
Microbiome Analysis of the Rhizosphere from Wilt Infected Pomegranate Reveals Complex Adaptations in Fusarium—A Preliminary Study



Abstract
:1. Introduction
2. Materials and Methods
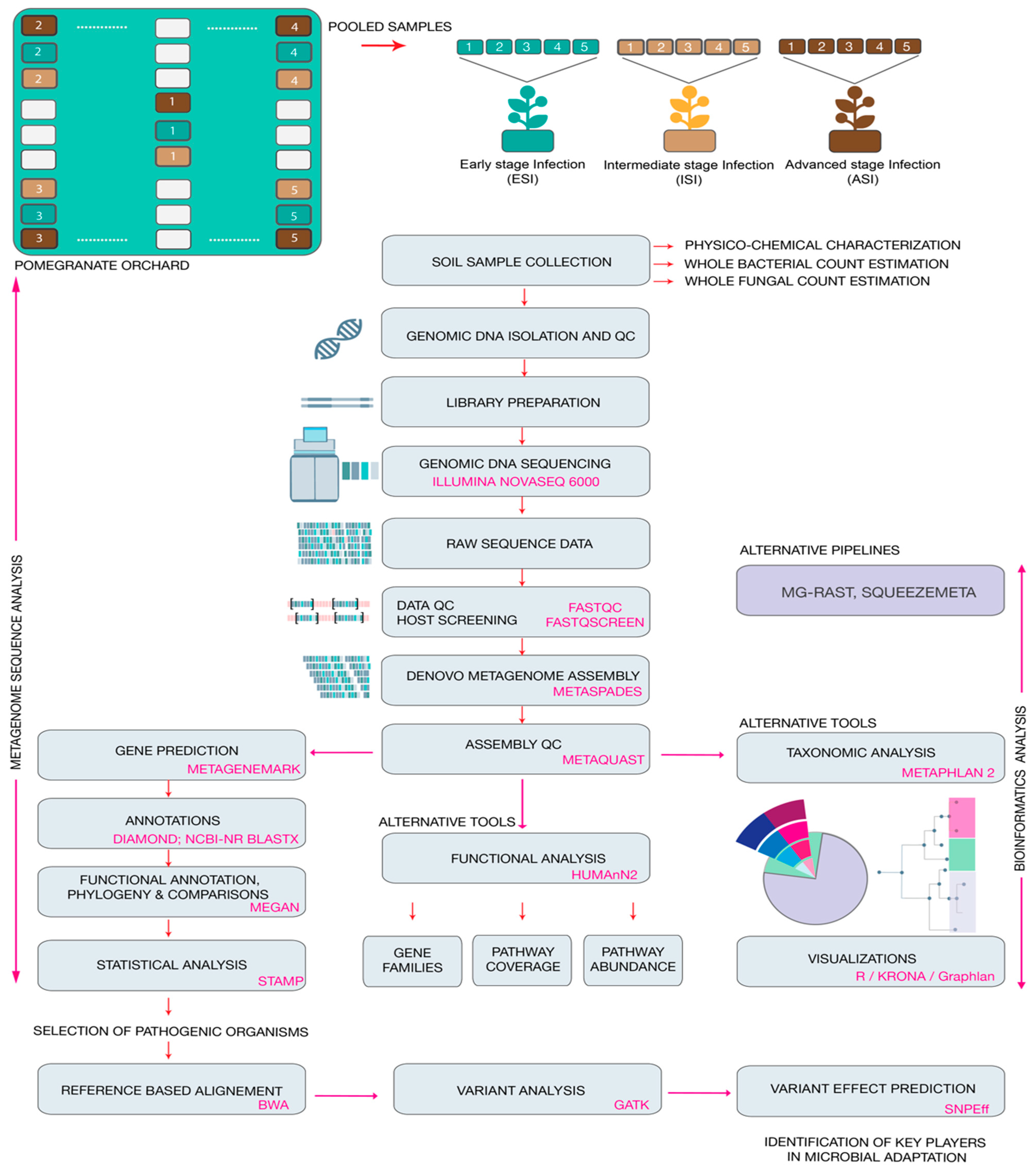
2.1. Site Description and Sampling
2.2. Physicochemical Characterization and Total Microbial Count Estimation
2.3. Isolation of Fusarium oxysporum, Aspergillus niger from Soil Samples
2.4. DNA Extraction and Quality Control
2.5. Library Construction and Quality Control
2.6. Whole Meta-Genome Sequencing
2.7. Data Analysis
2.8. Prediction of Protein Functions
2.9. Variant Analysis
2.10. Statistical Analysis
3. Results
3.1. Physical Examination
3.2. Physiochemical Properties
3.3. Total Microbial Counts
3.4. Sequence Information
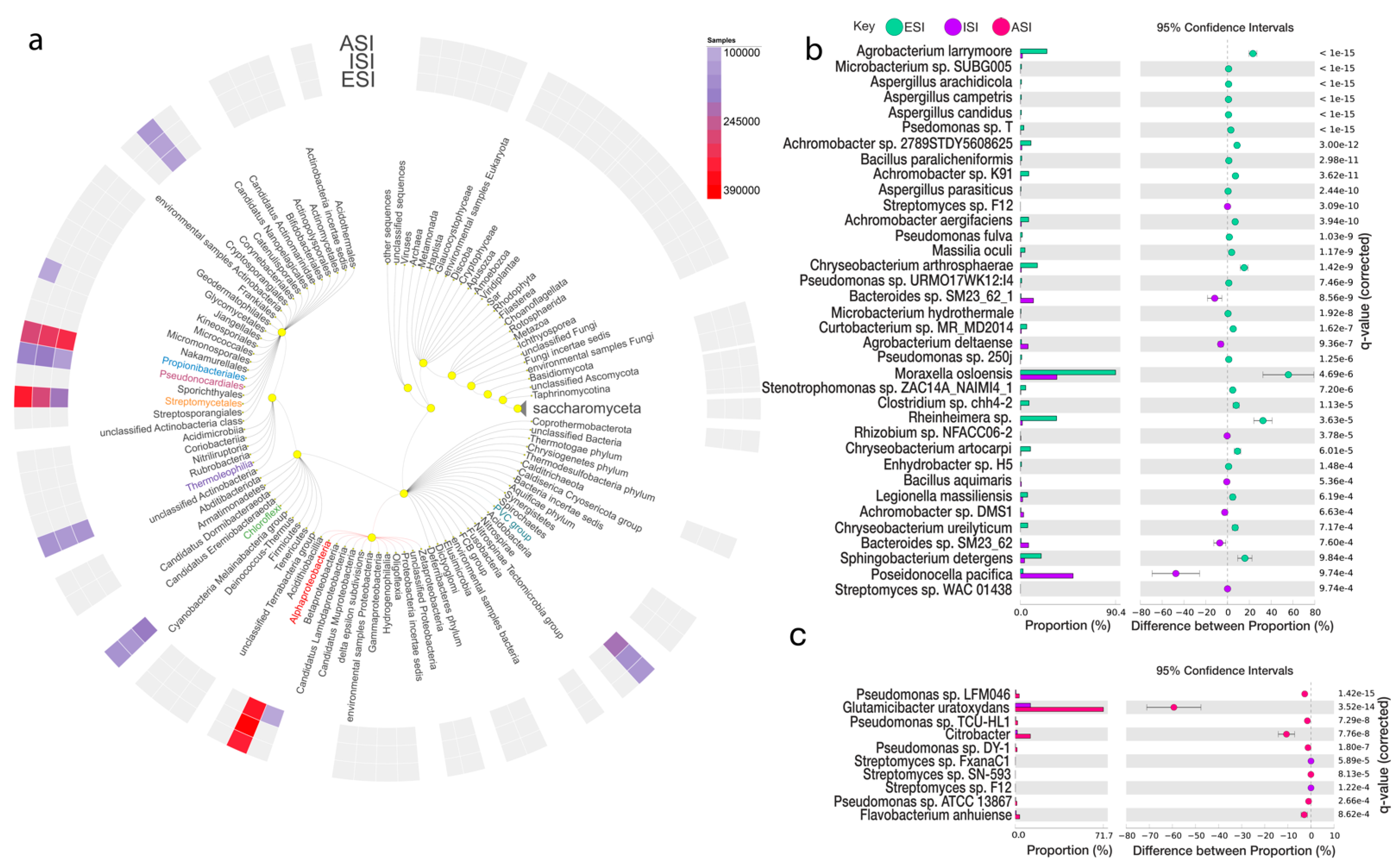
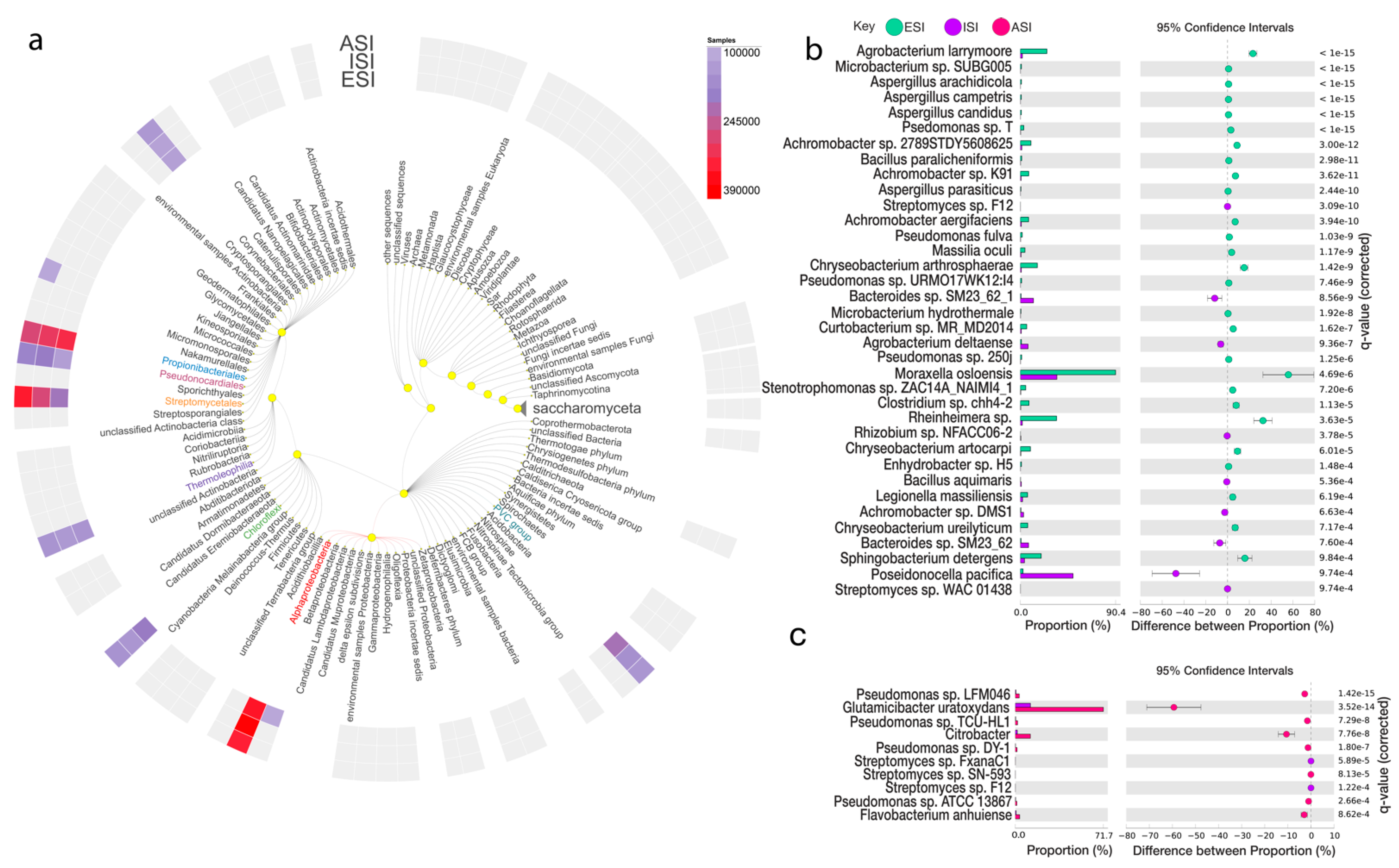
3.5. Microbial Abundance Analysis
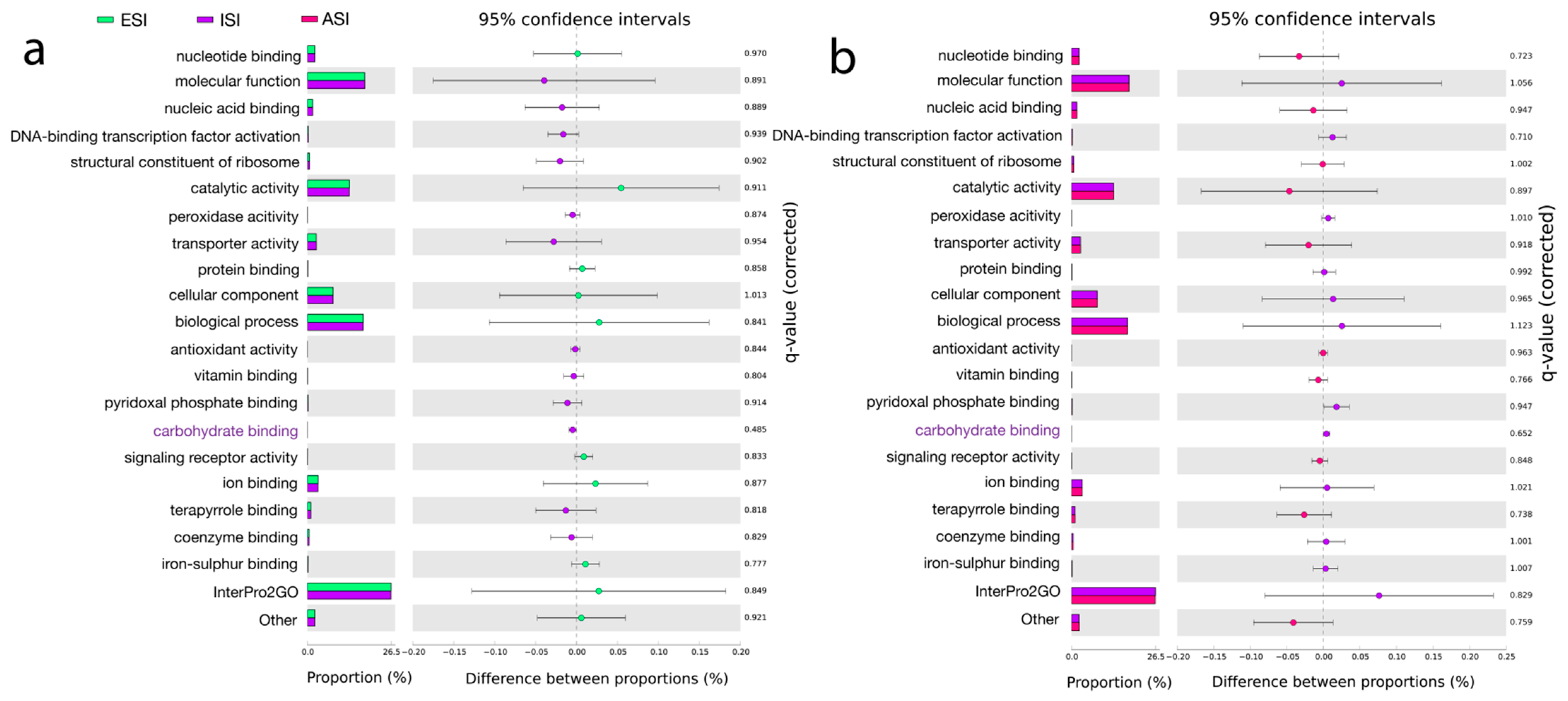
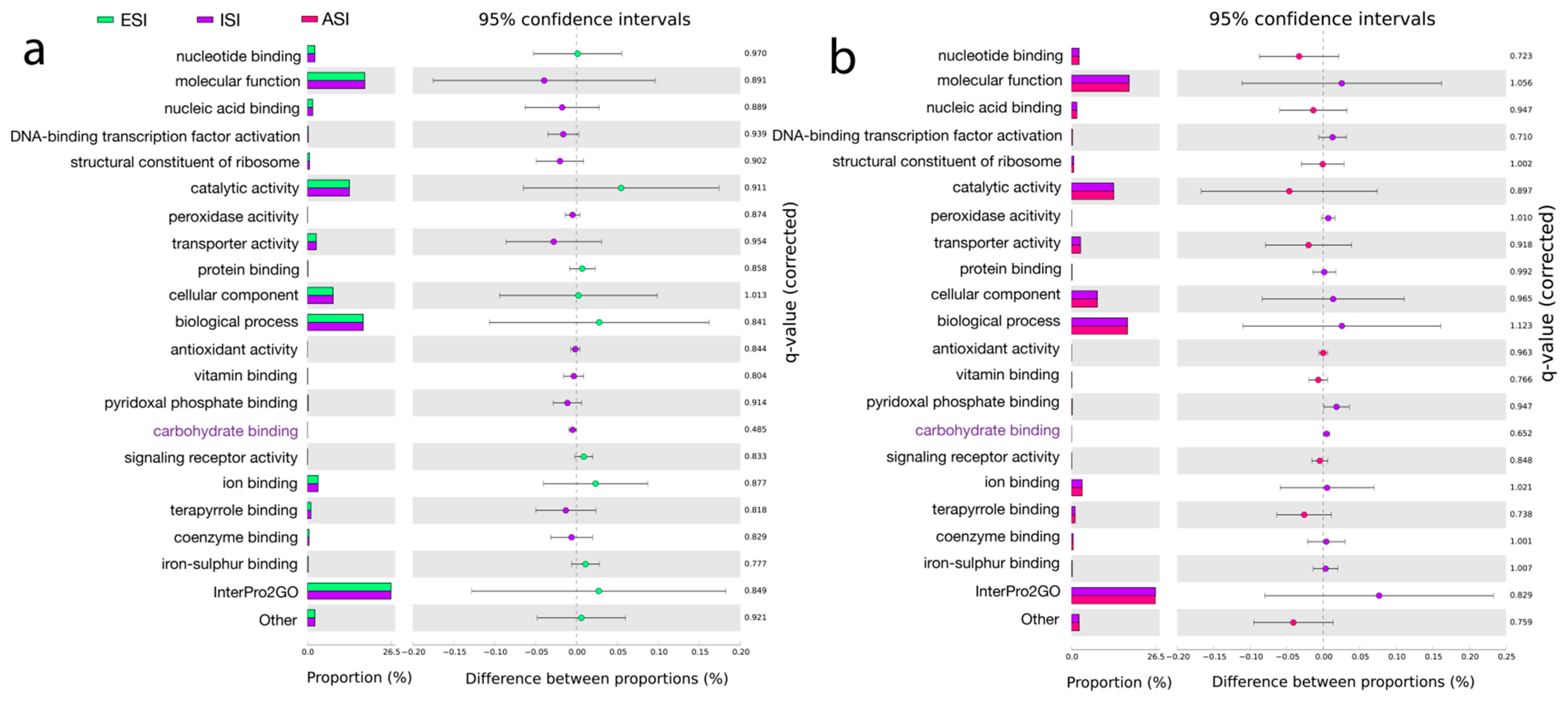
3.6. Pathway Predictions
3.7. Genome Resolved Metagenomics
3.8. Variant Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Teksur, P.K. Alternative technologies to control postharvest diseases of pomegranate. Stewart Postharvest Rev. 2015, 11, 1–7. [Google Scholar] [CrossRef]
- Middha, S.K.; Usha, T.; Pande, V. A Review on Antihyperglycemic and Antihepatoprotective Activity of eco-friendly Punica granatum peel waste. Evid. Based Complement. Alt Med. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, J.A.T.; Rana, T.S.; Narzary, D.; Verma, N.; Meshram, D.T.; Ranade, S.A. Scientia Horticulturae Pomegranate biology and biotechnology: A review. Sci. Hortic. 2013, 160, 85–107. [Google Scholar] [CrossRef]
- Chen, J.; Liao, C.; Ouyang, X.; Kahramanoğlu, I.; Gan, Y.; Li, M. Antimicrobial Activity of Pomegranate Peel and Its Applications on Food Preservation. J. Food Qual. 2020, 2020, 1–8. [Google Scholar] [CrossRef]
- Alexandre, E.; Silva, S.; Santos, S.; Silvestre, A.J.; Duarte, M.F.; Saraiva, J.A.; Pintado, M. Antimicrobial activity of pomegranate peel extracts performed by high pressure and enzymatic assisted extraction. Food Res. Int. 2018, 115, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Usha, T.; Middha, S.K.; Sidhalinghamurthy, K.R. Pomegranate peel and its anticancer activity: A mechanism-based review. In Plant-derived Bioactives: Chemistry and Mode of Action; Springer: Berlin/Heidelberg, Germany, 2020; ISBN 9789811523618. [Google Scholar]
