Effects of Ischemia-Reperfusion on Tubular Cell Membrane Transporters and Consequences in Kidney Transplantation


Abstract
:1. Introduction
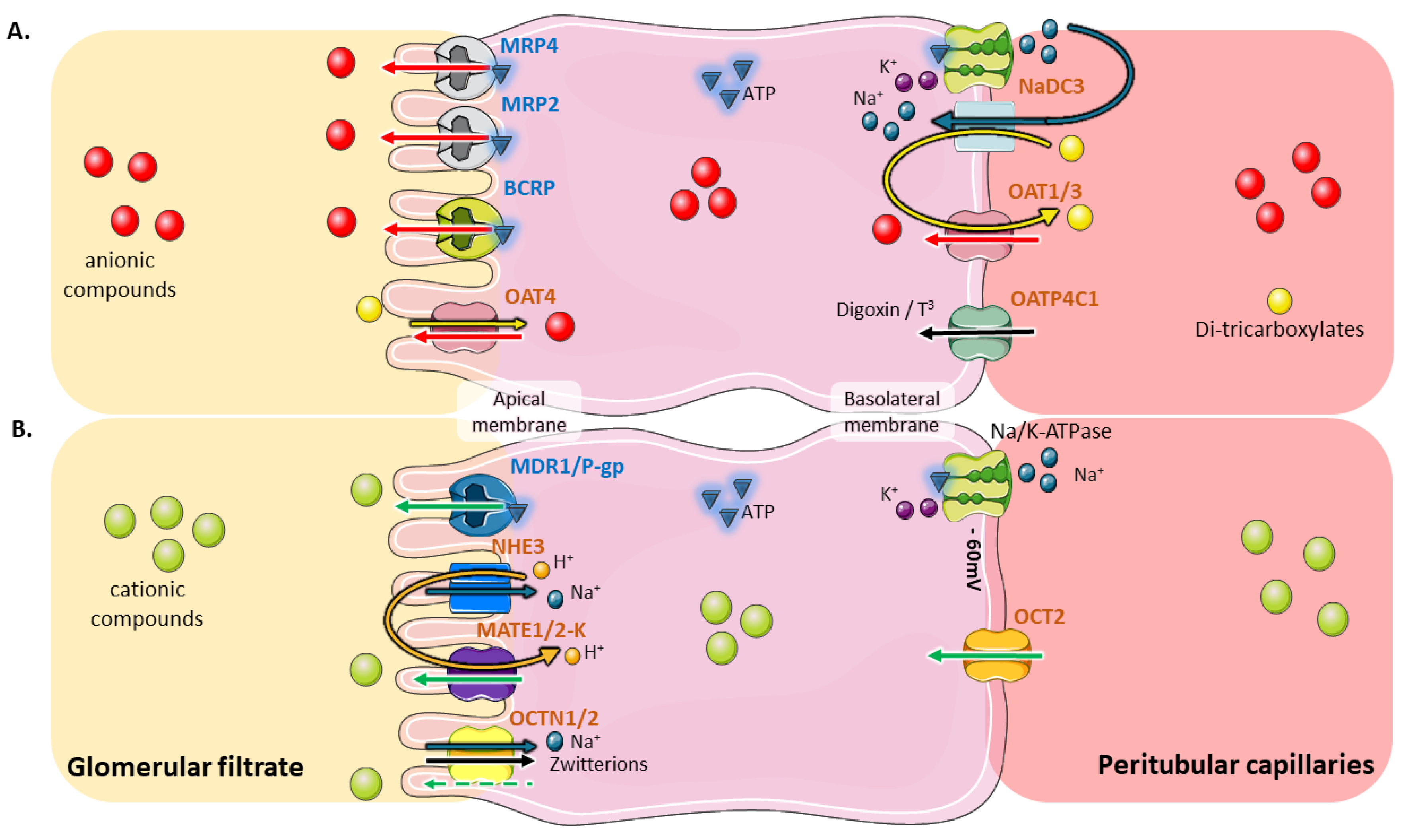
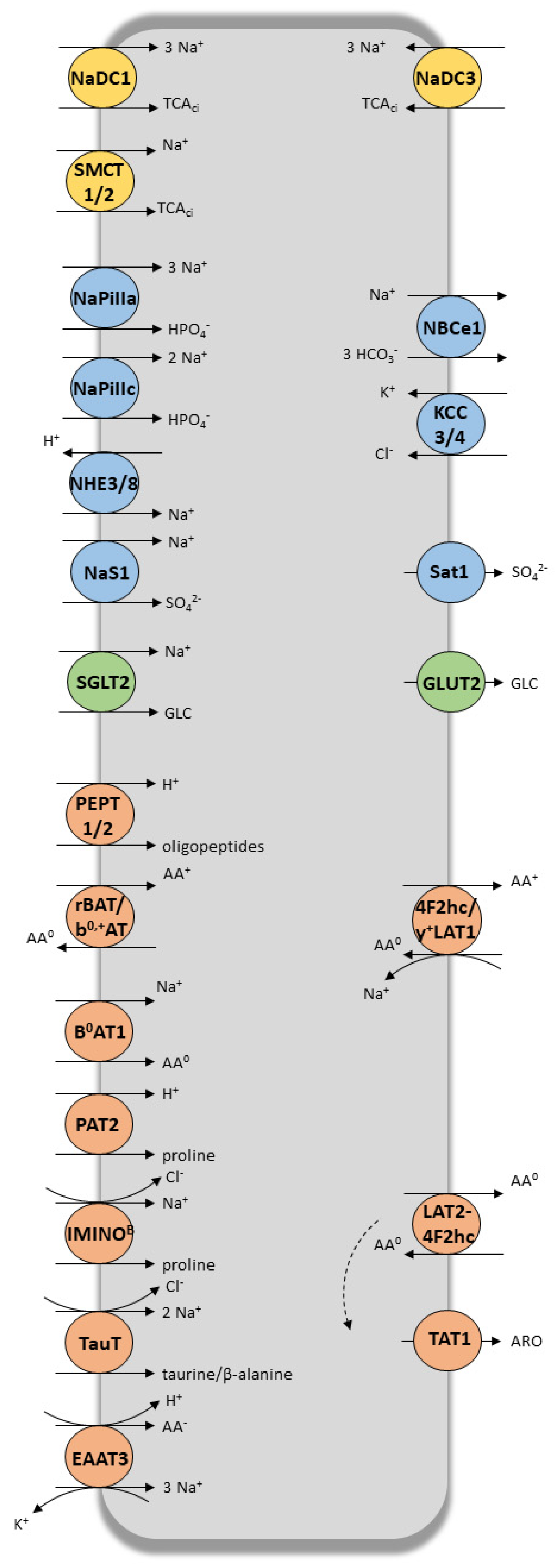
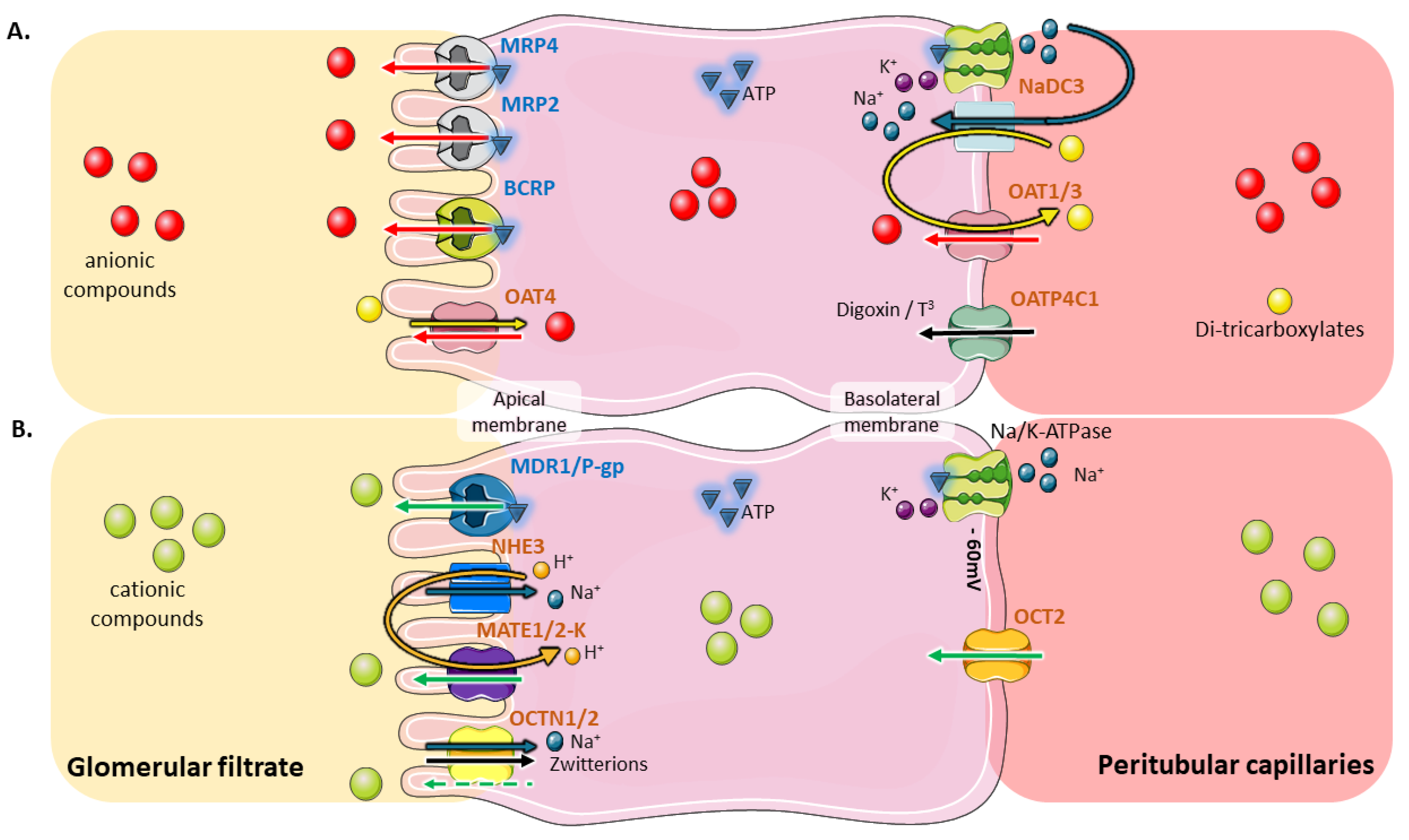
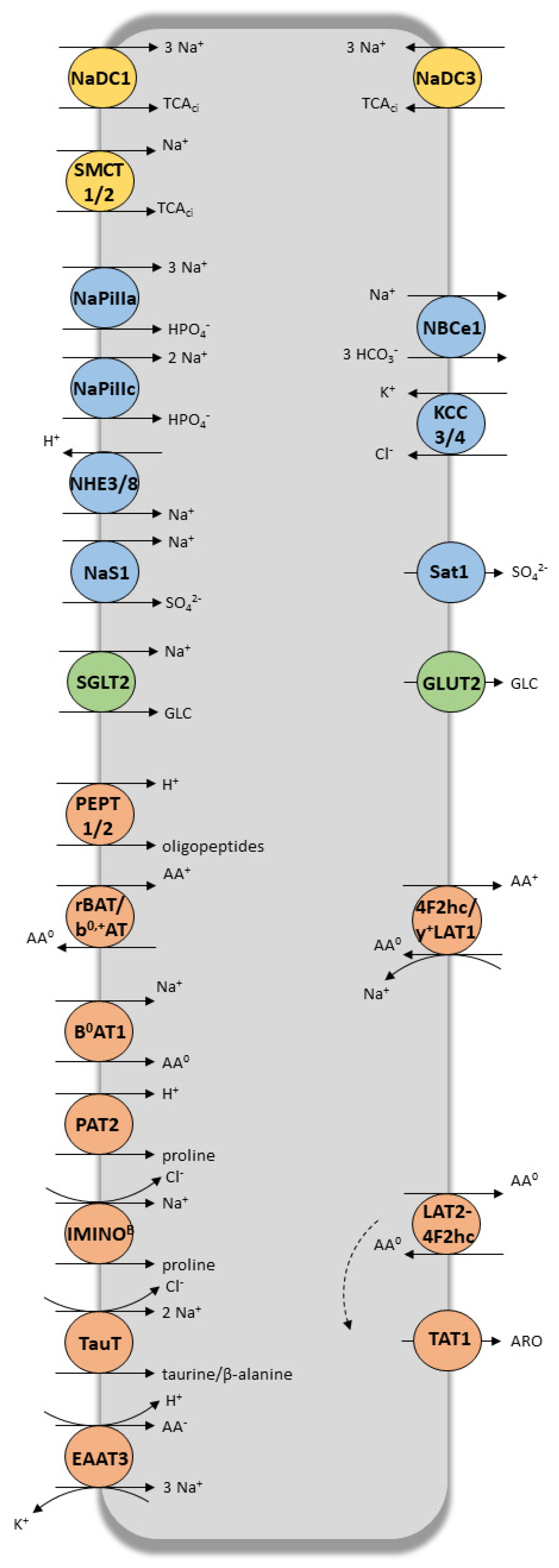
2. Coordinated and Bidirectional Transcellular Transport in Proximal Tubular Cells
3. Role of Tubular Transporters in Maintaining Renal Cells’ Equilibrium, Tissue Homeostasis, Detoxification Processes and Drug Elimination
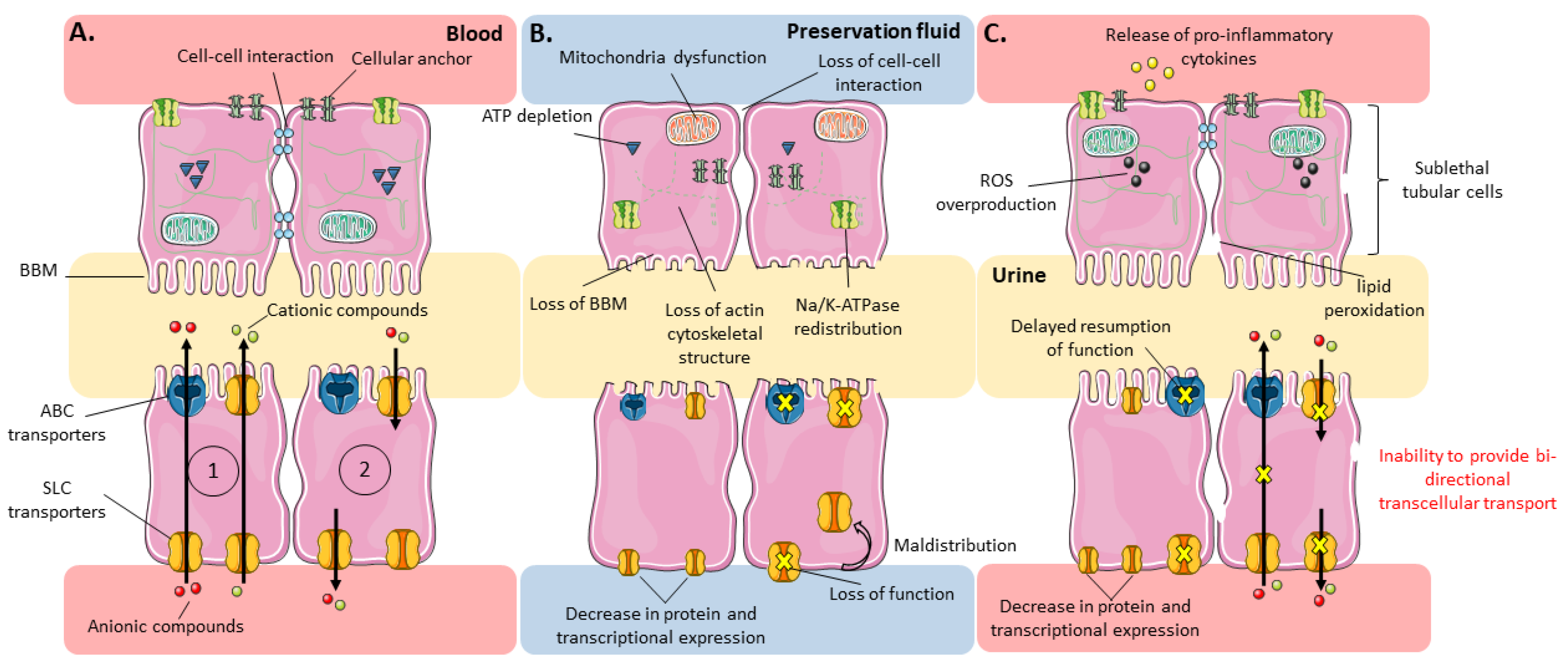
4. Effects of IRI on Renal SLC and ABC Proximal Tubular Transporters
4.1. Effects of Ischemia (I) and/or Ischemia-Reperfusion/Reoxygenation (IR) on Transporters’ Expression and/or Function
4.1.1. Effects on Solute Carriers (SLC)
4.1.2. Effects on ABC Transporters
4.1.3. Possible Mechanisms Underlying Membrane Transporter Dysfunction during I or I/R
4.2. Effects of I and/or IR on Tubular Transport Functions as Evaluated by Metabolomics Studies
5. Consequences of IR-Induced Modulation of Renal Transporters in the Post-Transplantation Period
5.1. Alteration of the Graft Itself
5.2. Accumulation of Toxic Substances
5.3. Impact on Drugs Used in Transplanted Patients
6. Modulation of Transporter’s Function for Improving Graft Outcome
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wong, G.; Teixeira-Pinto, A.; Chapman, J.R.; Craig, J.C.; Pleass, H.; McDonald, S.; Lim, W.H. The Impact of Total Ischemic Time, Donor Age and the Pathway of Donor Death on Graft Outcomes After Deceased Donor Kidney Transplantation. Transplantation 2017, 101, 1152–1158. [Google Scholar] [CrossRef]
- Chen, C.-C.; Chapman, W.C.; Hanto, D.W. Ischemia-reperfusion injury in kidney transplantation. Front. Biosci. (Elite Ed.) 2015, 7, 117–134. [Google Scholar]
- Menke, J.; Sollinger, D.; Schamberger, B.; Heemann, U.; Lutz, J. The effect of ischemia/reperfusion on the kidney graft. Curr. Opin. Organ Transplant. 2014, 19, 395–400. [Google Scholar] [CrossRef]
- Ponticelli, C. Ischaemia-reperfusion injury: A major protagonist in kidney transplantation. Nephrol. Dial. Transplant. 2014, 29, 1134–1140. [Google Scholar] [CrossRef]
- Salvadori, M.; Rosso, G.; Bertoni, E. Update on ischemia-reperfusion injury in kidney transplantation: Pathogenesis and treatment. World J. Transplant. 2015, 5, 52–67. [Google Scholar] [CrossRef]
