Current Insights in Genetics of Sarcoidosis: Functional and Clinical Impacts



Abstract
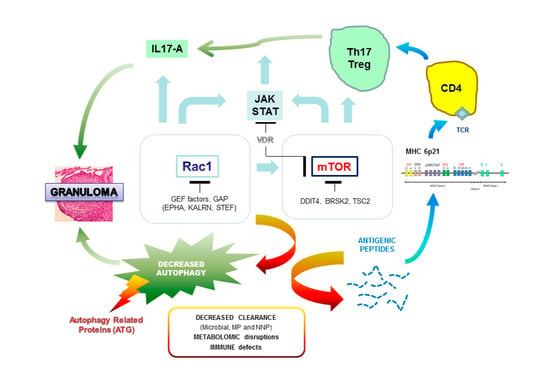
{kind=link}
{kind=link}
{kind=link}
1. Introduction
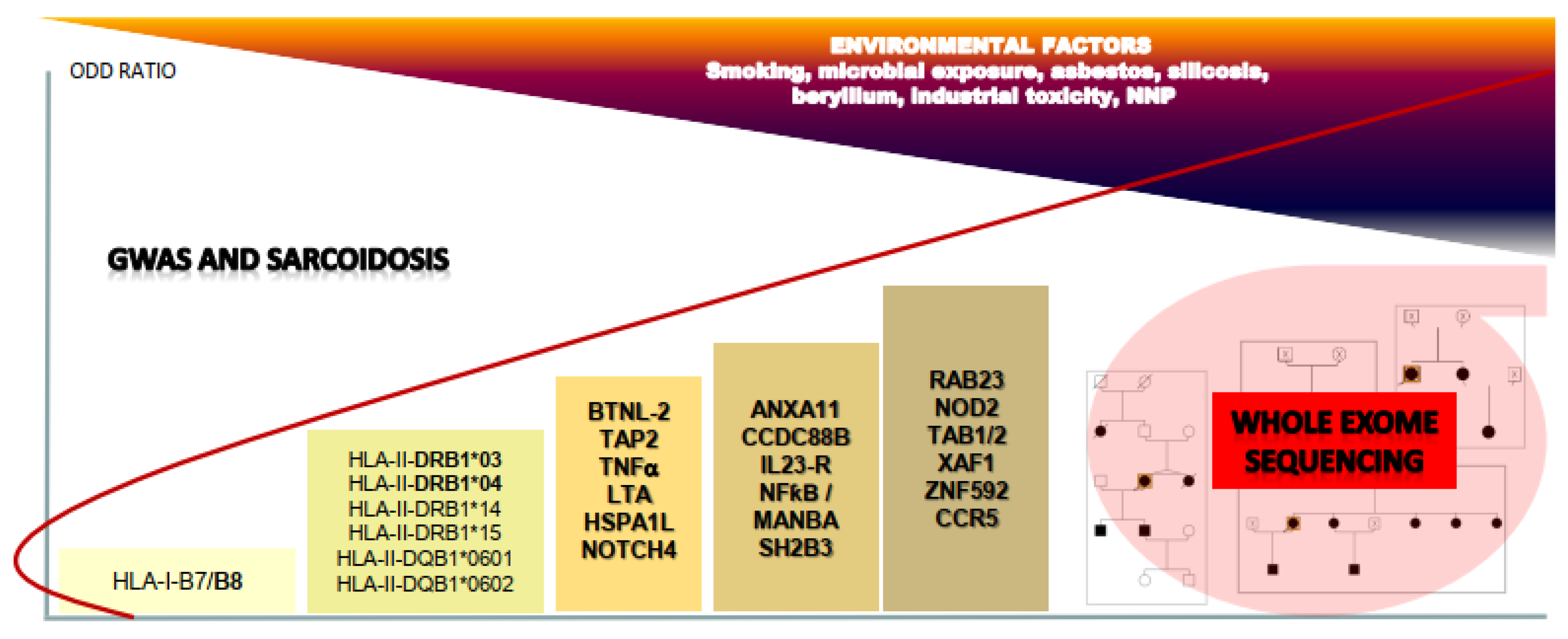
2. Genome Wide Association Studies (GWAS) in Sarcoidosis
2.1. GWAS: from Success to Disappointments
2.2. Contribution of GWAS to Understanding Immunogenetics of Sarcoidosis
2.3. Other Pathways Identified by GWAS in Sarcoidosis Etiology
3. Next Generation Sequencing Contributes to Genetic Research on Sarcoidosis
3.1. WES Screening of Familial and Sporadic Forms of Sarcoidosis
3.2. Are We Able to Integrate All the Results from the Genetic Studies?
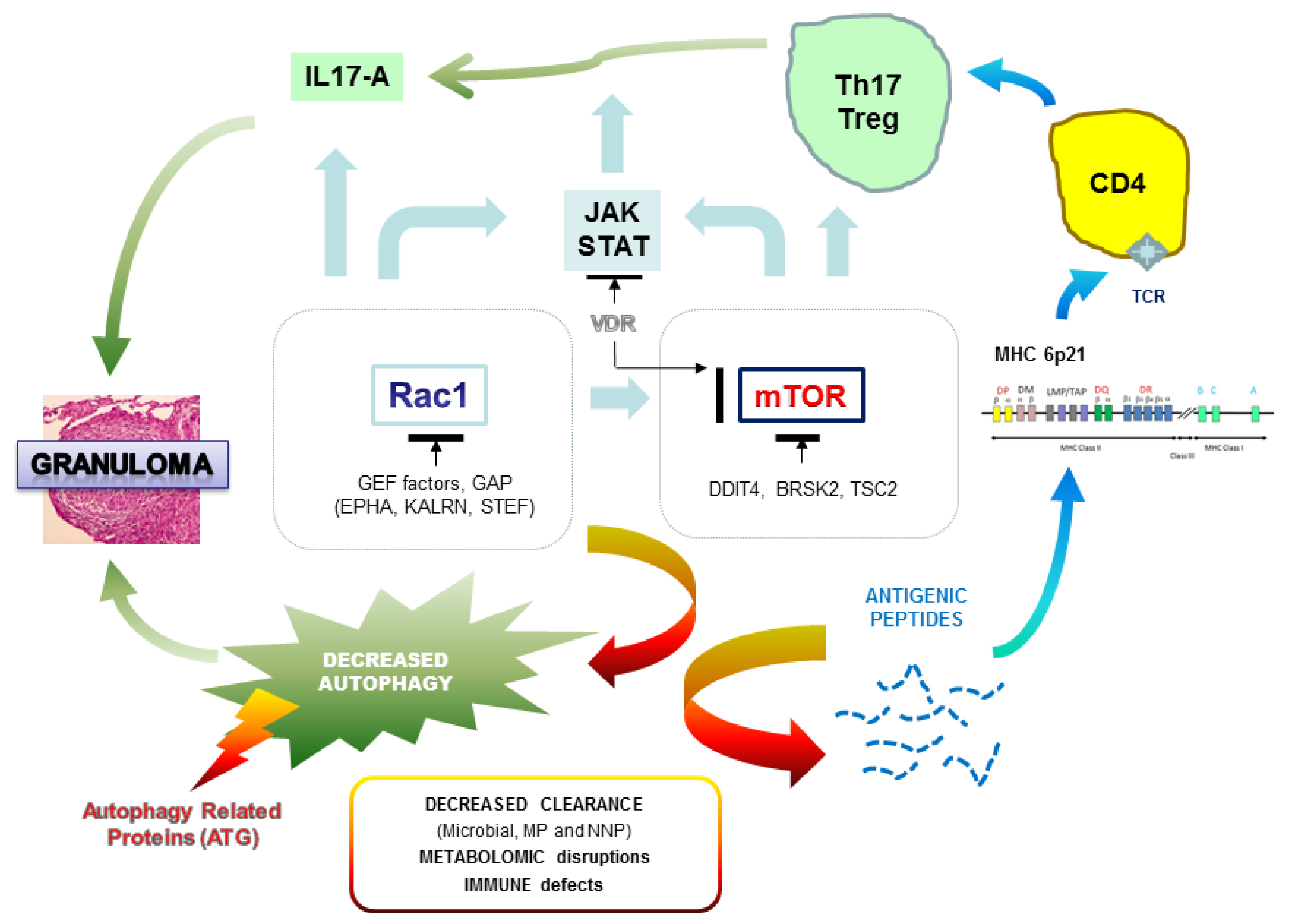
3.3. A Functional Link between Genomic and Transcriptome Analysis
4. Perspectives on the Clinical Utility of Genetics in the Management of Sarcoidosis Patients
Author Contributions
Funding
Conflicts of Interest
References
- Valeyre, D.; Prasse, A.; Nunes, H.; Uzunhan, Y.; Brillet, P.Y.; Müller-Quernheim, J. Sarcoidosis. Lancet 2014, 383, 1155–1167. [Google Scholar] [CrossRef]
- Grunewald, J.; Grutters, J.C.; Arkema, E.V.; Saketkoo, L.A.; Moller, D.R.; Müller-Quernheim, J. Sarcoidosis. Nat. Rev. Dis. Primers 2019, 5, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Wells, A. Advanced sarcoidosis. Curr. Opin. Pulm. Med. 2019, 25, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Rybicki, B.A.; Iannuzzi, M.C. Epidemiology of sarcoidosis: Recent advances and future prospects. Semin. Respir. Crit. Care Med. 2007, 28, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Westney, G.E.; Judson, M.A. Racial and ethnic disparities in sarcoidosis: From genetics to socioeconomics. Clin. Chest Med. 2006, 27, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Brito-Zerón, P.; Kostov, B.; Superville, D.; Baughman, R.P.; Ramos-Casals, M.; Autoimmune Big Data Study Group. Geoepidemiological big data approach to sarcoidosis: Geographical and ethnic determinants. Clin. Exp. Rheumatol. 2019, 37, 1052–1064. [Google Scholar]
- Terwiel, M.; van Moorsel, C.H.M. Clinical epidemiology of familial sarcoidosis: A systematic literature review. Respir. Med. 2019, 149, 36–41. [Google Scholar] [CrossRef]
- Rybicki, B.A.; Hirst, K.; Iyengar, S.K.; Barnard, J.G.; Judson, M.A.; Rose, C.S.; Donohue, J.F.; Kavuru, M.S.; Rabin, D.L.; Rossman, M.D.; et al. A sarcoidosis genetic linkage consortium: The sarcoidosis genetic analysis (SAGA) study. Sarcoidosis Vasc. Diffus. Lung Dis. 2005, 22, 115–122. [Google Scholar]
- Rybicki, B.A.; Sinha, R.; Iyengar, S.; Gray-McGuire, C.; Elston, R.C.; Iannuzzi, M.C. SAGA Study Consortium. Genetic linkage analysis of sarcoidosis phenotypes: The sarcoidosis genetic analysis (SAGA) study. Genes Immun. 2007, 8, 379–386. [Google Scholar] [CrossRef][Green Version]
- Iannuzzi, M.C. Genetics of sarcoidosis. Semin. Respir. Crit. Care Med. 2007, 28, 15–21. [Google Scholar] [CrossRef]
- Rossides, M.; Grunewald, J.; Eklund, A.; Kullberg, S.; Di Giuseppe, D.; Askling, J.; Arkema, E.V. Familial aggregation and heritability of sarcoidosis: A Swedish nested case-control study. Eur. Respir. J. 2018, 52, 1800385. [Google Scholar] [CrossRef] [PubMed]
- Dehghan, A. Genome-Wide Association Studies. Methods Mol. Biol. 2018, 1793, 37–49. [Google Scholar] [PubMed]
- Inshaw, J.R.J.; Cutler, A.J.; Burren, O.S.; Stefana, M.I.; Todd, J.A. Approaches and advances in the genetic causes of autoimmune disease and their implications. Nat. Immunol. 2018, 19, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Wang, Y.; Huang, R.; Skinner, C.; Thompson, T.; Pollard, L.; Wood, T.; Luo, F.; Stevenson, R.; Polimanti, R.; et al. Human Genome Meeting 2016: Houston, TX, USA, 28 February–2 March 2016. Hum. Genom. 2016, 10 (Suppl. 1), 12–42. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; MacCarthy, M.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef]
- Broekema, R.V.; Bakker, O.B.; Jonkers, I.H. A practical view of fine-mapping and gene prioritization in the post-genome-wide association era. Open Biol. 2020, 10, 190221. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, J. Review: Role of genetics in susceptibility and outcome of sarcoidosis. Semin. Respir. Crit. Care Med. 2010, 31, 380–389. [Google Scholar] [CrossRef]
- Fischer, A.; Grunewald, J.; Spagnolo, P.; Nebel, A.; Schreiber, S.; Müller-Quernheim, J. Genetics of sarcoidosis. Semin. Respir. Crit. Care Med. 2014, 35, 296–306. [Google Scholar] [CrossRef]