- Sharma, P.; McClees, S.F.; Afaq, F. Pomegranate for Prevention and Treatment of Cancer: An Update. Molecules 2017, 22, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, A.; Barbarossa, A.; Tassone, A.; Ceramella, J.; Carocci, A.; Catalano, A.; Basile, G.; Fazio, A.; Iacopetta, D.; Franchini, C.; et al. Pomegranate: Nutraceutical with Promising Benefits on Human Health. Appl. Sci. 2020, 10, 6915. [Google Scholar] [CrossRef]
- Elshafie, H.; Caputo, L.; de Martino, L.; Sakr, S.; de Feo, V.; Camele, I. Study of Bio-Pharmaceutical and Antimicrobial Properties of Pomegranate (Punica granatum L.) Leathery Exocarp Extract. Plants 2021, 10, 153. [Google Scholar] [CrossRef]
- Aslam, M.N.; Lansky, E.P.; Varani, J. Pomegranate as a cosmeceutical source: Pomegranate fractions promote proliferation and procollagen synthesis and inhibit matrix metalloproteinase-1 production in human skin cells. J. Ethnopharmacol. 2006, 103, 311–318. [Google Scholar] [CrossRef]
- Al-Zoreky, N. Antimicrobial activity of pomegranate (Punica granatum L.) fruit peels. Int. J. Food Microbiol. 2009, 134, 244–248. [Google Scholar] [CrossRef]
- Usha, T.; Goyal, A.K.; Lubna, S.; Prashanth, H.P.; Mohan, T.M.; Pande, V.; Middha, S.K. Identification of anti-cancer targets of eco-friendly waste Punica granatum peel by dual reverse virtual screening and binding analysis. Asian Pac. J. Cancer Prev. 2015, 15, 10345–10350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salgado, J.; Ferreira, T.R.B.; Biazotto, F.D.O.; Dias, C. Increased Antioxidant Content in Juice Enriched with Dried Extract of Pomegranate (Punica granatum) Peel. Plant Foods Hum. Nutr. 2012, 67, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Henning, S.M.; Yang, J.; Lee, R.-P.; Huang, J.; Hsu, M.; Thames, G.; Gilbuena, I.; Long, J.; Xu, Y.; Park, E.H.; et al. Pomegranate Juice and Extract Consumption Increases the Resistance to UVB-induced Erythema and Changes the Skin Microbiome in Healthy Women: A Randomized Controlled Trial. Sci. Rep. 2019, 9, 14528. [Google Scholar] [CrossRef] [PubMed]
- Bellé, C.; Moccellin, R.; Souza-Júnior, I.T.; Maich, S.L.P.; Neves, C.G.; Nascimento, M.B.; Barros, D.R. First Report of Colletotrichum acutatum Causing Flower Anthracnose on Pomegranate (Punica granatum) in Southern Brazil. Plant Dis. 2018, 102, 2373. [Google Scholar] [CrossRef]
- Kanetis, L.; Testempasis, S.; Goulas, V.; Samuel, S.; Myresiotis, C.; Karaoglanidis, G.S. Identification and mycotoxigenic capacity of fungi associated with pre- and postharvest fruit rots of pomegranates in Greece and Cyprus. Int. J. Food Microbiol. 2015, 208, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Cintora-Martínez, E.A.; Leyva-Mir, S.G.; Ayala-Escobar, V.; Quezada, G.D.A.; Camacho-Tapia, M.; Tovar-Pedraza, J.M. Pomegranate fruit rot caused by Pilidiella granati in Mexico. Australas. Plant Dis. Notes 2017, 12, 4. [Google Scholar] [CrossRef]