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Alam, A.; Soo, A.P.; George, A.J.T.; Ma, D. Ischemia-Reperfusion Injury Reduces Long Term Renal Graft Survival: Mechanism and Beyond. EBioMedicine 2018, 28, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Nieuwenhuijs-Moeke, G.J.; Pischke, S.E.; Berger, S.P.; Sanders, J.S.F.; Pol, R.A.; Struys, M.M.R.F.; Ploeg, R.J.; Leuvenink, H.G.D. Ischemia and Reperfusion Injury in Kidney Transplantation: Relevant Mechanisms in Injury and Repair. J. Clin. Med. 2020, 9, 253. [Google Scholar] [CrossRef] [Green Version]
- Nigam, S.K.; Wu, W.; Bush, K.T.; Hoenig, M.P.; Blantz, R.C.; Bhatnagar, V. Handling of Drugs, Metabolites, and Uremic Toxins by Kidney Proximal Tubule Drug Transporters. Clin. J. Am. Soc. Nephrol. 2015, 10, 2039–2049. [Google Scholar] [CrossRef] [Green Version]
- George, B.; You, D.; Joy, M.S.; Aleksunes, L.M. Xenobiotic transporters and kidney injury. Adv. Drug Deliv. Rev. 2017, 116, 73–91. [Google Scholar] [CrossRef] [Green Version]
- Risso, M.A.; Sallustio, S.; Sueiro, V.; Bertoni, V.; Gonzalez-Torres, H.; Musso, C.G. The Importance of Tubular Function in Chronic Kidney Disease. Int. J. Nephrol. Renov. Dis. 2019, 12, 257–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [PubMed] [Green Version]
- Sheridan, A.M.; Bonventre, J.V. Cell biology and molecular mechanisms of injury in ischemic acute renal failure. Curr. Opin. Nephrol. Hypertens. 2000, 9, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Masereeuw, R.; Russel, F.G.M. Regulatory pathways for ATP-binding cassette transport proteins in kidney proximal tubules. AAPS J. 2012, 14, 883–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skorecki, K.; Chertow, G.; Marsden, P.; Taal, M.; Yu, A. Brenner and Rector’s The Kidney, (2 Volume Set), 10th ed.; Elsevier: Philadelphia, PA, USA, 2015. [Google Scholar]
- El-Sheikh, A.A.K.; Masereeuw, R.; Russel, F.G.M. Mechanisms of renal anionic drug transport. Eur. J. Pharmacol. 2008, 585, 245–255. [Google Scholar] [CrossRef]
- Otani, N.; Ouchi, M.; Hayashi, K.; Jutabha, P.; Anzai, N. Roles of organic anion transporters (OATs) in renal proximal tubules and their localization. Anat. Sci. Int. 2017, 92, 200–206. [Google Scholar] [CrossRef]