- Moller, D.R.; Rybicki, B.A.; Hamzeh, N.Y.; Montgomery, C.G.; Chen, E.S.; Drake, W.; Fontenot, A.P. Genetic, Immunologic, and Environmental Basis of Sarcoidosis. Ann. Am. Thorac. Soc. 2017, 14 (Suppl. 6), S429–S436. [Google Scholar] [CrossRef]
- Valentonyte, R.; Hampe, J.; Huse, K.; Rosenstiel, P.; Albrecht, M.; Stenzel, A.; Nagy, M.; Gaede, K.I.; Franke, A.; Haesler, R.; et al. Sarcoidosis is associated with a truncating splice site mutation in BTNL2. Nat. Genet. 2005, 37, 357–364. [Google Scholar] [CrossRef]
- Fischer, A.; Ellinghaus, D.; Nutsua, M.; Hofmann, S.; Montgomery, C.G.; Iannuzzi, M.C.; Rybicki, B.A.; Petrek, M.; Mrazek, F.; Pabst, S.; et al. Identification of Immune-Relevant Factors Conferring Sarcoidosis Genetic Risk. Am. J. Respir. Crit. Care Med. 2015, 192, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Ma, Y.; Niu, X.; Yan, Z.; Liu, S.; Peng, B.; Peng, S.; Fan, H. The BTNL2 G16071A gene polymorphism increases granulomatous disease susceptibility: A meta-analysis including FPRP test of 8710 participants. Medicine (Baltim.) 2016, 95, e4325. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, Y.; Calender, A.; Israël-Biet, D.; Roy, P.; Lebecque, S.; Cottin, V.; Bouvry, D.; Nunes, H.; Sève, P.; Pérard, L.; et al. Familial vs. sporadic sarcoidosis: BTNL2 polymorphisms, clinical presentations, and outcomes in a French cohort. Orphanet J. Rare Dis. 2016, 11, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Chaperon, M.; Pacheco, Y.; Maucort-Boulch, D.; Iwaz, J.; Perard, L.; Broussolle, C.; Jamilloux, Y.; Burillon, C.; Kodjikian, L.; Calender, A.; et al. BTNL2 gene polymorphism and sarcoid uveitis. Br. J. Ophthalmol. 2019, 103, 1690–1694. [Google Scholar] [CrossRef]
- Morais, A.; Lima, B.; Alves, H.; Melo, N.; Mota, P.C.; Marques, A.; Delgado, L. Associations between sarcoidosis clinical course and ANXA11 rs1049550 C/T, BTNL2 rs2076530 G/A, and HLA class I and II alleles. Clin. Respir. J. 2018, 12, 532–537. [Google Scholar] [CrossRef]
- Rossman, M.D.; Thompson, B.; Frederick, M.; Maliarik, M.; Iannuzzi, M.C.; Rybicki, B.A.; Pandey, J.P.; Newman, L.S.; Magira, E.; Monos, D.; et al. HLA-DRB1*1101: A significant risk factor for sarcoidosis in blacks and whites. Am. J. Hum. Genet. 2003, 73, 720–735. [Google Scholar] [CrossRef]
- Sato, H.; Woodhead, F.A.; Ahmad, T.; Grutters, J.C.; Spagnolo, P.; van den Bosch, J.M.; Maier, L.A.; Newman, L.S.; Nagai, S.; Izumi, T.; et al. Sarcoidosis HLA class II genotyping distinguishes differences of clinical phenotype across ethnic groups. Hum. Mol. Genet. 2010, 19, 4100–4111. [Google Scholar] [CrossRef]
- Wennerström, A.; Pietinalho, A.; Lasota, J.; Salli, K.; Surakka, I.; Seppänen, M.; Selroos, O.; Lokki, M.L. Major histocompatibility complex class II and BTNL2 associations in sarcoidosis. Eur. Respir. J. 2013, 42, 550–553. [Google Scholar] [CrossRef]
- Fischer, A.; Rybicki, B.A. Granuloma genes in sarcoidosis: What is new? Curr. Opin. Pulm. Med. 2015, 21, 510–516. [Google Scholar] [CrossRef]
- Grunewald, J.; Idali, F.; Kockum, I.; Seddighzadeh, M.; Nisell, M.; Eklund, A.; Padyukov, L. Major histocompatibility complex class II transactivator gene polymorphism: Associations with Löfgren’s syndrome. Tissue Antigens 2010, 76, 96–101. [Google Scholar] [CrossRef]
- Zhou, Y.; Shen, L.; Zhang, Y.; Jiang, D.; Li, H. Human leukocyte antigen-A, -B, and -DRB1 alleles and sarcoidosis in Chinese Han subjects. Hum. Immunol. 2011, 72, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.M.; Adrianto, I.; Datta, I.; Iannuzzi, M.C.; Trudeau, S.; Li, J.; Drake, W.P.; Montgomery, C.G.; Rybicki, B.A. Association of HLA-DRB1 with Sarcoidosis Susceptibility and Progression in African Americans. Am. J. Respir. Cell Mol. Biol. 2015, 53, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Darlington, P.; Gabrielsen, A.; Sörensson, P.; Tallstedt, L.; Padyukov, L.; Eklund, A.; Grunewald, J. HLA-alleles associated with increased risk for extra-pulmonary involvement in sarcoidosis. Tissue Antigens 2014, 83, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Garman, L.; Pezant, N.; Pastori, A.; Savoy, K.A.; Li, C.; Levin, A.M.; Rybicki, B.A.; Adrianto, I.; Iannuzzi, M.C.; Montgomery, C.G. Genome-Wide Association Study of Ocular Sarcoidosis Confirms HLA Associations and Implicates Barrier Function and Autoimmunity in African Americans. Ocul. Immunol. Inflamm. 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Naruse, T.K.; Matsuzawa, Y.; Ota, M.; Katsuyama, Y.; Matsumori, A.; Hara, M.; Nagai, S.; Morimoto, S.; Sasayama, S.; Inoko, H. HLA-DQB1*0601 is primarily associated with the susceptibility to cardiac sarcoidosis. Tissue Antigens 2000, 56, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Hena, K.M.; Yip, J.; Jaber, N.; Goldfarb, D.; Fullam, K.; Cleven, K.; Moir, W.; Zeig-Owens, R.; Webber, M.P.; Spevack, D.M. FDNY Sarcoidosis Clinical Research Group. Clinical Course of Sarcoidosis in World Trade Center-Exposed Firefighters. Chest 2018, 153, 114–123. [Google Scholar] [CrossRef]
