Triage of Amyotrophic Lateral Sclerosis Patients during the COVID-19 Pandemic: An Application of the D50 Model

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Methods
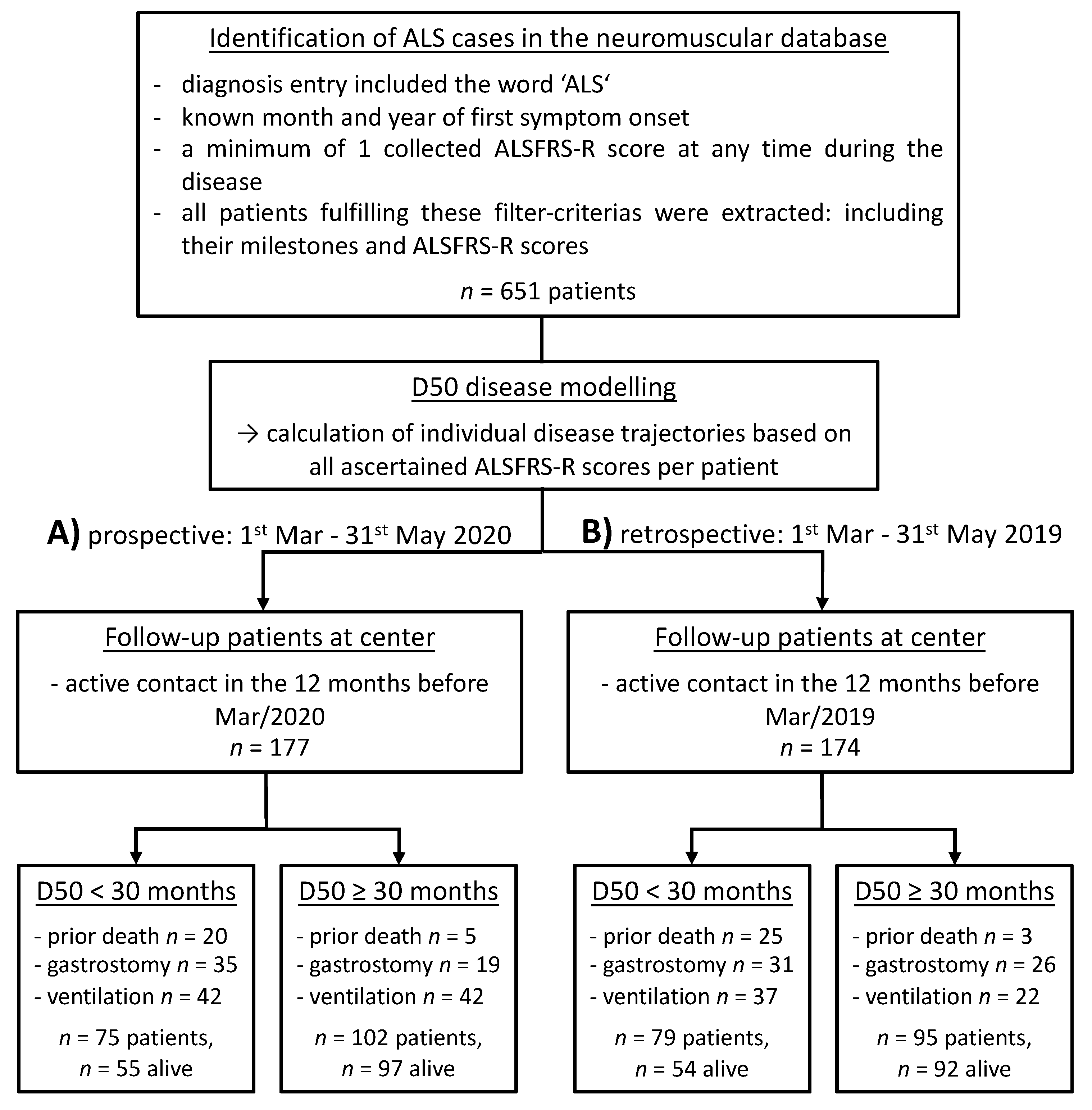
2.1. Design and Study Cohorts
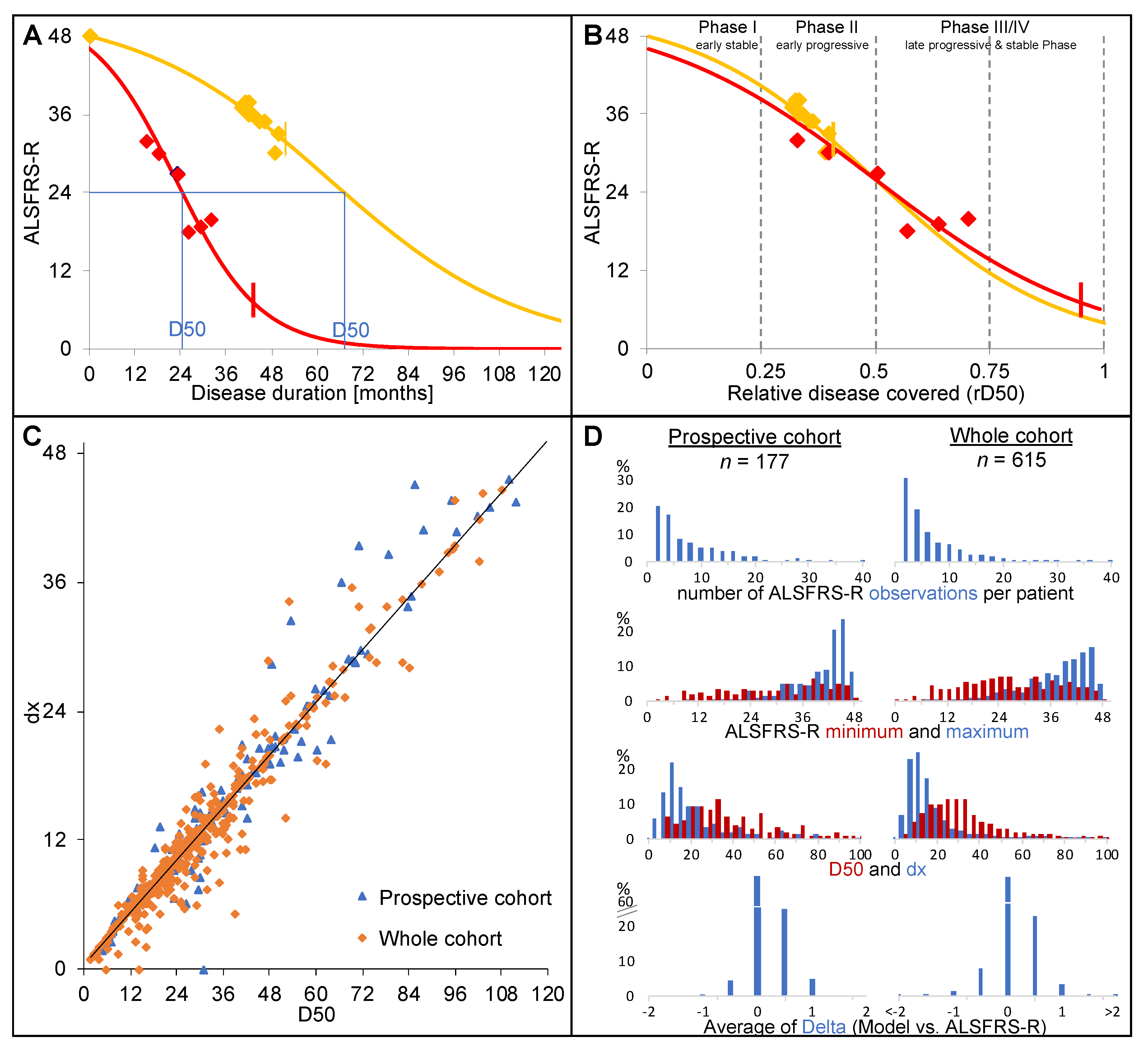
2.2. The D50 Disease Progression Model
2.3. Statistical Analyses
3. Results
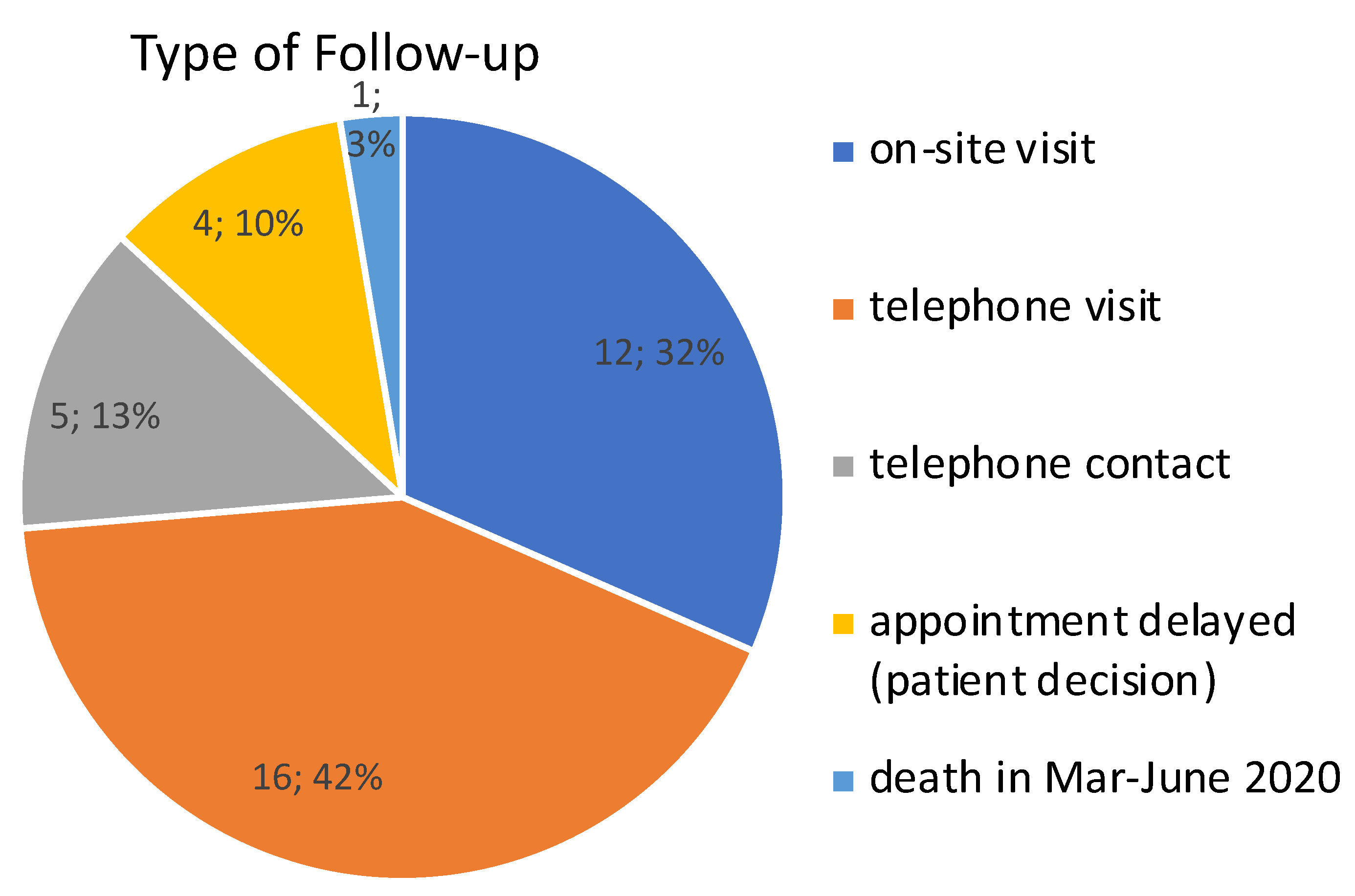
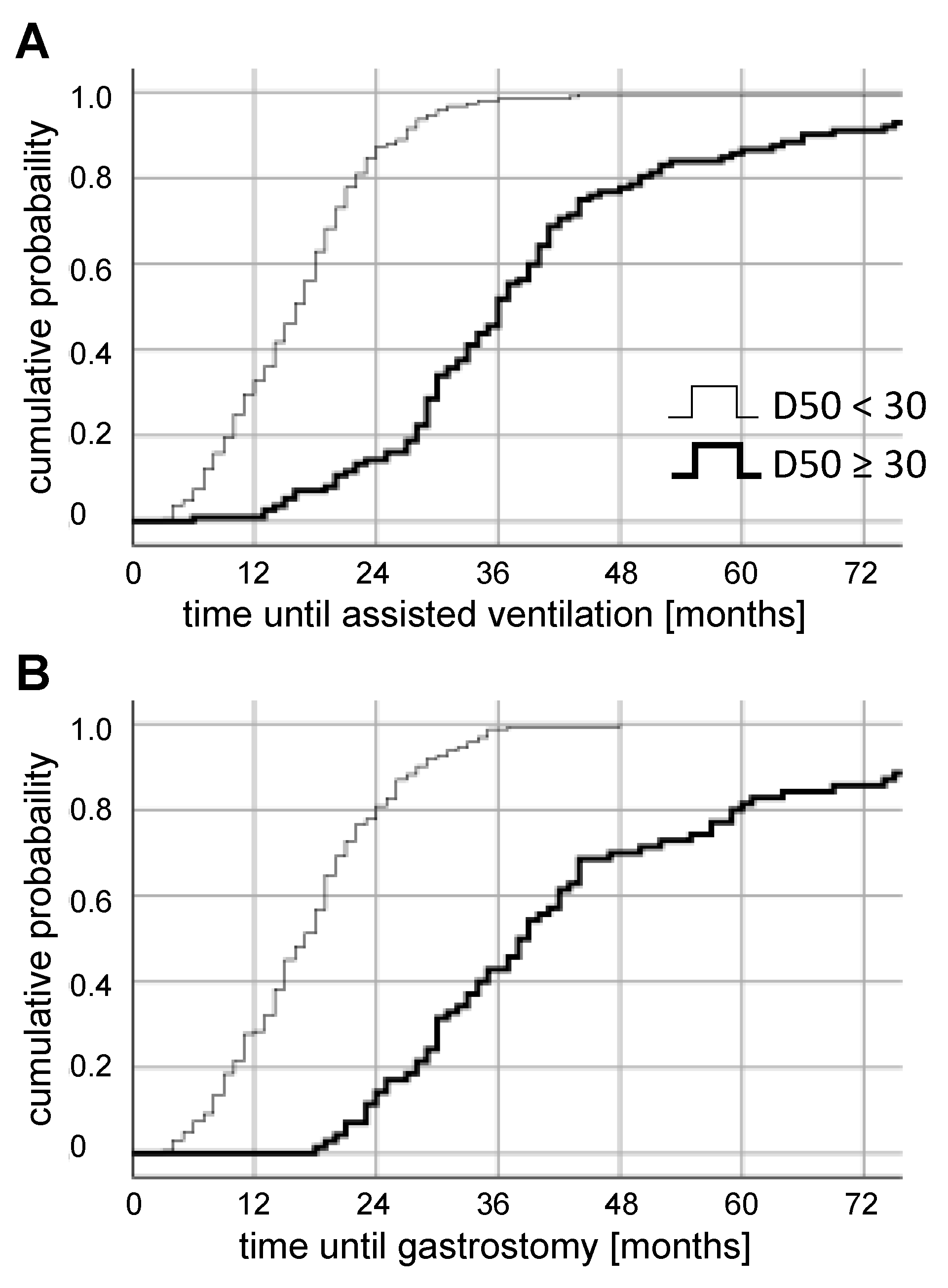
3.1. Triage in the Prospective Cohort
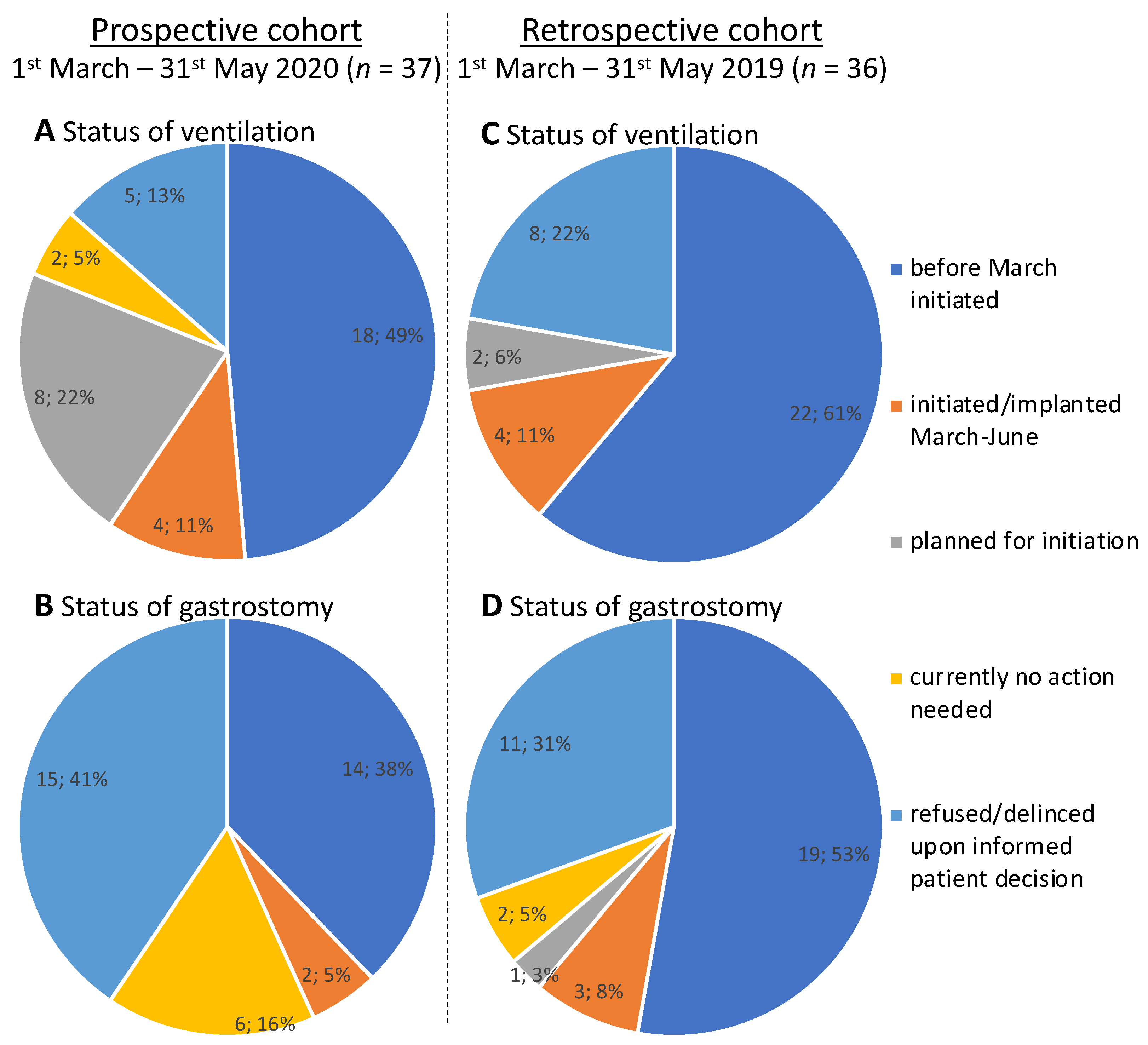
3.2. Outcomes in the Prospective Cohort
3.3. Outcomes in the Retrospective Cohort
3.4. Retrospective Long-Term Analysis Of D50 Subgroups
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Dorst, J.; Ludolph, A.C.; Huebers, A. Disease-modifying and symptomatic treatment of amyotrophic lateral sclerosis. Ther. Adv. Neurol. Disord. 2018, 11, 1756285617734734. [Google Scholar] [CrossRef] [PubMed]
- Westeneng, H.J.; Debray, T.P.A.; Visser, A.E.; Van Eijk, R.P.A.; Rooney, J.P.K.; Calvo, A.; Martin, S.; McDermott, C.J.; Thompson, A.G.; Pinto, S.; et al. Prognosis for patients with amyotrophic lateral sclerosis: Development and validation of a personalised prediction model. Lancet Neurol. 2018, 17, 423–433. [Google Scholar] [CrossRef]
- Simon, N.G.; Turner, M.R.; Vucic, S.; Al-Chalabi, A.; Shefner, J.; Lomen-Hoerth, C.; Kiernan, M.C. Quantifying disease progression in amyotrophic lateral sclerosis. Ann. Neurol. 2014, 76, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Mitchell, J.D.; Lyon, M.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2007, CD001447. [Google Scholar] [CrossRef]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef]
- Hobson, E.V.; McDermott, C.J. Supportive and symptomatic management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2016, 12, 526–538. [Google Scholar] [CrossRef]
