; Hashim, Z.; Kosugi, A. Effect of decomposing oil palm trunk fibers on plant growth and soil microbial community composition. J. Environ. Manag. 2021, 295, 113050. [Google Scholar] [CrossRef] [PubMed]
- Ozimek, E.; Hanaka, A. Mortierella Species as the Plant Growth-Promoting Fungi Present in the Agricultural Soils. Agriculture 2020, 11, 7. [Google Scholar] [CrossRef]
- Ainsworth, T.D.; Krause, L.; Bridge, T.; Torda, G.; Raina, J.-B.; Zakrzewski, M.; Gates, R.D.; Padilla-Gamiño, J.L.; Spalding, H.L.; Smith, C.; et al. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 2015, 9, 2261–2274. [Google Scholar] [CrossRef] [PubMed]
- Shade, A.; Handelsman, J. Beyond the Venn diagram: The hunt for a core microbiome. Environ. Microbiol. 2011, 14, 4–12. [Google Scholar] [CrossRef]
- Kouřilová, X.; Schwarzerová, J.; Pernicová, I.; Sedlář, K.; Mrázová, K.; Krzyžánek, V.; Nebesářová, J.; Obruča, S. The First Insight into Polyhydroxyalkanoates Accumulation in Multi-Extremophilic Rubrobacter xylanophilus and Rubrobacter spartanus. Microorganisms 2021, 9, 909. [Google Scholar] [CrossRef]
- Cui, Y.; Wang, X.; Zhang, X.; Ju, W.; Duan, C.; Guo, X.; Wang, Y.; Fang, L. Soil moisture mediates microbial carbon and phosphorus metabolism during vegetation succession in a semiarid region. Soil Biol. Biochem. 2020, 147, 107814. [Google Scholar] [CrossRef]
- Lopes, L.D.; Hao, J.; Schachtman, D.P. Alkaline soil pH affects bulk soil, rhizosphere and root endosphere microbiomes of plants growing in a Sandhills ecosystem. FEMS Microbiol. Ecol. 2021, 97, fiab028. [Google Scholar] [CrossRef]
- Qiang, W.; He, L.; Zhang, Y.; Liu, B.; Liu, Y.; Liu, Q.; Pang, X. Aboveground vegetation and soil physicochemical properties jointly drive the shift of soil microbial community during subalpine secondary succession in southwest China. CATENA 2021, 202, 105251. [Google Scholar] [CrossRef]
- He, M.; Xiong, X.; Wang, L.; Hou, D.; Bolan, N.S.; Ok, Y.S.; Rinklebe, J.; Tsang, D.C. A critical review on performance indicators for evaluating soil biota and soil health of biochar-amended soils. J. Hazard. Mater. 2021, 414, 125378. [Google Scholar] [CrossRef]
- Xiong, J.; Liu, Y.; Lin, X.; Zhang, H.; Zeng, J.; Hou, J.; Yang, Y.; Yao, T.; Knight, R.; Chu, H. Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ. Microbiol. 2012, 14, 2457–2466. [Google Scholar] [CrossRef]
- Tripathi, B.M.; Stegen, J.C.; Kim, M.; Dong, K.; Adams, J.M.; Lee, Y.K. Soil pH mediates the balance between stochastic and deterministic assembly of bacteria. ISME J. 2018, 12, 1072–1083. [Google Scholar] [CrossRef]
- Maestre, F.T.; Delgado-Baquerizo, M.; Jeffries, T.C.; Eldridge, D.J.; Ochoa, V.; Gozalo, B.; Quero, J.L.; García-Gómez, M.; Gallardo, A.; Ulrich, W.; et al. Increasing aridity reduces soil microbial diversity and abundance in global drylands. Proc. Natl. Acad. Sci. USA 2015, 112, 15684–15689. [Google Scholar] [CrossRef]
- Zhou, Z.; Yu, M.; Ding, G.; Gao, G.; He, Y.; Wang, G. Effects of Hedysarum leguminous plants on soil bacterial communities in the Mu Us Desert, northwest China. Ecol. Evol. 2020, 10, 11423–11439. [Google Scholar] [CrossRef]