- Cleven, K.L.; Ye, K.; Zeig-Owens, R.; Hena, K.M.; Montagna, C.; Shan, J.; Hosgood, H.D.; Jaber, N.; Weiden, M.D.; Colbeth, H.L.; et al. Genetic Variants Associated with FDNY WTC-Related Sarcoidosis. Int. J. Environ. Res. Public Health 2019, 16, 1830. [Google Scholar] [CrossRef]
- Starshinova, A.A.; Malkova, A.M.; Basantsova, N.Y.; Zinchenko, Y.S.; Kudryavtsev, I.V.; Ershov, G.A.; Soprun, L.A.; Mayevskaya, V.A.; Churilov, L.P.; Yablonskiy, P.K. Sarcoidosis as an Autoimmune Disease. Front. Immunol. 2020, 10, 2933. [Google Scholar] [CrossRef]
- Wahlström, J.; Dengjel, J.; Winqvist, O.; Targoff, I.; Persson, B.; Duyar, H.; Rammensee, H.G.; Eklund, A.; Weissert, R.; Grunewald, J. Autoimmune T cell responses to antigenic peptides presented by bronchoalveolar lavage cell HLA-DR molecules in sarcoidosis. Clin. Immunol. 2009, 133, 353–363. [Google Scholar] [CrossRef]
- Adrianto, I.; Lin, C.P.; Hale, J.J.; Levin, A.M.; Datta, I.; Parker, R.; Adler, A.; Kelly, J.A.; Kaufman, K.M.; Lessard, C.J. Genome-wide association study of African and European Americans implicates multiple shared and ethnic specific loci in sarcoidosis susceptibility. PLoS ONE 2012, 7, e43907. [Google Scholar] [CrossRef]
- Wolin, A.; Lahtela, E.L.; Anttila, V.; Petrek, M.; Grunewald, J.; van Moorsel, C.H.M.; Eklund, A.; Grutters, J.C.; Kolek, V.; Mrazek, F.; et al. SNP Variants in Major Histocompatibility Complex Are Associated with Sarcoidosis Susceptibility-A Joint Analysis in Four European Populations. Front. Immunol. 2017, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Radtke, F.; MacDonald, H.R.; Tacchini-Cottier, F. Regulation of innate and adaptive immunity by Notch. Nat. Rev. Immunol. 2013, 13, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Foley, P.J.; Lympany, P.A.; Puscinska, E.; Zielinski, J.; Welsh, K.I.; du Bois, R.M. Analysis of MHC encoded antigen-processing genes TAP1 and TAP2 polymorphisms in sarcoidosis. Am. J. Respir. Crit. Care Med. 1999, 160, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Hulpke, S.; Tomioka, M.; Kremmer, E.; Ueda, K.; Abele, R.; Tampé, R. Direct evidence that the N-terminal extensions of the TAP complex act as autonomous interaction scaffolds for the assembly of the MHC I peptide-loading complex. Cell. Mol. Life Sci. 2012, 69, 3317–3327. [Google Scholar] [CrossRef] [PubMed]
- Song, G.G.; Kim, J.H.; Lee, Y.H. Associations between TNF-α -308 A/G and lymphotoxin-α +252 A/G polymorphisms and susceptibility to sarcoidosis: A meta-analysis. Mol. Biol. Rep. 2014, 41, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Ghosh, B.; Sharma, S.K. Association of TNF polymorphisms with sarcoidosis, its prognosis and tumour necrosis factor (TNF)-alpha levels in Asian Indians. Clin. Exp. Immunol. 2008, 151, 251–259. [Google Scholar] [CrossRef]
- Wijnen, P.A.; Cremers, J.P.; Nelemans, P.J.; Erckens, R.J.; Hoitsma, E.; Jansen, T.L.; Bekers, O.; Drent, M. Association of the TNF-α G-308A polymorphism with TNF-inhibitor response in sarcoidosis. Eur. Respir. J. 2014, 43, 1730–1739. [Google Scholar] [CrossRef]
- Decock, A.; Van Assche, G.; Vermeire, S.; Wuyts, W.; Ferrante, M. Sarcoidosis-Like Lesions: Another Paradoxical Reaction to Anti-TNF Therapy? J. Crohns Colitis 2017, 11, 378–383. [Google Scholar] [CrossRef]
- Amber, K.T.; Bloom, R.; Mrowietz, U.; Hertl, M. TNF-α: A treatment target or cause of sarcoidosis? J. Eur. Acad. Dermatol. Venereol. 2015, 29, 2104–2111. [Google Scholar] [CrossRef]
- Ruddle, N.H. Lymphotoxin and TNF: How it all began-a tribute to the travelers. Cytokine Growth Factor Rev. 2014, 25, 83–89. [Google Scholar] [CrossRef]
- McDougal, K.E.; Fallin, M.D.; Moller, D.R.; Song, Z.; Cutler, D.J.; Steiner, L.L.; Cutting, G.R.; ACCESS Research Group. Variation in the lymphotoxin-alpha/tumor necrosis factor locus modifies risk of erythema nodosum in sarcoidosis. J. Investig. Dermatol. 2009, 129, 1921–1926. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Ruiz-Argüello, M.B.; Pontejo, S.M.; Fernández de Marco, M.D.M.; Saraiva, M.; Hernáez, B.; Alcamí, A. Chemokines cooperate with TNF to provide protective anti-viral immunity and to enhance inflammation. Nat. Commun. 2018, 9, 1790. [Google Scholar] [CrossRef] [PubMed]