- Meyer, T.; Kettemann, D.; Maier, A.; Grehl, T.; Weyen, U.; Grosskreutz, J.; Steinbach, R.; Norden, J.; George, A.; Hermann, A.; et al. Symptomatic pharmacotherapy in ALS: Data analysis from a platform-based medication management programme. J. Neurol. Neurosurg. Psychiatry 2020, 91, 783–785. [Google Scholar] [CrossRef]
- Foley, G.; Timonen, V.; Hardiman, O. Patients’ perceptions of services and preferences for care in amyotrophic lateral sclerosis: A review. Amyotroph. Lateral Scler. 2012, 13, 11–24. [Google Scholar] [CrossRef]
- Seeber, A.A.; Pols, A.J.; Hijdra, A.; Grupstra, H.F.; Willems, D.L.; de Visser, M. Advance care planning in progressive neurological diseases: Lessons from ALS. BMC Palliat. Care 2019, 18, 50. [Google Scholar] [CrossRef]
- Poesen, K.; De Schaepdryver, M.; Stubendorff, B.; Gille, B.; Muckova, P.; Wendler, S.; Prell, T.; Ringer, T.M.; Rhode, H.; Stevens, O.; et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology 2017, 88, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, R.; Batyrbekova, M.; Gaur, N.; Voss, A.; Stubendorff, B.; Mayer, T.E.; Gaser, C.; Witte, O.W.; Prell, T.; Grosskreutz, J. Applying the D50 disease progression model to gray and white matter pathology in amyotrophic lateral sclerosis. Neuroimage Clin. 2020, 25, 102094. [Google Scholar] [CrossRef] [PubMed]
- Prell, T.; Gaur, N.; Steinbach, R.; Witte, O.W.; Grosskreutz, J. Modelling disease course in amyotrophic lateral Sclerosis: Pseudo-longitudinal insights from cross-sectional health-related quality of life data. Health Qual. Life Outcomes 2020, 18, 117. [Google Scholar] [CrossRef] [PubMed]
- Khamankar, N.; Coan, G.; Weaver, B.; Mitchell, C.S. Associative Increases in Amyotrophic Lateral Sclerosis Survival Duration With Non-invasive Ventilation Initiation and Usage Protocols. Front. Neurol. 2018, 9, 578. [Google Scholar] [CrossRef]