- Palou, L.; Crisosto, C.H.; Garner, D. Combination of postharvest antifungal chemical treatments and controlled atmosphere storage to control gray mold and improve storability of ‘Wonderful’ pomegranates. Postharvest Biol. Technol. 2007, 43, 133–142. [Google Scholar] [CrossRef]
- Thomidis, T. Pathogenicity and characterization of Pilidiella granati causing pomegranate diseases in Greece. Eur. J. Plant Pathol. 2014, 141, 45–50. [Google Scholar] [CrossRef]
- Mondal, K.K.; Mani, C. ERIC-PCR-Generated Genomic Fingerprints and Their Relationship with Pathogenic Variability of Xanthomonas campestris pv. punicae, the Incitant of Bacterial Blight of Pomegranate. Curr. Microbiol. 2009, 59, 616–620. [Google Scholar] [CrossRef]
- Grote, D.; Olmos, A.; Kofoet, A.; Tuset, J.; Bertolini, E.; Cambra, M. Specific and Sensitive Detection of Phytophthora nicotianae By Simple and Nested-PCR. Eur. J. Plant Pathol. 2002, 108, 197–207. [Google Scholar] [CrossRef]
- Yang, X.; Hameed, U.; Zhang, A.-F.; Zang, H.-Y.; Gu, C.-Y.; Chen, Y.; Xu, Y.-L. Development of a nested-PCR assay for the rapid detection of Pilidiella granati in pomegranate fruit. Sci. Rep. 2017, 7, 40954. [Google Scholar] [CrossRef] [Green Version]
- She, X.; Yu, L.; Lan, G.; Tang, Y.; He, Z. Identification and Genetic Characterization of Ralstonia solanacearum Species Complex Isolates from Cucurbita maxima in China. Front. Plant Sci. 2017, 8, 1794. [Google Scholar] [CrossRef] [Green Version]
- Suhaimi, N.S.M.; Goh, S.-Y.; Ajam, N.; Othman, R.Y.; Chan, K.-G.; Thong, K.L. Diversity of microbiota associated with symptomatic and non-symptomatic bacterial wilt-diseased banana plants determined using 16S rRNA metagenome sequencing. World J. Microbiol. Biotechnol. 2017, 33, 168. [Google Scholar] [CrossRef] [PubMed]
- Ts, A.; Sg, B. Bacterial root bark necrosis and wilt of pomegranate, hereto a new disease. J. Appl. Biotechnol. Bioeng. 2018, 5, 329–332. [Google Scholar] [CrossRef]
- Shen, Z.; Penton, C.R.; Lv, N.; Xue, C.; Yuan, X.; Ruan, Y.; Li, R.; Shen, Q. Banana Fusarium Wilt Disease Incidence Is Influenced by Shifts of Soil Microbial Communities Under Different Monoculture Spans. Microb. Ecol. 2017, 75, 739–750. [Google Scholar] [CrossRef]
- Tian, X.; Zhang, Q.; Chen, G.; Mao, Z.; Yang, J.; Xie, B. Diversity of bacteria associated with pine wood nematode revealed by metagenome. Acta Microbiol. Sin. 2010, 50, 909–916. [Google Scholar]