- Allie, N.; Keeton, R.; Court, N.; Abel, B.; Fick, L.; Vasseur, V.; Vacher, R.; Olleros, M.L.; Drutskaya, M.S.; Guler, R.; et al. Limited role for lymphotoxin α in the host immune response to Mycobacterium tuberculosis. J. Immunol. 2010, 185, 4292–4301. [Google Scholar] [CrossRef] [PubMed]
- Mirsaeidi, M.; Gidfar, S.; Vu, A.; Schraufnagel, D. Annexins family: Insights into their functions and potential role in pathogenesis of sarcoidosis. J. Transl. Med. 2016, 14, 89. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Franke, A.; Fischer, A.; Jacobs, G.; Nothnagel, M.; Gaede, K.I.; Schürmann, M.; Müller-Quernheim, J.; Krawczak, M.; Rosenstiel, P.; et al. Genome-wide association study identifies ANXA11 as a new susceptibility locus for sarcoidosis. Nat. Genet. 2008, 40, 1103–1106. [Google Scholar] [CrossRef]
- Mirsaeidi, M.; Vu, A.; Zhang, W.; Arbieva, Z.; Zhang, C.; Abbasi, T.; Hakim, A.; Schraufnagel, D.; Sweiss, N.; Baughman, R.; et al. Annexin A11 is associated with pulmonary fibrosis in African American patients with sarcoidosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2016, 33, 418–422. [Google Scholar]
- Levin, A.M.; Iannuzzi, M.C.; Montgomery, C.G.; Trudeau, S.; Datta, I.; McKeigue, P.; Fischer, A.; Nebel, A.; Rybicki, B.A. Association of ANXA11 Genetic Variation with Sarcoidosis in African Americans and European Americans. Genes Immun. 2013, 14, 13–18. [Google Scholar] [CrossRef][Green Version]
- Sikorova, K.; Kishore, A.; Rapti, A.; Adam, K.; Kocourkova, L.; Zizkova, V.; Charikiopoulou, M.; Kalianos, A.; Bouros, E.; Bouros, D.; et al. Association of TGF-β3 and ANXA11 With Pulmonary Sarcoidosis in Greek Population. Expert Rev. Respir. Med. 2020, 18. [Google Scholar] [CrossRef]
- Davoudi, S.; Chang, V.S.; Navarro-Gomez, D.; Stanwyck, L.K.; Sevgi, D.D.; Papavasileiou, E.; Ren, A.; Uchiyama, E.; Sullivan, L.; Lobo, A.M.; et al. Association of genetic variants in RAB23 and ANXA11 with uveitis in sarcoidosis. Mol. Vis. 2018, 24, 59–74. [Google Scholar]
- Elíes, J.; Yáñez, M.; Pereira, T.M.C.; Gil-Longo, J.; MacDougall, D.A.; Campos-Toimil, M. An Update to Calcium Binding Proteins. Adv. Exp. Med. Biol. 2020, 1131, 183–213. [Google Scholar] [CrossRef]
- Smith, B.N.; Topp, S.D.; Fallini, C.; Shibata, H.; Chen, H.J.; Troakes, C.; King, A.; Ticozzi, N.; Kenna, K.P.; Soragia-Gkazi, A.; et al. Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaad9157. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, E.; Beltrán, S.; Labrador, L.; Manque, P.; Nassif, M.; Woehlbier, U. Implications of Selective Autophagy Dysfunction for ALS Pathology. Cells 2020, 9, 381. [Google Scholar] [CrossRef] [PubMed]
- Veress, B.; Malmsköld, K. The distribution of S100 and lysozyme immunoreactive cells in the various phases of granuloma development in sarcoidosis. Sarcoidosis 1987, 4, 33–37. [Google Scholar]
- Zhou, H.; Diao, M.; Zhang, M. The Association between ANXA11 Gene Polymorphisms and Sarcoidosis: A Meta-Analysis and systematic review. Sarcoidosis Vasc. Diffus. Lung Dis. 2016, 33, 102–111. [Google Scholar]
- Fischer, A.; Schmid, B.; Ellinghaus, D.; Nothnagel, M.; Gaede, K.I.; Schürmann, M.; Lipinski, S.; Rosenstiel, P.; Zissel, G.; Höhne, K.; et al. A novel sarcoidosis risk locus for Europeans on chromosome 11q13.1. Am. J. Respir. Crit. Care Med. 2012, 186, 877–885. [Google Scholar] [CrossRef]
- Fodil, N.; Moradin, N.; Leung, V.; Olivier, J.F.; Radovanovic, I.; Jeyakumar, T.; Flores Molina, M.; McFarquhar, A.; Cayrol, R.; Bozec, D. CCDC88B is required for pathogenesis of inflammatory bowel disease. Nat. Commun. 2017, 8, 932. [Google Scholar] [CrossRef]
- Kennedy, J.M.; Fodil, N.; Torre, S.; Bongfen, S.E.; Olivier, J.F.; Leung, V.; Langlais, D.; Meunier, C.; Berghout, J.; Langat, P. CCDC88B is a novel regulator of maturation and effector functions of T cells during pathological inflammation. J. Exp. Med. 2014, 211, 2519–2535. [Google Scholar] [CrossRef]
- Kim, H.S.; Choi, D.; Lim, L.L.; Allada, G.; Smith, J.R.; Austin, C.R.; Doyle, T.M.; Goodwin, K.A.; Rosenbaum, J.T.; Martin, T.M. Association of interleukin 23 receptor gene with sarcoidosis. Dis. Markers 2011, 31, 17–24. [Google Scholar] [CrossRef]
- Revu, S.; Wu, J.; Henkel, M.; Rittenhouse, N.; Menk, A.; Delgoffe, G.M.; Poholek, A.C.; McGeachy, M.J. IL-23 and IL-1β Drive Human Th17 Cell Differentiation and Metabolic Reprogramming in Absence of CD28 Costimulation. Cell Rep. 2018, 22, 2642–2653. [Google Scholar] [CrossRef]