- Yin, J.; Wang, J. Renal drug transporters and their significance in drug–drug interactions. Acta Pharm. Sin. B 2016, 6, 363. [Google Scholar] [CrossRef] [Green Version]
- Ekaratanawong, S.; Anzai, N.; Jutabha, P.; Miyazaki, H.; Noshiro, R.; Takeda, M.; Kanai, Y.; Sophasan, S.; Endou, H. Human organic anion transporter 4 is a renal apical organic anion/dicarboxylate exchanger in the proximal tubules. J. Pharmacol. Sci. 2004, 94, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Motohashi, H.; Sakurai, Y.; Saito, H.; Masuda, S.; Urakami, Y.; Goto, M.; Fukatsu, A.; Ogawa, O.; Inui, K. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J. Am. Soc. Nephrol. 2002, 13, 866–874. [Google Scholar]
- Koepsell, H.; Endou, H. The SLC22 drug transporter family. Pflug. Arch. 2004, 447, 666–676. [Google Scholar] [CrossRef]
- Koepsell, H. The SLC22 family with transporters of organic cations, anions and zwitterions. Mol. Asp. Med. 2013, 34, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Pelis, R.M.; Wright, S.H. SLC22, SLC44, and SLC47 transporters-organic anion and cation transporters: Molecular and cellular properties. Curr. Top. Membr. 2014, 73, 233–261. [Google Scholar] [PubMed]
- Farrow, E.G.; White, K.E. Recent Advances in Renal Phosphate Handling. Nat. Rev. Nephrol. 2010, 6, 207–217. [Google Scholar] [CrossRef]
- Srikant, S.; Gaudet, R. Mechanics and pharmacology of substrate selection and transport by eukaryotic ABC exporters. Nat. Struct. Mol. Biol. 2019, 26, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Aperia, A.; Fryckstedt, J.; Holtbäck, U.; Belusa, R.; Cheng, X.J.; Eklöf, A.C.; Li, D.; Wang, Z.M.; Ohtomo, Y. Cellular mechanisms for bi-directional regulation of tubular sodium reabsorption. Kidney Int. 1996, 49, 1743–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masereeuw, R.; Mutsaers, H.A.M.; Toyohara, T.; Abe, T.; Jhawar, S.; Sweet, D.H.; Lowenstein, J. The kidney and uremic toxin removal: Glomerulus or tubule? Semin. Nephrol. 2014, 34, 191–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Bush, K.T.; Nigam, S.K. Key Role for the Organic Anion Transporters, OAT1 and OAT3, in the in vivo Handling of Uremic Toxins and Solutes. Sci. Rep. 2017, 7, 4939. [Google Scholar] [CrossRef] [Green Version]
- Ivanyuk, A.; Livio, F.; Biollaz, J.; Buclin, T. Renal Drug Transporters and Drug Interactions. Clin. Pharm. 2017, 56, 825–892. [Google Scholar] [CrossRef]