- Brockett, B.F.; Prescott, C.E.; Grayston, S.J. Soil moisture is the major factor influencing microbial community structure and enzyme activities across seven biogeoclimatic zones in western Canada. Soil Biol. Biochem. 2012, 44, 9–20. [Google Scholar] [CrossRef]
- Chen, J.; Shi, W.; Cao, J. Effects of Grazing on Ecosystem CO2 Exchange in a Meadow Grassland on the Tibetan Plateau during the Growing Season. Environ. Manag. 2014, 55, 347–359. [Google Scholar] [CrossRef]
- Iovieno, P.; Bã¥Ã¥Th, E. Effect of drying and rewetting on bacterial growth rates in soil. FEMS Microbiol. Ecol. 2008, 65, 400–407. [Google Scholar] [CrossRef]
- Wang, T.; Wu, Y.; Li, Z.; Sha, X. Potential impact of active substances in non-thermal discharge plasma process on microbial community structures and enzymatic activities in uncontaminated soil. J. Hazard. Mater. 2020, 393, 122489. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, D.; Cheng, H.; Ren, L.; Jin, X.; Fang, W.; Yan, D.; Li, Y.; Wang, Q.; Cao, A. Organic fertilizers activate soil enzyme activities and promote the recovery of soil beneficial microorganisms after dazomet fumigation. J. Environ. Manag. 2022, 309, 114666. [Google Scholar] [CrossRef]
- Li, T.F.; Liu, C.Y.; Jin, X.M.; Cao, X.Y.; Lin, Z.Q.; Lu, Q.; Long, M.X.; He, S.B. Effects of different cultivation strategies on soil nutrients and bacterial diversity in kiwifruit orchards. Eur. J. Hortic. Sci. 2022, 87, 1–8. [Google Scholar] [CrossRef]
- Yuan, Z.; Liu, H.; Han, J.; Sun, J.; Wu, X.; Yao, J. Monitoring Soil Microbial Activities in Different Cropping Systems Using Combined Methods. Pedosphere 2017, 27, 138–146. [Google Scholar] [CrossRef]
- Wang, G.; Jin, Z.; Wang, X.; George, T.S.; Feng, G.; Zhang, L. Simulated root exudates stimulate the abundance of Saccharimonadales to improve the alkaline phosphatase activity in maize rhizosphere. Appl. Soil Ecol. 2021, 170, 104274. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Plot | Moisture Content | pH | TN (%) | TC (%) | TH (%) | TS (%) | C/N | C/H | TP (μg/g) | OP (μg/g) | IOP (μg/g) |
| ND | 8.622 ± 3.926a | 5.680 ± 0.256c | 0.682 ± 0.121a | 7.209 ± 1.251a | 0.677 ± 0.080a | 0.051 ± 0.011a | 10.583 ± 0.165 | 10.577 ± 0.863a | 1660.536 ± 123.743a | 1434.619 ± 43.680a | 225.917 ± 80.831 |
| SD | 2.611 ± 1.950b | 6.360 ± 0.425b | 0.472 ± 0.036b | 4.967 ± 0.515b | 0.567 ± 0.073ab | 0.037 ± 0.003ab | 10.510 ± 0.307 | 8.792 ± 0.282ab | 1029.349 ± 114.512b | 830.352 ± 136.978b | 198.997 ± 68.542 |
| LD | 3.922 ± 2.366ab | 5.980 ± 0.298bc | 0.392 ± 0.056b | 4.303 ± 0.490b | 0.553 ± 0.028b | 0.030 ± 0.008bc | 11.024 ± 0.326 | 7.760 ± 0.578b | 861.068 ± 244.441b | 669.989 ± 341.562b | 191.079 ± 110.578 |
| MD | 2.100 ± 0.852b | 7.753 ± 0.118a | 0.105 ± 0.017c | 0.904 ± 0.121c | 0.237 ± 0.033c | 0.009 ± 0.001d | 8.618 ± 0.152 | 3.836 ± 0.322c | 238.449 ± 104.807c | 174.052 ± 125.931c | 64.397 ± 25.675 |