- Hou, S.; Liao, D.; Zhang, J.; Fang, J.; Chen, L.; Qi, J.; Zhang, Q.; Liu, Y.; Bai, L.; Zhou, Y.; et al. Genetic variations of IL17F and IL23A show associations with Behçet’s disease and Vogt-Koyanagi-Harada syndrome. Ophthalmology 2015, 122, 518–523. [Google Scholar] [CrossRef]
- Rivera, N.V.; Patasova, K.; Kullberg, S.; Diaz-Gallo, L.M.; Iseda, T.; Bengtsson, C.; Alfredsson, L.; Eklund, A.; Kockum, I.; Grunewald, J.; et al. A Gene-Environment Interaction between Smoking and Gene polymorphisms Provides a High Risk of Two Subgroups of Sarcoidosis. Sci. Rep. 2019, 9, 18633. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Mizoguchi, I.; Chiba, Y.; Ohashi, M.; Xu, M.; Yoshimoto, T. Expanding Diversity in Molecular Structures and Functions of the IL-6/IL-12 Heterodimeric Cytokine Family. Front. Immunol. 2016, 7, 479. [Google Scholar] [CrossRef] [PubMed]
- Bloch, Y.; Bouchareychas, L.; Merceron, R.; Składanowska, K.; Van den Bossche, L.; Detry, S.; Govindarajan, S.; Elewaut, D.; Haerynck, F.; Dullaers, M.; et al. Structural Activation of Pro-inflammatory Human Cytokine IL-23 by Cognate IL-23 Receptor Enables Recruitment of the Shared Receptor IL-12Rβ1. Immunity 2018, 48, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.L.; Miao, Y.; et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2016, 375, 1946–1960. [Google Scholar] [CrossRef]
- Zhao, X.; Ross, E.J.; Wang, Y.; Horwitz, B.H. Nfkb1 inhibits LPS-induced IFN-β and IL-12 p40 production in macrophages by distinct mechanisms. PLoS ONE 2012, 7, e32811. [Google Scholar] [CrossRef] [PubMed]
- Devallière, J.; Charreau, B. The adaptor Lnk (SH2B3): An emerging regulator in vascular cells and a link between immune and inflammatory signaling. Biochem. Pharmacol. 2011, 82, 1391–1402. [Google Scholar] [CrossRef]
- Hofmann, S.; Fischer, A.; Till, A.; Müller-Quernheim, J.; Häsler, R.; Franke, A.; Gäde, K.I.; Schaarschmidt, H.; Rosenstiel, P.; Nebel, A.; et al. A genome-wide association study reveals evidence of association with sarcoidosis at 6p12.1. Eur. Respir. J. 2011, 38, 1127–1135. [Google Scholar] [CrossRef]
- Zheng, L.Q.; Chi, S.M.; Li, C.X. Rab23’s genetic structure, function and related diseases: A review. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef]
- Ao, X.; Zou, L.; Wu, Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014, 21, 348–358. [Google Scholar] [CrossRef]
- Abe, J.I.; Ko, K.A.; Kotla, S.; Wang, Y.; Paez-Mayorga, J.; Shin, I.J.; Guzman, L.; Abe, R.J.; Taunton, J.; Thomas, T.N.; et al. MAGI1 as a link between endothelial activation and ER stress drives atherosclerosis. JCI Insight 2019, 4, e125570. [Google Scholar] [CrossRef]
- Bello, G.A.; Adrianto, I.; Dumancas, G.G.; Levin, A.M.; Iannuzzi, M.C.; Rybicki, B.A.; Montgomery, C. Role of NOD2 Pathway Genes in Sarcoidosis Cases with Clinical Characteristics of Blau Syndrome. Am. J. Respir. Crit. Care Med. 2015, 192, 1133–1135. [Google Scholar] [CrossRef] [PubMed]
- Al Nabhani, Z.; Dietrich, G.; Hugot, J.P.; Barreau, F. Nod2: The intestinal gate keeper. PLoS Pathog. 2017, 13, e1006177. [Google Scholar] [CrossRef] [PubMed]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Mihaly, S.R.; Morioka, S.; Ninomiya-Tsuji, J.; Takaesu, G. Activated macrophage survival is coordinated by TAK1 binding proteins. PLoS ONE 2014, 9, e94982. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.H.; Picco, M.F. Crohn’s Disease: Genetics Update. Gastroenterol. Clin. N. Am. 2017, 46, 449–461. [Google Scholar] [CrossRef]
- Miceli-Richard, C.; Lesage, S.; Rybojad, M.; Prieur, A.M.; Manouvrier-Hanu, S.; Häfner, R.; Chamaillard, M.; Zouali, H.; Thomas, G.; Hugot, J.P. CARD15 mutations in Blau syndrome. Nat. Genet. 2001, 29, 19–20. [Google Scholar] [CrossRef]
- Besnard, V.; Calender, A.; Bouvry, D.; Pacheco, Y.; Chapelon-Abric, C.; Jeny, F.; Nunes, H.; Planès, C.; Valeyre, D. G908R NOD2 variant in a family with sarcoidosis. Respir. Res. 2018, 19, 44. [Google Scholar] [CrossRef]
- Davoudi, S.; Navarro-Gomez, D.; Shen, L.; Ung, C.; Ren, A.; Sullivan, L.; Kwong, M.; Janessian, M.; Comander, J.; Gai, X.; et al. NOD2 genetic variants and sarcoidosis-associated uveitis. Am. J. Ophthalmol. Case Rep. 2016, 3, 39–42. [Google Scholar] [CrossRef]
- Mentzer, A.; Nayee, S.; Omar, Y.; Hullah, E.; Taylor, K.; Goel, R.; Bye, H.; Shembesh, T.; Elliott, T.R.; Campbell, H.; et al. Genetic Association Analysis Reveals Differences in the Contribution of NOD2 Variants to the Clinical Phenotypes of Orofacial Granulomatosis. Inflamm. Bowel. Dis. 2016, 22, 1552–1558. [Google Scholar] [CrossRef]