- Burckhardt, G. Drug transport by Organic Anion Transporters (OATs). Pharmacol. Ther. 2012, 136, 106–130. [Google Scholar] [CrossRef]
- Koepsell, H. Organic Cation Transporters in Health and Disease. Pharmacol. Rev. 2020, 72, 253–319. [Google Scholar] [CrossRef]
- Sharom, F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X. SLC Family Transporters. Adv. Exp. Med. Biol. 2019, 1141, 101–202. [Google Scholar] [PubMed]
- Fenton, R.A.; Poulsen, S.B.; de la Mora Chavez, S.; Soleimani, M.; Dominguez Rieg, J.A.; Rieg, T. Renal tubular NHE3 is required in the maintenance of water and sodium chloride homeostasis. Kidney Int. 2017, 92, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, M.; Burnham, C.E. Physiologic and molecular aspects of the Na+:HCO3− cotransporter in health and disease processes. Kidney Int. 2000, 57, 371–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercado, A.; Vázquez, N.; Song, L.; Cortés, R.; Enck, A.H.; Welch, R.; Delpire, E.; Gamba, G.; Mount, D.B. NH2-terminal heterogeneity in the KCC3 K+-Cl- cotransporter. Am. J. Physiol. Ren. Physiol. 2005, 289, F1246–F1261. [Google Scholar] [CrossRef]
- Markovich, D. Physiological roles of renal anion transporters NaS1 and Sat1. Am. J. Physiol. Ren. Physiol. 2011, 300, F1267–F1270. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, C.; Loo, D.D.F.; Wright, E.M. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 2018, 61, 2087–2097. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.E.; Clémençon, B.; Hediger, M.A. Proton-coupled oligopeptide transporter family SLC15: Physiological, pharmacological and pathological implications. Mol. Asp. Med. 2013, 34, 323–336. [Google Scholar] [CrossRef] [Green Version]
- Verrey, F.; Singer, D.; Ramadan, T.; Vuille-dit-Bille, R.N.; Mariotta, L.; Camargo, S.M.R. Kidney amino acid transport. Pflug. Arch. 2009, 458, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Turner, R.J. beta-Amino acid transport across the renal brush-border membrane is coupled to both Na and Cl. J. Biol. Chem. 1986, 261, 16060–16066. [Google Scholar]