- Paipa, A.J.; Povedano, M.; Barcelo, A.; Dominguez, R.; Saez, M.; Turon, J.; Prats, E.; Farrero, E.; Virgili, N.; Martinez, J.A.; et al. Survival benefit of multidisciplinary care in amyotrophic lateral sclerosis in Spain: Association with noninvasive mechanical ventilation. J. Multidiscip. Healthc. 2019, 12, 465–470. [Google Scholar] [CrossRef]
- Bond, L.; Bowen, G.; Mertens, B.; Denson, K.; Jordan, K.; Vidakovic, B.; Mitchell, C.S. Associations of Patient Mood, Modulators of Quality of Life, and Pharmaceuticals with Amyotrophic Lateral Sclerosis Survival Duration. Behav. Sci. 2020, 10, 33. [Google Scholar] [CrossRef]
- Bond, L.; Ganguly, P.; Khamankar, N.; Mallet, N.; Bowen, G.; Green, B.; Mitchell, C.S. A Comprehensive Examination of Percutaneous Endoscopic Gastrostomy and its Association with Amyotrophic Lateral Sclerosis Patient Outcomes. Brain Sci. 2019, 9, 223. [Google Scholar] [CrossRef]
- Burkhardt, C.; Neuwirth, C.; Sommacal, A.; Andersen, P.M.; Weber, M. Is survival improved by the use of NIV and PEG in amyotrophic lateral sclerosis (ALS)? A post-mortem study of 80 ALS patients. PLoS ONE 2017, 12, e0177555. [Google Scholar] [CrossRef]
- Mishra-Kalyani, P.S.; Johnson, B.A.; Glass, J.D.; Long, Q. Estimating the palliative effect of percutaneous endoscopic gastrostomy in an observational registry using principal stratification and generalized propensity scores. Sci. Rep. 2016, 6, 33431. [Google Scholar] [CrossRef]
- Cedarbaum, J.M.; Stambler, N.; Malta, E.; Fuller, C.; Hilt, D.; Thurmond, B.; Nakanishi, A. The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J. Neurol. Sci. 1999, 169, 13–21. [Google Scholar] [CrossRef]