| HD | 2.622 ± 0.264b | 7.990 ± 0.022a | 0.106 ± 0.006c | 1.154 ± 0.301c | 0.233 ± 0.003c | 0.011 ± 0.002cd | 10.849 ± 2.458 | 4.962 ± 1.274c | 341.048 ± 63.545c | 239.702 ± 68.063c | 101.346 ± 29.285 |
| ED | 2.589 ± 0.594b | 8.160 ± 0.071a | 0.094 ± 0.028c | 1.113 ± 0.397c | 0.230 ± 0.042c | 0.016 ± 0.014cd | 11.597 ± 1.766 | 4.684 ± 1.111c | 429.370 ± 137.407c | 212.954 ± 59.998c | 216.416 ± 84.442 |
| Sampling plot | AP (μmoL/g) | NN (μg/g) | AN (μg/g) | AK (μg/g) | SOM (%) | SC (mg/d/g) | UE (μg/d/g) | CAT (μmoL/d/g) | ACP (μmoL/d/g) | NP (μmoL/d/g) | AKP (μmol/d/g) |
| ND | 2.118 ± 0.909 | 3.785 ± 1.595 | 53.628 ± 22.620 | 75.478 ± 23.495 | 3.344 ± 0.780 | 135.551 ± 16.357a | 456.620 ± 65.620 | 124.001 ± 3.170a | 7.050 ± 0.853a | 6.669 ± 0.210a | 3.419 ± 0.354ab |
| SD | 2.406 ± 1.281 | 3.622 ± 1.106 | 70.126 ± 17.547 | 69.712 ± 13.614 | 3.034 ± 0.518 | 162.207 ± 38.405a | 489.420 ± 131.136 | 122.412 ± 1.960a | 5.922 ± 1.137a | 6.665 ± 0.129a | 3.787 ± 0.026a |
| LD | 1.787 ± 0.650 | 2.606 ± 0.714 | 39.042 ± 2.948 | 83.691 ± 19.261 | 3.584 ± 0.184 | 158.168 ± 67.895a | 490.732 ± 75.857 | 123.355 ± 3.178a | 7.604 ± 1.124a | 6.029 ± 0.227a | 2.433 ± 0.946bc |
| MD | 2.243 ± 0.378 | 0.486 ± 0.519 | 47.492 ± 24.455 | 80.314 ± 40.631 | 3.557 ± 0.528 | 32.286 ± 10.785b | 495.105 ± 120.924 | 116.635 ± 0.482b | 3.590 ± 0.627b | 1.896 ± 0.487b | 2.043 ± 0.621cd |
| HD | 1.798 ± 0.106 | 1.000 ± 0.641 | 61.944 ± 9.427 | 50.926 ± 3.694 | 3.297 ± 0.475 | 28.138 ± 2.816b | 504.726 ± 37.677 | 116.383 ± 0.277b | 2.097 ± 0.326bc | 0.444 ± 0.058c | 1.867 ± 0.083cd |
| ED | 3.286 ± 0.722 | 4.909 ± 3.561 | 80.648 ± 4.008 | 73.192 ± 1.043 | 3.056 ± 0.441 | 25.051 ± 7.731b | 366.529 ± 77.990 | 115.079 ± 0.538b | 1.543 ± 0.305c | 0.658 ± 0.448c | 1.095 ± 0.197d |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chao, L.; Ma, X.; Tsetsegmaa, M.; Zheng, Y.; Qu, H.; Dai, Y.; Li, J.; Bao, Y. Response of Soil Microbial Community Composition and Diversity at Different Gradients of Grassland Degradation in Central Mongolia. Agriculture 2022, 12, 1430. https://doi.org/10.3390/agriculture12091430
Chao L, Ma X, Tsetsegmaa M, Zheng Y, Qu H, Dai Y, Li J, Bao Y. Response of Soil Microbial Community Composition and Diversity at Different Gradients of Grassland Degradation in Central Mongolia. Agriculture. 2022; 12(9):1430. https://doi.org/10.3390/agriculture12091430
Chicago/Turabian StyleChao, Lumeng, Xiaodan Ma, Munkhzul Tsetsegmaa, Yaxin Zheng, Hanting Qu, Yuan Dai, Jingpeng Li, and Yuying Bao. 2022. "Response of Soil Microbial Community Composition and Diversity at Different Gradients of Grassland Degradation in Central Mongolia" Agriculture 12, no. 9: 1430. https://doi.org/10.3390/agriculture12091430
APA StyleChao, L., Ma, X., Tsetsegmaa, M., Zheng, Y., Qu, H., Dai, Y., Li, J., & Bao, Y. (2022). Response of Soil Microbial Community Composition and Diversity at Different Gradients of Grassland Degradation in Central Mongolia. Agriculture, 12(9), 1430. https://doi.org/10.3390/agriculture12091430