- Park, S.Y.; Kim, J.-K.; Kim, I.J.; Choi, B.K.; Jung, K.Y.; Lee, S.; Park, K.J.; Chairoungdua, A.; Kanai, Y.; Endou, H.; et al. Reabsorption of neutral amino acids mediated by amino acid transporter LAT2 and TAT1 in the basolateral membrane of proximal tubule. Arch. Pharm. Res. 2005, 28, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Vilches, C.; Boiadjieva-Knöpfel, E.; Bodoy, S.; Camargo, S.; López de Heredia, M.; Prat, E.; Ormazabal, A.; Artuch, R.; Zorzano, A.; Verrey, F.; et al. Cooperation of Antiporter LAT2/CD98hc with Uniporter TAT1 for Renal Reabsorption of Neutral Amino Acids. J. Am. Soc. Nephrol. 2018, 29, 1624–1635. [Google Scholar] [CrossRef]
- Mora, C.; Chillarón, J.; Calonge, M.J.; Forgo, J.; Testar, X.; Nunes, V.; Murer, H.; Zorzano, A.; Palacín, M. The rBAT gene is responsible for L-cystine uptake via the b0,(+)-like amino acid transport system in a “renal proximal tubular” cell line (OK cells). J. Biol. Chem. 1996, 271, 10569–10576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, E.; Carrascal, M.; Rousaud, F.; Abián, J.; Zorzano, A.; Palacín, M.; Chillarón, J. rBAT-b(0,+)AT heterodimer is the main apical reabsorption system for cystine in the kidney. Am. J. Physiol. Ren. Physiol. 2002, 283, F540–F548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrents, D.; Estévez, R.; Pineda, M.; Fernández, E.; Lloberas, J.; Shi, Y.B.; Zorzano, A.; Palacín, M. Identification and characterization of a membrane protein (y+L amino acid transporter-1) that associates with 4F2hc to encode the amino acid transport activity y+L. A candidate gene for lysinuric protein intolerance. J. Biol. Chem. 1998, 273, 32437–32445. [Google Scholar] [CrossRef] [Green Version]
- Bergeron, M.J.; Clémençon, B.; Hediger, M.A.; Markovich, D. SLC13 family of Na+-coupled di- and tri-carboxylate/sulfate transporters. Mol. Asp. Med. 2013, 34, 299–312. [Google Scholar] [CrossRef]
- Gopal, E.; Umapathy, N.S.; Martin, P.M.; Ananth, S.; Gnana-Prakasam, J.P.; Becker, H.; Wagner, C.A.; Ganapathy, V.; Prasad, P.D. Cloning and functional characterization of human SMCT2 (SLC5A12) and expression pattern of the transporter in kidney. Biochim. Biophys. Acta 2007, 1768, 2690–2697. [Google Scholar] [CrossRef] [Green Version]
- Eraly, S.A.; Vallon, V.; Vaughn, D.A.; Gangoiti, J.A.; Richter, K.; Nagle, M.; Monte, J.C.; Rieg, T.; Truong, D.M.; Long, J.M.; et al. Decreased Renal Organic Anion Secretion and Plasma Accumulation of Endogenous Organic Anions in OAT1 Knock-out Mice. Available online: http://www.jbc.org (accessed on 7 May 2019).