- Capozzo, R.; Zoccolella, S.; Musio, M.; Barone, R.; Accogli, M.; Logroscino, G. Telemedicine is a useful tool to deliver care to patients with Amyotrophic Lateral Sclerosis during COVID-19 pandemic: Results from Southern Italy. Amyotroph. Lateral Scler. Front. Degener. 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Cantello, R.; Ambrosini, S.; Mazzini, L.; Group, C.S. Telemedicine and technological devices for amyotrophic lateral sclerosis in the era of COVID-19. Neurol. Sci. 2020, 41, 1365–1367. [Google Scholar] [CrossRef] [PubMed]
- Helleman, J.; Kruitwagen, E.T.; van den Berg, L.H.; Visser-Meily, J.M.A.; Beelen, A. The current use of telehealth in ALS care and the barriers to and facilitators of implementation: A systematic review. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Sole, G.; Salort-Campana, E.; Pereon, Y.; Stojkovic, T.; Wahbi, K.; Cintas, P.; Adams, D.; Laforet, P.; Tiffreau, V.; Desguerre, I.; et al. Guidance for the care of neuromuscular patients during the COVID-19 pandemic outbreak from the French Rare Health Care for Neuromuscular Diseases Network. Rev. Neurol. 2020, 176, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Schoser, B.; Baum, P.; Boentert, M.; Dillmann, K.U.; Emmer, A.; Knauss, S.; Enax-Krumova, E.; Grosskreutz, J.; Güttsches, A.K.; Hellwig, K.; et al. SARS-CoV-2/COVID-19 und neuromuskuläre Erkrankungen. DGNeurologie 2020, 3, 310–320. [Google Scholar] [CrossRef]
- Andrews, J.A.; Berry, J.D.; Baloh, R.H.; Carberry, N.; Cudkowicz, M.E.; Dedi, B.; Glass, J.; Maragakis, N.J.; Miller, T.M.; Paganoni, S.; et al. Amyotrophic lateral sclerosis care and research in the United States during the COVID-19 pandemic: Challenges and opportunities. Muscle Nerve 2020, 62, 182–186. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Borges do Nascimento, I.J.; Cacic, N.; Abdulazeem, H.M.; von Groote, T.C.; Jayarajah, U.; Weerasekara, I.; Esfahani, M.A.; Civile, V.T.; Marusic, A.; Jeroncic, A.; et al. Novel Coronavirus Infection (COVID-19) in Humans: A Scoping Review and Meta-Analysis. J. Clin. Med. 2020, 9, 941. [Google Scholar] [CrossRef]
- Rosenbohm, A.; Liu, M.; Nagel, G.; Peter, R.S.; Cui, B.; Li, X.; Kassubek, J.; Rothenbacher, D.; Lule, D.; Cui, L.; et al. Phenotypic differences of amyotrophic lateral sclerosis (ALS) in China and Germany. J. Neurol. 2018, 265, 774–782. [Google Scholar] [CrossRef]
- Niedermeyer, S.; Murn, M.; Choi, P.J. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155, 401–408. [Google Scholar] [CrossRef]
- Prem, K.; Liu, Y.; Russell, T.W.; Kucharski, A.J.; Eggo, R.M.; Davies, N.; Jit, M.; Klepac, P.; Flasche, S.; Clifford, S.; et al. The effect of control strategies to reduce social mixing on outcomes of the COVID-19 epidemic in Wuhan, China: A modelling study. Lancet Public Health 2020, 5, e261–e270. [Google Scholar] [CrossRef]
- Clark, A.; Jit, M.; Warren-Gash, C.; Guthrie, B.; Wang, H.H.X.; Mercer, S.W.; Sanderson, C.; McKee, M.; Troeger, C.; Ong, K.L.; et al. Global, regional, and national estimates of the population at increased risk of severe COVID-19 due to underlying health conditions in 2020: A modelling study. Lancet Glob. Health 2020, 8, e1003–e1017. [Google Scholar] [CrossRef]
- Gromicho, M.; Figueiral, M.; Uysal, H.; Grosskreutz, J.; Kuzma-Kozakiewicz, M.; Pinto, S.; Petri, S.; Madeira, S.; Swash, M.; de Carvalho, M. Spreading in ALS: The relative impact of upper and lower motor neuron involvement. Ann. Clin. Transl. Neurol. 2020, 7, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Calvo, A.; Moglia, C.; Mazzini, L.; Mora, G. Phenotypic heterogeneity of amyotrophic lateral sclerosis: A population based study. J. Neurol. Neurosurg. Psychiatry 2011, 82, 740–746. [Google Scholar] [CrossRef]
- EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis; Andersen, P.M.; Abrahams, S.; Borasio, G.D.; de Carvalho, M.; Chio, A.; Van Damme, P.; Hardiman, O.; Kollewe, K.; Morrison, K.E.; et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force. Eur. J. Neurol. 2012, 19, 360–375. [Google Scholar] [CrossRef]
- Miller, R.G.; Jackson, C.E.; Kasarskis, E.J.; England, J.D.; Forshew, D.; Johnston, W.; Kalra, S.; Katz, J.S.; Mitsumoto, H.; Rosenfeld, J.; et al. Practice parameter update: The care of the patient with amyotrophic lateral sclerosis: Drug, nutritional, and respiratory therapies (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009, 73, 1218–1226. [Google Scholar] [CrossRef]
- De Carvalho, M.; Gooch, C.L. The yin and yang of gastrostomy in the management of ALS: Friend or foe? Neurology 2017, 89, 1435–1436. [Google Scholar] [CrossRef]
- Andersen, P.M.; Kuzma-Kozakiewicz, M.; Keller, J.; Aho-Oezhan, H.E.A.; Ciecwierska, K.; Szejko, N.; Vazquez, C.; Bohm, S.; Badura-Lotter, G.; Meyer, T.; et al. Therapeutic decisions in ALS patients: Cross-cultural differences and clinical implications. J. Neurol. 2018, 265, 1600–1606. [Google Scholar] [CrossRef]
- Greenaway, L.P.; Martin, N.H.; Lawrence, V.; Janssen, A.; Al-Chalabi, A.; Leigh, P.N.; Goldstein, L.H. Accepting or declining non-invasive ventilation or gastrostomy in amyotrophic lateral sclerosis: Patients’ perspectives. J. Neurol. 2015, 262, 1002–1013. [Google Scholar] [CrossRef]
- Martin, N.H.; Lawrence, V.; Murray, J.; Janssen, A.; Higginson, I.; Lyall, R.; Burman, R.; Leigh, P.N.; Al-Chalabi, A.; Goldstein, L.H. Decision Making About Gastrostomy and Noninvasive Ventilation in Amyotrophic Lateral Sclerosis. Qual. Health Res. 2016, 26, 1366–1381. [Google Scholar] [CrossRef]