- Reddy, T.; Thomas, A.D.; Stamatis, D.; Bertsch, J.; Isbandi, M.; Jansson, J.; Mallajosyula, J.; Pagani, I.; Lobos, E.A.; Kyrpides, N.C. The Genomes OnLine Database (GOLD) v.5: A metadata management system based on a four level (meta)genome project classification. Nucleic Acids Res. 2014, 43, D1099–D1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruitt, K.D.; Tatusova, T.; Brown, G.R.; Maglott, D.R. NCBI Reference Sequences (RefSeq): Current status, new features and genome annotation policy. Nucleic Acids Res. 2011, 40, D130–D135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.; Euzéby, J.; Amann, R.; Rossello-Mora, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Genet. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Singer, E.; Bushnell, B.; Coleman-Derr, D.; Bowman, B.; Bowers, R.M.; Levy, A.; Gies, E.A.; Cheng, J.-F.; Copeland, A.; Klenk, H.-P.; et al. High-resolution phylogenetic microbial community profiling. ISME J. 2016, 10, 2020–2032. [Google Scholar] [CrossRef]
- Santos, S.S.; Nielsen, T.; Hansen, L.H.; Winding, A. Comparison of three DNA extraction methods for recovery of soil protist DNA. J. Microbiol. Methods 2015, 115, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Medinger, R.; Nolte, V.; Pandey, R.V.; Jost, S.; Ottenwälder, B.; Schlötterer, C.; Boenigk, J. Diversity in a hidden world: Potential and limitation of next-generation sequencing for surveys of molecular diversity of eukaryotic microorganisms. Mol. Ecol. 2010, 19, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Dong, J.; Liu, X.; Massana, R. Extremely High Copy Numbers and Polymorphisms of the rDNA Operon Estimated from Single Cell Analysis of Oligotrich and Peritrich Ciliates. Protist 2013, 164, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Kunin, V.; Engelbrektson, A.; Ochman, H.; Hugenholtz, P. Wrinkles in the rare biosphere: Pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 2010, 12, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Herbold, C.; Polson, S.; Wommack, K.E.; Williamson, S.J.; McDonald, I.; Cary, S. Groundtruthing Next-Gen Sequencing for Microbial Ecology–Biases and Errors in Community Structure Estimates from PCR Amplicon Pyrosequencing. PLoS ONE 2012, 7, e44224. [Google Scholar] [CrossRef] [Green Version]
- Pawlowski, J.W.; Christen, R.; Lecroq, B.; Bachar, D.; Shahbazkia, H.R.; Amaral-Zettler, L.; Guillou, L. Eukaryotic Richness in the Abyss: Insights from Pyrotag Sequencing. PLoS ONE 2011, 6, e18169. [Google Scholar] [CrossRef]