- Burckhardt, G.; Burckhardt, B.C. In vitro and in vivo evidence of the importance of organic anion transporters (OATs) in drug therapy. Handb. Exp. Pharmacol. 2011, 201, 29–104. [Google Scholar]
- Bush, K.T.; Wu, W.; Lun, C.; Nigam, S.K. The drug transporter OAT3 (SLC22A8) and endogenous metabolite communication via the gut-liver-kidney axis. J. Biol. Chem. 2017, 292, 15789–15803. [Google Scholar] [CrossRef] [Green Version]
- Uwai, Y.; Motohashi, H.; Tsuji, Y.; Ueo, H.; Katsura, T.; Inui, K. Interaction and transport characteristics of mycophenolic acid and its glucuronide via human organic anion transporters hOAT1 and hOAT3. Biochem. Pharmacol. 2007, 74, 161–168. [Google Scholar] [CrossRef]
- Russel, F.G.M.; Koenderink, J.B.; Masereeuw, R. Multidrug resistance protein 4 (MRP4/ABCC4): A versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol. Sci. 2008, 29, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Van de Water, F.M.; Masereeuw, R.; Russel, F.G.M. Function and regulation of multidrug resistance proteins (MRPs) in the renal elimination of organic anions. Drug Metab. Rev. 2005, 37, 443–471. [Google Scholar] [CrossRef] [PubMed]
- Liu, X. ABC Family Transporters. Adv. Exp. Med. Biol. 2019, 1141, 13–100. [Google Scholar] [PubMed]
- Mutsaers, H.A.M.; Caetano-Pinto, P.; Seegers, A.E.M.; Dankers, A.C.A.; van den Broek, P.H.H.; Wetzels, J.F.M.; van den Brand, J.A.J.G.; van den Heuvel, L.P.; Hoenderop, J.G.; Wilmer, M.J.G.; et al. Proximal tubular efflux transporters involved in renal excretion of p-cresyl sulfate and p-cresyl glucuronide: Implications for chronic kidney disease pathophysiology. Toxicol. In Vitro 2015, 29, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Dehghan, A.; Köttgen, A.; Yang, Q.; Hwang, S.-J.; Kao, W.L.; Rivadeneira, F.; Boerwinkle, E.; Levy, D.; Hofman, A.; Astor, B.C.; et al. Association of three genetic loci with uric acid concentration and risk of gout: A genome-wide association study. Lancet 2008, 372, 1953–1961. [Google Scholar] [CrossRef] [Green Version]
- Kimura, N.; Masuda, S.; Katsura, T.; Inui, K. Transport of guanidine compounds by human organic cation transporters, hOCT1 and hOCT2. Biochem. Pharmacol. 2009, 77, 1429–1436. [Google Scholar] [CrossRef]
- Bacher, P.; Giersiefer, S.; Bach, M.; Fork, C.; Schömig, E.; Gründemann, D. Substrate discrimination by ergothioneine transporter SLC22A4 and carnitine transporter SLC22A5: Gain-of-function by interchange of selected amino acids. Biochim. Biophys. Acta 2009, 1788, 2594–2602. [Google Scholar] [CrossRef] [Green Version]
- Nies, A.T.; Damme, K.; Kruck, S.; Schaeffeler, E.; Schwab, M. Structure and function of multidrug and toxin extrusion proteins (MATEs) and their relevance to drug therapy and personalized medicine. Arch. Toxicol. 2016, 90, 1555–1584. [Google Scholar] [CrossRef]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of Acute Kidney Injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar]
- Guellec, C.B.-L.; Largeau, B.; Bon, D.; Marquet, P.; Hauet, T. Ischemia/reperfusion-associated tubular cells injury in renal transplantation: Can metabolomics inform about mechanisms and help identify new therapeutic targets? Pharmacol. Res. 2018, 129, 34–43. [Google Scholar] [CrossRef]
- Du, J.; Zhang, L.; Yang, Y.; Li, W.; Chen, L.; Ge, Y.; Sun, C.; Zhu, Y.; Gu, L. ATP depletion-induced actin rearrangement reduces cell adhesion via p38 MAPK-HSP27 signaling in renal proximal tubule cells. Cell. Physiol. Biochem. 2010, 25, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Molitoris, B.A.; Dahl, R.; Geerdes, A. Cytoskeleton disruption and apical redistribution of proximal tubule Na(+)-K(+)-ATPase during ischemia. Am. J. Physiol. 1992, 263, F488–F495. [Google Scholar] [CrossRef] [PubMed]
- Khundmiri, S.J.; Asghar, M.; Khan, F.; Salim, S.; Yusufi, A.N. Effect of reversible and irreversible ischemia on marker enzymes of BBM from renal cortical PT subpopulations. Am. J. Physiol. 1997, 273, F849–F856. [Google Scholar] [CrossRef] [PubMed]
- Collard, C.D.; Gelman, S. Pathophysiology, clinical manifestations, and prevention of ischemia-reperfusion injury. Anesthesiology 2001, 94, 1133–1138. [Google Scholar] [CrossRef] [Green Version]