- Boentert, M.; Brenscheidt, I.; Glatz, C.; Young, P. Effects of non-invasive ventilation on objective sleep and nocturnal respiration in patients with amyotrophic lateral sclerosis. J. Neurol. 2015, 262, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Prell, T.; Ringer, T.M.; Wullenkord, K.; Garrison, P.; Gunkel, A.; Stubendorff, B.; Witte, O.W.; Grosskreutz, J. Assessment of pulmonary function in amyotrophic lateral sclerosis: When can polygraphy help evaluate the need for non-invasive ventilation? J. Neurol. Neurosurg. Psychiatry 2016, 87, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.; Lerner, B. Insights into Amyotrophic Lateral Sclerosis from a Machine Learning Perspective. J. Clin. Med. 2019, 8, 1578. [Google Scholar] [CrossRef] [PubMed]
- Demetriou, C.A.; Hadjivasiliou, P.M.; Kleopa, K.A.; Christou, Y.P.; Leonidou, E.; Kyriakides, T.; Zamba-Papanicolaou, E. Retrospective longitudinal study of ALS in Cyprus: Clinical characteristics, management and survival. PLoS ONE 2019, 14, e0220246. [Google Scholar] [CrossRef] [PubMed]
- Logroscino, G.; Marin, B.; Piccininni, M.; Arcuti, S.; Chio, A.; Hardiman, O.; Rooney, J.; Zoccolella, S.; Couratier, P.; Preux, P.M.; et al. Referral bias in ALS epidemiological studies. PLoS ONE 2018, 13, e0195821. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| High Aggressiveness (D50 < 30) | Low Aggressiveness (D50 ≥ 30) | p | |
|---|---|---|---|
| n | 55 | 97 | |
| Age At Symptom Onset [In Years] | 65.3 ± 13.6 (62.3 – 67.6) | 59.6 ± 15.0 (57.8 – 62.3) | 0.015* |
| Symptom Duration In Mar-2020 [In Months] | 20.0 ± 18.0 (17 – 27) | 46.0 ± 39.0 (40 – 54) | <0.001* |
| rD50 In Mar-2020 | 0.55 ± 0.52 (0.46 – 0.69) | 0.34 ± 0.36 (0.28 – 0.39) | <0.001* |