- Abraham, B.S.; Caglayan, D.; Carrillo, N.V.; Chapman, M.C.; Hagan, C.T.; Hansen, S.T.; Jeanty, R.O.; Klimczak, A.A.; Klingler, M.J.; Kutcher, T.P.; et al. Shotgun metagenomic analysis of microbial communities from the Loxahatchee nature preserve in the Florida Everglades. Environ. Microbiome 2020, 15, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babalola, O.O.; Fadiji, A.E.; Ayangbenro, A.S. Shotgun metagenomic data of root endophytic microbiome of maize (Zea mays L.). Data Brief 2020, 31, 105893. [Google Scholar] [CrossRef]
- Das, A.J.; Ravinath, R.; Shilpa, B.R.; Rohith, B.S.; Goyal, A.K.; Ramesh, N.; Ekambaram, H.; Prasannakumar, M.K.; Usha, T.; Middha, S.K. Microbiomics and cloud-based analytics advance sustainable soil management. Front. Biosci. 2021, 26, 478–495. [Google Scholar] [CrossRef] [PubMed]
- Weston, W.H. Soil and Plant Analysis. A Laboratory Manual of Methods for the Examination of Soils and the Determination of the Inorganic Constituents of Plants. C. S. Piper. Q. Rev. Biol. 1945, 20, 272–273. [Google Scholar] [CrossRef]
- Bureau of Indian Standards Microbiology. General Guidance for the Enumeration of Micro-organisms Colony Count Technique at 30 °C; Bureau of Indian Standards Microbiology: New Delhi, Indian, 2002. [Google Scholar]
- Bureau of Indian Standards Microbiology. Method for Yeast and Mould Count of Foodstuffs and Animal Feeds; Bureau of Indian Standards Microbiology: New Delhi, Indian, 1999. [Google Scholar]
- Amorim, J.H.; Macena, T.; Lacerda-Junior, G.; Rezende, R.; Dias, J.; Brendel, M.; Cascardo, J. An improved extraction protocol for metagenomic DNA from a soil of the Brazilian Atlantic Rainforest. Genet. Mol. Res. 2008, 7, 1226–1232. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laslett, D. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R.; Wheeler, T. HMMER-biosequence analysis using profile hidden Markov models. Howard Hughes Medical Institute 2015. [Google Scholar]
- Wu, Y.-W.; Simmons, B.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2015, 32, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Sieber, C.; Probst, A.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, R.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.B.; Daly, M.J.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Tyson, G.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Beiko, R.G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 2010, 26, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camon, E.B.; Barrell, D.G.; Dimmer, E.C.; Lee, V.; Magrane, M.; Maslen, J.; Binns, D.; Apweiler, R. An evaluation of GO annotation retrieval for BioCreAtIvE and GOA. BMC Bioinform. 2005, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2018, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overbeek, R.A.