- Pochineni, V.; Rondon-Berrios, H. Electrolyte and Acid-Base Disorders in the Renal Transplant Recipient. Front. Med. (Lausanne) 2018, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Khundmiri, S.J.; Asghar, M.; Banday, A.A.; Khan, F.; Salim, S.; Levi, M.; Yusufi, A.N.K. Effect of ischemia reperfusion on sodium-dependent phosphate transport in renal brush border membranes. Biochim. Biophys. Acta 2005, 1716, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Di Sole, F.; Hu, M.-C.; Zhang, J.; Babich, V.; Bobulescu, I.A.; Shi, M.; McLeroy, P.; Rogers, T.E.; Moe, O.W. The reduction of Na/H exchanger-3 protein and transcript expression in acute ischemia-reperfusion injury is mediated by extractable tissue factor(s). Kidney Int. 2011, 80, 822–831. [Google Scholar] [CrossRef] [Green Version]
- Kwon, T.H.; Frøkiaer, J.; Han, J.S.; Knepper, M.A.; Nielsen, S. Decreased abundance of major Na(+) transporters in kidneys of rats with ischemia-induced acute renal failure. Am. J. Physiol. Ren. Physiol. 2000, 278, F925–F939. [Google Scholar] [CrossRef] [Green Version]
- Johnston, P.A.; Rennke, H.; Levinsky, N.G. Recovery of proximal tubular function from ischemic injury. Am. J. Physiol. 1984, 246, F159–F166. [Google Scholar] [CrossRef]
- Molitoris, B.A.; Kinne, R. Ischemia induces surface membrane dysfunction. Mechanism of altered Na+-dependent glucose transport. J. Clin. Investig. 1987, 80, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Saito, H. Pathophysiological regulation of renal SLC22A organic ion transporters in acute kidney injury: Pharmacological and toxicological implications. Pharmacol. Ther. 2010, 125, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischoff, A.; Bucher, M.; Gekle, M.; Sauvant, C. PAH clearance after renal ischemia and reperfusion is a function of impaired expression of basolateral Oat1 and Oat3. Physiol. Rep. 2014, 2, e00243. [Google Scholar] [CrossRef]
- Schneider, R.; Sauvant, C.; Betz, B.; Otremba, M.; Fischer, D.; Holzinger, H.; Wanner, C.; Galle, J.; Gekle, M. Downregulation of organic anion transporters OAT1 and OAT3 correlates with impaired secretion of para-aminohippurate after ischemic acute renal failure in rats. Am. J. Physiol. Ren. Physiol. 2007, 292, F1599–F1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, T.; Watanabe, H.; Yoshitome, K.; Morisaki, T.; Hamada, A.; Nonoguchi, H.; Kohda, Y.; Tomita, K.; Inui, K.; Saito, H. Downregulation of organic anion transporters in rat kidney under ischemia/reperfusion-induced acute [corrected] renal failure. Kidney Int. 2007, 71, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Sauvant, C.; Schneider, R.; Holzinger, H.; Renker, S.; Wanner, C.; Gekle, M. Implementation of an in vitro model system for investigation of reperfusion damage after renal ischemia. Cell. Physiol. Biochem. 2009, 24, 567–576. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Morisaki, T.; Sugimoto, W.; Yokoo, K.; Sato, D.; Nonoguchi, H.; Tomita, K.; Terada, T.; Inui, K.; Hamada, A.; et al. Altered pharmacokinetics of cationic drugs caused by down-regulation of renal rat organic cation transporter 2 (Slc22a2) and rat multidrug and toxin extrusion 1 (Slc47a1) in ischemia/reperfusion-induced acute kidney injury. Drug Metab. Dispos. 2008, 36, 649–654. [Google Scholar] [CrossRef] [Green Version]
- Schneider, R.; Meusel, M.; Betz, B.; Kersten, M.; Möller-Ehrlich, K.; Wanner, C.; Koepsell, H.; Sauvant, C. Nitric oxide-induced regulation of renal organic cation transport after renal ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2011, 301, F997–F1004. [Google Scholar] [CrossRef] [Green Version]
- Ciarimboli, G.; Schröter, R.; Neugebauer, U.; Vollenbröker, B.; Gabriëls, G.; Brzica, H.; Sabolić, I.; Pietig, G.; Pavenstädt, H.; Schlatter, E.; et al. Kidney transplantation down-regulates expression of organic cation transporters, which translocate β-blockers and fluoroquinolones. Mol. Pharm. 2013, 10, 2370–2380. [Google Scholar] [CrossRef]
- Corrigan, G.; Ramaswamy, D.; Kwon, O.; Sommer, F.G.; Alfrey, E.J.; Dafoe, D.C.; Olshen, R.A.; Scandling, J.D.; Myers, B.D. PAH extraction and estimation of plasma flow in human postischemic acute renal failure. Am. J. Physiol. 1999, 277, F312–F318. [Google Scholar] [CrossRef]
- Kwon, O.; Hong, S.-M.; Blouch, K. Alteration in renal organic anion transporter 1 after ischemia/reperfusion in cadaveric renal allografts. J. Histochem. Cytochem. 2007, 55, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Huls, M.; van den Heuvel, J.J.M.W.; Dijkman, H.B.P.M.; Russel, F.G.M.; Masereeuw, R. ABC transporter expression profiling after ischemic reperfusion injury in mouse kidney. Kidney Int. 2006, 69, 2186–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, R.; Meusel, M.; Betz, B.; Held, C.; Möller-Ehrlich, K.; Büttner-Herold, M.; Wanner, C.; Gekle, M.; Sauvant, C. Oat1/3 restoration protects against renal damage after ischemic AKI. Am. J. Physiol. Ren. Physiol. 2015, 308, F198–F208. [Google Scholar] [CrossRef] [Green Version]