| Gender [Male/Female] | 26/29; 47.3%/52.7% | 56/41; 57.7%/42.3% | 1 |
| Onset-Type [Bulbar/Limb/Generalized/ Respiratory] | 22/29/1/3; 40.0%/52.7%/1.8%/5.5% | 16/79/1/1; 16.5%/81.4%/1%/1% | 0.005* |
| Phase In Mar-2020 [I/II/III&IV] | 1/24/30 1.8%/43.6%/54.5% | 32/36/59 33%/37.1%/29.9% | <0.001* |
| Prior Assisted Ventilation In Mar-2020 [No/Yes] | 32/23; 58.2%/41.8% | 59/38; 60.8%/39.2% | 0.191 |
| Prior Gastrostomy In Mar-2020 [No/Yes] | 31/24; 56.4%/43.6% | 81/16; 83.5%/16.5% | 0.002* |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinbach, R.; Prell, T.; Gaur, N.; Stubendorff, B.; Roediger, A.; Ilse, B.; Witte, O.W.; Grosskreutz, J. Triage of Amyotrophic Lateral Sclerosis Patients during the COVID-19 Pandemic: An Application of the D50 Model. J. Clin. Med. 2020, 9, 2873. https://doi.org/10.3390/jcm9092873
Steinbach R, Prell T, Gaur N, Stubendorff B, Roediger A, Ilse B, Witte OW, Grosskreutz J. Triage of Amyotrophic Lateral Sclerosis Patients during the COVID-19 Pandemic: An Application of the D50 Model. Journal of Clinical Medicine. 2020; 9(9):2873. https://doi.org/10.3390/jcm9092873
Chicago/Turabian StyleSteinbach, Robert, Tino Prell, Nayana Gaur, Beatrice Stubendorff, Annekathrin Roediger, Benjamin Ilse, Otto W. Witte, and Julian Grosskreutz. 2020. "Triage of Amyotrophic Lateral Sclerosis Patients during the COVID-19 Pandemic: An Application of the D50 Model" Journal of Clinical Medicine 9, no. 9: 2873. https://doi.org/10.3390/jcm9092873
APA StyleSteinbach, R., Prell, T., Gaur, N., Stubendorff, B., Roediger, A., Ilse, B., Witte, O. W., & Grosskreutz, J. (2020). Triage of Amyotrophic Lateral Sclerosis Patients during the COVID-19 Pandemic: An Application of the D50 Model. Journal of Clinical Medicine, 9(9), 2873. https://doi.org/10.3390/jcm9092873