- Levin, A.M.; Iannuzzi, M.C.; Montgomery, C.G.; Trudeau, S.; Datta, I.; Adrianto, I.; Chitale, D.A.; McKeigue, P.; Rybicki, B.A. Admixture fine-mapping in African Americans implicates XAF1 as a possible sarcoidosis risk gene. PLoS ONE 2014, 9, e92646. [Google Scholar] [CrossRef]
- Sun, P.H.; Zhu, L.M.; Qiao, M.M.; Zhang, Y.P.; Jiang, S.H.; Wu, Y.L.; Tu, S.P. The XAF1 tumor suppressor induces autophagic cell death via upregulation of Beclin-1 and inhibition of Akt pathway. Cancer Lett. 2011, 310, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.I.; Kim, J.W.; Ko, K.P.; Ryu, B.K.; Lee, M.G.; Kim, H.J.; Chi, S.G. XAF1 forms a positive feedback loop with IRF-1 to drive apoptotic stress response and suppress tumorigenesis. Cell Death Dis. 2018, 9, 806. [Google Scholar] [CrossRef] [PubMed]
- Lareau, C.A.; Adrianto, I.; Levin, A.M.; Iannuzzi, M.C.; Rybicki, B.A.; Montgomery, C.G. Fine mapping of chromosome 15q25 implicates ZNF592 in neurosarcoidosis patients. Ann. Clin. Transl. Neurol. 2015, 2, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, E.; Poitelon, Y.; Chouery, E.; Salem, N.; Levy, N.; Mégarbané, A.; Delague, V. CAMOS, a nonprogressive, autosomal recessive, congenital cerebellar ataxia, is caused by a mutant zinc-finger protein, ZNF592. Eur. J. Hum. Genet. 2010, 18, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Petrek, M.; Drábek, J.; Kolek, V.; Zlámal, J.; Welsh, K.I.; Bunce, M.; Weigl, E.; Du Bois, R. CC chemokine receptor gene polymorphisms in Czech patients with pulmonary sarcoidosis. Am. J. Respir. Crit. Care Med. 2000, 162, 1000–1003. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Renzoni, E.A.; Wells, A.U.; Copley, S.J.; Desai, S.R.; Sato, H.; Grutters, J.C.; Abdallah, A.; Taegtmeyer, A.; Welsh, K.I.; et al. C-C chemokine receptor 5 gene variants in relation to lung disease in sarcoidosis. Am. J. Respir. Crit. Care Med. 2005, 172, 721–728. [Google Scholar] [CrossRef]
- Palchevskiy, V.; Hashemi, N.; Weigt, S.S.; Xue, Y.Y.; Derhovanessian, A.; Keane, M.P.; Strieter, R.M.; Fishbein, M.C.; Deng, J.C.; Lynch, J.P., III. Immune response CC chemokines CCL2 and CCL5 are associated with pulmonary sarcoidosis. Fibrogenesis Tissue Repair. 2011, 4. [Google Scholar] [CrossRef]
- Sato, H.; Silveira, L.; Spagnolo, P.; Gillespie, M.; Gottschall, E.B.; Welsh, K.I.; du Bois, R.M.; Newman, L.S.; Maier, L.A. CC chemokine receptor 5 gene polymorphisms in beryllium disease. Eur. Respir. J. 2010, 36, 331–338. [Google Scholar] [CrossRef]
- Ghanem, M.; Naccache, J.M.; Bonneterre, V.; L’huillier, J.P.; Guillaud Segard, B.; Lazor, R.; Tazi, A.; Gondouin, A.; Israel-Biet, D.; Marquignon, M.F.; et al. Diagnostic difficulties of chronic pulmonary berylliosis in France. Rev. Mal. Respir. 2020, 37, 364–368. [Google Scholar] [CrossRef]
- Greaves, S.A.; Atif, S.M.; Fontenot, A.P. Adaptive Immunity in Pulmonary Sarcoidosis and Chronic Beryllium Disease. Front. Immunol. 2020, 11, 474. [Google Scholar] [CrossRef]
- Fontenot, A.P. Immunologic Effects of Beryllium Exposure. Ann. Am. Thorac. Soc. 2018, 15 (Suppl. 2), S81–S85. [Google Scholar] [CrossRef] [PubMed]
- Sverrild, A.; Backer, V.; Kyvik, K.O.; Kaprio, J.; Milman, N.; Svendsen, C.B.; Thomsen, S.F. Heredity in sarcoidosis: A registry-based twin study. Thorax 2008, 63, 894–896. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, D.; Bohra, A.; Thudi, M.; Varshney, R.K. Fine mapping and gene cloning in the post-NGS era: Advances and prospects. Theor. Appl. Genet. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Calender, A.; Rollat Farnier, P.A.; Buisson, A.; Pinson, S.; Bentaher, A.; Lebecque, S.; Corvol, H.; Abou Taam, R.; Houdouin, V.; Bardel, C.; et al. Whole exome sequencing in three families segregating a pediatric case of sarcoidosis. BMC Med. Genom. 2018, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Calender, A.; Lim, C.X.; Weichhart, T.; Buisson, A.; Besnard, V.; Rollat-Farnier, P.A.; Bardel, C.; Roy, P.; Cottin, V.; Devouassoux, G.; et al. Exome sequencing and pathogenicity-network analysis of five French families implicate mTOR signalling and autophagy in familial sarcoidosis. Eur. Respir. J. 2019, 54, 1900430. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7, 7–20. [Google Scholar] [CrossRef]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef]