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.; Gerdes, S.; Parrello, B.D.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2013, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusov, R.L.; Natale, D.A.; Garkavtsev, I.V.; Tatusova, T.A.; Shankavaram, U.T.; Rao, B.S.; Kiryutin, B.; Galperin, M.Y.; Fedorova, N.D.; Koonin, E.V. The COG database. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Que, Y.; Yue, X.; Yang, N.; Xu, Z.; Tang, S.; Wang, C.; Lv, W.; Xu, L.; Talbot, N.J.; Wang, Z. Leucine biosynthesis is required for infection-related morphogenesis and pathogenicity in the rice blast fungus Magnaporthe oryzae. Curr. Genet. 2019, 66, 155–171. [Google Scholar] [CrossRef]
- Sun, R.-L.; Jing, Y.-L.; de Boer, W.; Guo, R.-J.; Li, S.-D. Dominant hyphae-associated bacteria of Fusarium oxysporum f. sp. cucumerinum in different cropping systems and insight into their functions. Appl. Soil Ecol. 2021, 165, 103977. [Google Scholar] [CrossRef]
- Vijay, K.; Devi, T.S.; Sree, K.K.; Elgorban, A.; Kumar, P.; Govarthanan, M.; Kavitha, T. In vitro screening and in silico prediction of antifungal metabolites from rhizobacterium Achromobacter kerstersii JKP9. Arch. Microbiol. 2020, 202, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Blackman, L.M.; Cullerne, D.P.; Hardham, A.R. Bioinformatic characterisation of genes encoding cell wall degrading enzymes in the Phytophthora parasitica genome. BMC Genom. 2014, 15, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, D.M.; King, B.; Hayes, M.L.; Bergstrom, G. Plant pathogens as a source of diverse enzymes for lignocellulose digestion. Curr. Opin. Microbiol. 2011, 14, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Jorge, I.; Navas-Cortes, J.; Jimenezdiaz, R.M.J.M.; Tena, M. Cell wall degrading enzymes in fusarium wilt of chickpea: Correlation between pectinase and xylanase activities and disease development in plants infected with two pathogenic races of Fusarium oxysporum f. sp. ciceris. Can. J. Bot. 2006, 84, 1395–1404. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | pH | EC | N | P | K | OC | Cl | Fe | Cu | Mn | Zn | B | Microbial Count/g | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (μs/cm) | % | ppm | Bacterial (cfu) | Fungal (cfu) | ||||||||||
| ESI | 7.73 | 135 | 0.20 | 0.0084 | 0.011 | 0.92 | 15 | 0.98 | 26.9 | 9.5 | 24.8 | 3.4 | 1968 | 154 |
| ISI | 6.35 | 139 | 0.191 | 0.010 | 0.011 | 0.93 | 18 | 0.93 | 29.4 | 9.1 | 30.9 | 4.1 | 2240 | 170 |
| ASI | 6.63 | 180 | 0.20 | 0.011 | 0.014 | 0.97 | 21 | 0.98 | 31.4 | 9.6 | 33.2 | 4.3 | 2126 | 154 |
| Sample | Raw Reads | Raw Data (Gb) | Sequence Count | BioProject | BioSample | SRA |