- Linke, M.; Pham, H.T.; Katholnig, K.; Schnöller, T.; Miller, A.; Demel, F.; Schütz, B.; Rosner, M.; Kovacic, B.; Sukhbaatar, N.; et al. Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat. Immunol. 2017, 18, 293–302. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, H.; Wang, N.; Fang, C.L.; Jing, X.Y.; Guo, J.; Sun, W.; Yu, C.; Yang, X.Y.; Xu, Z.J. Whole Exome Sequencing and Analysis of a Chinese Family with Familial Pulmonary Sarcoidosis. Zhonghua Jie He He Hu Xi Za Zhi 2020, 43, 525–531. [Google Scholar]
- Nagahama, Y.; Shimoda, M.; Mao, G.; Singh, S.K.; Kozakai, Y.; Sun, X.; Motooka, D.; Nakamura, S.; Tanaka, H.; Satoh, T.; et al. Regnase-1 Controls Colon Epithelial Regeneration via Regulation of mTOR and Purine Metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, 11036–11041. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Arima, Y.; Kamimura, D.; Tanaka, Y.; Takahashi, N.; Uehata, T.; Maeda, K.; Satoh, T.; Murakami, M.; Akira, S. Phosphorylation-dependent Regnase-1 Release From Endoplasmic Reticulum Is Critical in IL-17 Response. J. Exp. Med. 2019, 216, 1431–1449. [Google Scholar] [CrossRef] [PubMed]
- Kishore, A.; Petersen, B.S.; Nutsua, M.; Müller-Quernheim, J.; Franke, A.; Fischer, A.; Schreiber, S.; Petrek, M. Whole-exome sequencing identifies rare genetic variations in German families with pulmonary sarcoidosis. Hum. Genet. 2018, 137, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Lahtela, E.; Kankainen, M.; Sinisalo, J.; Selroos, O.; Lokki, M.L. Exome Sequencing Identifies Susceptibility Loci for Sarcoidosis Prognosis. Front. Immunol. 2019, 10, 2964. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, Y.; Lim, C.X.; Weichhart, T.; Valeyre, D.; Bentaher, A.; Calender, A. Sarcoidosis and the mTOR, Rac1, and Autophagy Triad. Trends Immunol. 2020, 41, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Schuldt, A. Cell growth: RAC1 sizes up mTOR. Nat. Rev. Mol. Cell Biol. 2011, 12, 343–354. [Google Scholar] [CrossRef]
- Shin, J.Y.; Wey, M.; Umutesi, H.G.; Sun, X.; Simecka, J.; Heo, J. Thiopurine Prodrugs Mediate Immunosuppressive Effects by Interfering with Rac1 Protein Function. J. Biol. Chem. 2016, 291, 13699–13713. [Google Scholar] [CrossRef]
- Al-Awqati, Q. Kidney growth and hypertrophy: The role of mTOR and vesicle trafficking. J. Clin. Investig. 2015, 125, 2267–2270. [Google Scholar] [CrossRef]
- Pleyer, U.; Thurau, S.R. Sirolimus for the treatment of noninfectious uveitis. Expert Opin. Pharmacother. 2016, 17, 127–135. [Google Scholar] [CrossRef]
- Silverpil, E.; Wright, A.K.; Hansson, M.; Jirholt, P.; Henningsson, L.; Smith, M.E.; Gordon, S.B.; Iwakura, Y.; Gjertsson, I.; Glader, P.; et al. Negative feedback on IL-23 exerted by IL-17A during pulmonary inflammation. Innate Immun. 2013, 19, 479–492. [Google Scholar] [CrossRef]
- Kurdi, A.T.; Bassil, R.; Olah, M.; Wu, C.; Xiao, S.; Taga, M.; Frangieh, M.; Buttrick, T.; Orent, W.; Bradshaw, E.M. Tiam1/Rac1 complex controls Il17a transcription and autoimmunity. Nat. Commun. 2016, 7, 13048. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Mett, I.; Bhunia, A.K.; Bowman, J.; Perez, M.; Zhang, L.; Gandjeva, A.; Zhen, L.; Chukwueke, U.; Mao, T.; et al. Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema. Nat. Med. 2010, 16, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Bucova, M.; Suchankova, M.; Tibenska, E.; Tedlova, E.; Demian, J.; Majer, I.; Novosadova, H.; Tedla, M. TREM-2 Receptor Expression Increases with 25(OH)D Vitamin Serum Levels in Patients with Pulmonary Sarcoidosis. Mediat. Inflamm. 2015, 181986. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Janovcik, J.; Ray, M.; Sweiss, N.; Lower, E.E. Calcium and vitamin D metabolism in sarcoidosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2013, 30, 113–120. [Google Scholar]
- Wang, H.; Wang, J.; Qu, H.; Wei, H.; Ji, B.; Yang, Z.; Wu, J.; He, Q.; Luo, Y.; Liu, D.; et al. In vitro and in vivo inhibition of mTOR by 1,25-dihydroxyvitamin D3 to improve early diabetic nephropathy via the DDIT4/TSC2/mTOR pathway. Endocrine 2016, 54, 348–359. [Google Scholar] [CrossRef]
- Tong, Y.; Song, F. Intracellular calcium signaling regulates autophagy via calcineurin-mediated TFEB dephosphorylation. Autophagy 2015, 11, 1192–1195. [Google Scholar] [CrossRef]
- Schupp, J.C.; Vukmirovic, M.; Kaminski, N.; Prasse, A. Transcriptome profiles in sarcoidosis and their potential role in disease prediction. Curr. Opin. Pulm. Med. 2017, 23, 487–492. [Google Scholar] [CrossRef]
- Damsky, W.; Thakral, D.; Emeagwali, N.; Galan, A.; King, B. Tofacitinib Treatment and Molecular Analysis of Cutaneous Sarcoidosis. N. Engl. J. Med. 2018, 379, 2540–2546. [Google Scholar] [CrossRef]
- Koth, L.L.; Solberg, O.D.; Peng, J.C.; Bhakta, N.R.; Nguyen, C.P.; Woodruff, P.G. Sarcoidosis blood transcriptome reflects lung inflammation and overlaps with tuberculosis. Am. J. Respir. Crit. Care Med. 2011, 184, 1153–1163. [Google Scholar] [CrossRef]
- Talreja, J.; Farshi, P.; Alazizi, A.; Luca, F.; Pique-Regi, R.; Samavati, L. RNA-sequencing Identifies Novel Pathways in Sarcoidosis Monocytes. Sci. Rep. 2017, 7, 2720. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, X.; Xu, G.; Guo, Y.; Sun, L.; Zhang, C.; Li, X.; Li, B. Arsenic Induces mTOR-dependent Autophagy, Whereas It Impairs the Autophagy-Lysosome Pathway and the Potential Role of TFEB in Cultured Dendritic Cells. Metallomics 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Ruf, S.; Heberle, A.M.; Langelaar-Makkinje, M.; Gelino, S.; Wilkinson, D.; Gerbeth, C.; Schwarz, J.J.; Holzwarth, B.; Warscheid, B.; Meisinger, C.; et al. PLK1 (Polo Like Kinase 1) Inhibits MTOR Complex 1 and Promotes Autophagy. Autophagy 2017, 13, 486–505. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.C. The phagocyte respiratory burst: Historical perspectives and recent advances. Immunol. Lett. 2017, 192, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Talreja, J.; Talwar, H.; Bauerfeld, C.; Grossman, L.I.; Zhang, K.; Tranchida, P.; Samavati, L. HIF-1α Regulates IL-1β and IL-17 in Sarcoidosis. Elife 2019, 8, e44519. [Google Scholar] [CrossRef] [PubMed]
- Racanelli, A.C.; Kikkers, S.A.; Choi, A.M.K.; Cloonan, S.M. Autophagy and inflammation in chronic respiratory disease. Autophagy 2018, 14, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR Function in Response to Hypoxia by REDD1 and the TSC1/TSC2 Tumor Suppressor Complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef]
- Seong, M.; Lee, J.; Kang, H. Hypoxia-induced regulation of mTOR signaling by miR-7 targeting REDD1. J. Cell. Biochem. 2019, 120, 4523–4532. [Google Scholar] [CrossRef]
- Pattnaik, B.; Sryma, P.B.; Mittal, S.; Agrawal, A.; Guleria, R.; Madan, K. MicroRNAs in pulmonary sarcoidosis: A systematic review. Respir. Investig. 2020, 58, 232–238. [Google Scholar] [CrossRef]
- Wang, R.; Jiao, H.; Zhao, J.; Wang, X.; Lin, H. Glucocorticoids Enhance Muscle Proteolysis through a Myostatin-Dependent Pathway at the Early Stage. PLoS ONE 2016, 11, e0156225. [Google Scholar] [CrossRef]
- Xu, X.; Sun, J.; Song, R.; Doscas, M.E.; Williamson, A.J.; Zhou, J.; Sun, J.; Jiao, X.; Liu, X.; Li, Y. Inhibition of p70 S6 kinase (S6K1) activity by A77 1726, the active metabolite of leflunomide, induces autophagy through TAK1-mediated AMPK and JNK activation. Oncotarget 2017, 8, 30438–30454. [Google Scholar] [CrossRef]
- Wei, J.J.; Kallenbach, L.R.; Kreider, M.; Leung, T.H.; Rosenbach, M. Resolution of cutaneous sarcoidosis after Janus kinase inhibitor therapy for concomitant polycythemia vera. JAAD Case Rep. 2019, 5, 360–361. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, S.; Duhamel, F.; Coulombe, P.; Popoff, M.R.; Meloche, S. Rho family GTPases are required for activation of Jak/STAT signaling by G protein-coupled receptors. Mol. Cell. Biol. 2003, 23, 1316–1333. [Google Scholar] [CrossRef]
- Saleiro, D.; Platanias, L.C. Intersection of mTOR and STAT signaling in immunity. Trends Immunol. 2015, 36, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Rossman, M.D.; Thompson, B.; Frederick, M.; Iannuzzi, M.C.; Rybicki, B.A.; Pandey, J.P.; Newman, L.S.; Rose, C.; Magira, E.; Weinberger, S.E.; et al. HLA and environmental interactions in sarcoidosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2008, 25, 125–132. [Google Scholar]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calender, A.; Weichhart, T.; Valeyre, D.; Pacheco, Y. Current Insights in Genetics of Sarcoidosis: Functional and Clinical Impacts. J. Clin. Med. 2020, 9, 2633. https://doi.org/10.3390/jcm9082633
Calender A, Weichhart T, Valeyre D, Pacheco Y. Current Insights in Genetics of Sarcoidosis: Functional and Clinical Impacts. Journal of Clinical Medicine. 2020; 9(8):2633. https://doi.org/10.3390/jcm9082633
Chicago/Turabian StyleCalender, Alain, Thomas Weichhart, Dominique Valeyre, and Yves Pacheco. 2020. "Current Insights in Genetics of Sarcoidosis: Functional and Clinical Impacts" Journal of Clinical Medicine 9, no. 8: 2633. https://doi.org/10.3390/jcm9082633
APA StyleCalender, A., Weichhart, T., Valeyre, D., & Pacheco, Y. (2020). Current Insights in Genetics of Sarcoidosis: Functional and Clinical Impacts. Journal of Clinical Medicine, 9(8), 2633. https://doi.org/10.3390/jcm9082633