|---|---|---|---|---|---|---|
| ESI | 44869321 | 13.5 | 5,924,482 | PRJNA701747 | SAMN17910186 | SRR13705840 |
| ISI | 44773336 | 13.4 | 5,165,924 | SAMN17910187 | SRR13705839 | |
| ASI | 44063752 | 13.2 | 5,379,446 | SAMN17910188 | SRR13705838 |
| Taxonomic Hits Distribution Domain Level Microbial Abundance | |||
|---|---|---|---|
| ESI Percent of Reads | ISI Percent of Reads | ASI Percent of Reads | |
| Bacteria | 2,076,360 (96.54%) | 2,192,380 (96.08%) | 2,348,985 (97.61%) |
| Eukaryota | 63,248 (2.94%) | 79,978 (3.50%) | 43,462 (1.81%) |
| Archaea | 8979 (0.42%) | 7089 (0.31%) | 10,928 (0.45%) |
| Unclassified sequences | 1311 (0.06%) | 1496 (0.07%) | 2003 (0.08%) |
| Viruses | 935 (0.04%) | 939 (0.04%) | 1001 (0.04%) |
| Phylum level Microbial Abundance | |||
| Actinobacteria | 1,026,641 (57.52%) | 995,904 (52.92%) | 955,903 (49.62%) |
| Proteobacteria | 450,739 (25.25%) | 517,416 (27.49%) | 562,154 (29.18%) |
| Plantomycetes | 55,347 (3.10%) | 72,331 (3.84%) | 102,778 (5.34%) |
| Ascomycota | 52,339 (2.93%) | 69,904 (3.71%) | 34,814 (1.81%) |
| Chloroflexi | 37,935 (2.13%) | 39,681 (2.11%) | 54,360 (2.82%) |
| Bacteroidetes | 33,727 (1.89%) | 38,024 (2.02%) | 36,944 (1.92%) |
| Firmicutes | 29,671 (1.66%), | 33,232 (1.77%) | 37,538 (1.95%) |
| Verrucomicrobia | 21,659 (1.21%) | 28,599 (1.52%) | 33,042 (1.72%) |
| Acidobacteria | 20,072 (1.12%) | 25,715 (1.37%) | 33,599 (1.74%) |
| Cyanobacteria | 11,919 (0.67%) | 13,685 (0.73%) | 18,085 (0.94%) |
| Unclassified (from Bacteria) | 7962 (0.45%) | 9248 (0.49%) | 11,289 (0.59%) |
| Gemmatimonadetes | 7030 (0.39%), | 9358 (0.50%) | 8945 (0.46%) |
| Deinococcus-Thermus | 4694 (0.26%) | 4978 (0.26%) | 6399 (0.33%) |
| Euryarchaeota | 4231 (0.24%). | 4290 (0.23%) | 5509 (0.29%) |
| Species | Parent Sequence Count | Relative Frequency % | p-Values | Effect Size | |||||
|---|---|---|---|---|---|---|---|---|---|
| ESI | ISI | ESI | ISI | ESI | ISI | PVal | corrected | ||
| Achromobacter sp. 2789STDY5608625 | 299 | 8 | 2975 | 588 | 10.05 | 1.36 | 5.90 × 10−16 | 3.00 × 10−12 | 8.69 |
| Achromobacter sp. K91 | 236 | 4 | 2975 | 588 | 7.93 | 0.68 | 9.17 × 10−15 | 3.62 × 10−11 | 7.25 |
| Achromobacter aegrifaciens | 233 | 5 | 2975 | 588 | 7.83 | 0.85 | 1.33 × 10−13 | 3.94 × 10−10 | 6.98 |
| Microbacterium sp. SUBG005 | 196 | 6 | 19,463 | 13,719 | 1.01 | 0.04 | 1.22 × 10−37 | 2.17 × 10−33 | 0.96 |
| Agrobacterium larrymoorei | 174 | 14 | 691 | 741 | 25.18 | 1.89 | 6.24 × 10−44 | 2.22 × 10−39 | 23.29 |
| Curtobacterium sp. MR_MD2014 | 153 | 6 | 2534 | 695 | 6.04 | 0.86 | 8.66 × 10−11 | 1.62 × 10−7 | 5.17 |
| Pseudomonas sp. T | 140 | 7 | 4318 | 3374 | 3.24 | 0.21 | 2.75 × 10−27 | 1.63 × 10−23 | 3.03 |
| Moraxella osloensis | 132 | 9 | 146 | 26 | 90.41 | 34.62 | 2.90 × 10−9 | 4.69 × 10−6 | 55.80 |
| Pathway Predictions | |||||
|---|---|---|---|---|---|
| InterPro2GO [60] | KEGG [63] | SEED [62] | COG [64] | Pfam [65] | eggNOC [61] |
| GO: 0030246; Carbohydrate binding. GO: 0046906; Tetrapyrrole binding. GO: 0030170; Pyridoxal phosphate binding. | K03088; RNA polymerase sigma-70 factor ECF sub-family. K12132; Eukaryotic-like serine/threonine Protein kinase. K01990; ABC-2 type transport system ATP-binding protein. | Acyl carrier protein. Stress response, defense virulence. | ENOG410XNMH; Histidine kinase. COG1012; NAD-dependent aldehyde dehydrogenases. COG1960; Acyl-CoA Dehydrogenases. COG0515; Serine/threonine Protein kinase. | PF00005; ABC transporter. PF07690; Major Facilitator superfamily. PF00528; Binding protein-dependent Transport system Inner membrane component. | ISP *: Transcription. Replication, recombination, and repair. CSP +: Cell wall/Membrane/Envelope biogenesis, signal transduction mechanisms. Metabolism: Amino acid transport and metabolism, carbohydrate transport and metabolism, energy production and conversion. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, A.J.; Ravinath, R.; Usha, T.; Rohith, B.S.; Ekambaram, H.; Prasannakumar, M.K.; Ramesh, N.; Middha, S.K. Microbiome Analysis of the Rhizosphere from Wilt Infected Pomegranate Reveals Complex Adaptations in Fusarium—A Preliminary Study. Agriculture 2021, 11, 831. https://doi.org/10.3390/agriculture11090831
Das AJ, Ravinath R, Usha T, Rohith BS, Ekambaram H, Prasannakumar MK, Ramesh N, Middha SK. Microbiome Analysis of the Rhizosphere from Wilt Infected Pomegranate Reveals Complex Adaptations in Fusarium—A Preliminary Study. Agriculture. 2021; 11(9):831. https://doi.org/10.3390/agriculture11090831
Chicago/Turabian StyleDas, Anupam J., Renuka Ravinath, Talambedu Usha, Biligi Sampgod Rohith, Hemavathy Ekambaram, Mothukapalli Krishnareddy Prasannakumar, Nijalingappa Ramesh, and Sushil Kumar Middha. 2021. "Microbiome Analysis of the Rhizosphere from Wilt Infected Pomegranate Reveals Complex Adaptations in Fusarium—A Preliminary Study" Agriculture 11, no. 9: 831. https://doi.org/10.3390/agriculture11090831
APA StyleDas, A. J., Ravinath, R., Usha, T., Rohith, B. S., Ekambaram, H., Prasannakumar, M. K., Ramesh, N., & Middha, S. K. (2021). Microbiome Analysis of the Rhizosphere from Wilt Infected Pomegranate Reveals Complex Adaptations in Fusarium—A Preliminary Study. Agriculture, 11(9), 831. https://doi.org/10.3390/agriculture11090831