- Hagos, Y.; Schley, G.; Schödel, J.; Krick, W.; Burckhardt, G.; Willam, C.; Burckhardt, B.C. α-Ketoglutarate-related inhibitors of HIF prolyl hydroxylases are substrates of renal organic anion transporters 1 (OAT1) and 4 (OAT4). Pflug. Arch. 2012, 464, 367–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulusma, C.C.; Kothe, M.J.; Bakker, C.T.; Bosma, P.J.; van Bokhoven, I.; van Marle, J.; Bolder, U.; Tytgat, G.N.; Oude Elferink, R.P. Zonal down-regulation and redistribution of the multidrug resistance protein 2 during bile duct ligation in rat liver. Hepatology 2000, 31, 684–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Hill, P.; Shukla, D.; Tran, M.G.B.; Aragones, J.; Cook, H.T.; Carmeliet, P.; Maxwell, P.H. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2008, 19, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Conde, E.; Alegre, L.; Blanco-Sánchez, I.; Sáenz-Morales, D.; Aguado-Fraile, E.; Ponte, B.; Ramos, E.; Sáiz, A.; Jiménez, C.; Ordoñez, A.; et al. Hypoxia inducible factor 1-alpha (HIF-1 alpha) is induced during reperfusion after renal ischemia and is critical for proximal tubule cell survival. PLoS ONE 2012, 7, e33258. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, S.; Zou, J.; Zhong, Y.; Ding, X. Silencing HIF-1α aggravates growth inhibition and necrosis of proximal renal tubular epithelial cell under hypoxia. Ren. Fail. 2016, 38, 1726–1734. [Google Scholar] [CrossRef]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar]
- Zapata-Morales, J.R.; Galicia-Cruz, O.G.; Franco, M.; Martinez, Y.; Morales, F. Hypoxia-inducible factor-1α (HIF-1α) protein diminishes sodium glucose transport 1 (SGLT1) and SGLT2 protein expression in renal epithelial tubular cells (LLC-PK1) under hypoxia. J. Biol. Chem. 2014, 289, 346–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Prokopenko, O.; Wong, L.; Inouye, M.; Mirochnitchenko, O. IRIP, a new ischemia/reperfusion-inducible protein that participates in the regulation of transporter activity. Mol. Cell. Biol. 2005, 25, 6496–6508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Yang, H.; Peng, X.; Guo, D.; Dong, Z.; Polli, J.E.; Shu, Y. Ischemia/Reperfusion-inducible protein modulates the function of organic cation transporter 1 and multidrug and toxin extrusion 1. Mol. Pharm. 2013, 10, 2578–2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prokopenko, O.; Mirochnitchenko, O. Ischemia-reperfusion-inducible protein modulates cell sensitivity to anticancer drugs by regulating activity of efflux transporter. Am. J. Physiol. Cell Physiol. 2009, 296, C1086–C1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, O.; Wang, W.-W.; Miller, S. Renal organic anion transporter 1 is maldistributed and diminishes in proximal tubule cells but increases in vasculature after ischemia and reperfusion. Am. J. Physiol. Ren. Physiol. 2008, 295, F1807–F1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, N.A.; Thies, K.; Kuhnke, N.; Reid, G.; Friedrich, B.; Lang, F.; Burckhardt, G. Protein kinase C activation downregulates human organic anion transporter 1-mediated transport through carrier internalization. J. Am. Soc. Nephrol. 2003, 14, 1959–1968. [Google Scholar] [CrossRef] [Green Version]
- Preising, C.; Schneider, R.; Bucher, M.; Gekle, M.; Sauvant, C. Regulation of Expression of Renal Organic Anion Transporters OAT1 and OAT3 in a Model of Ischemia/Reperfusion Injury. Cell. Physiol. Biochem. 2015, 37, 1–13. [Google Scholar] [CrossRef]
- Malagrino, P.A.; Venturini, G.; Yogi, P.S.; Dariolli, R.; Padilha, K.; Kiers, B.; Gois, T.C.; Motta-Leal-Filho, J.M.; Takimura, C.K.; Girardi, A.C.C.; et al. Metabolomic characterization of renal ischemia and reperfusion in a swine model. Life Sci. 2016, 156, 57–67. [Google Scholar] [CrossRef]
- Wei, Q.; Xiao, X.; Fogle, P.; Dong, Z. Changes in metabolic profiles during acute kidney injury and recovery following ischemia/reperfusion. PLoS ONE 2014, 9, e106647. [Google Scholar] [CrossRef] [Green Version]
- Jouret, F.; Leenders, J.; Poma, L.; Defraigne, J.-O.; Krzesinski, J.-M.; de Tullio, P. Nuclear Magnetic Resonance Metabolomic Profiling of Mouse Kidney, Urine and Serum Following Renal Ischemia/Reperfusion Injury. PLoS ONE 2016, 11, e0163021. [Google Scholar] [CrossRef] [Green Version]
- Chihanga, T.; Ma, Q.; Nicholson, J.D.; Ruby, H.N.; Edelmann, R.E.; Devarajan, P.; Kennedy, M.A. NMR spectroscopy and electron microscopy identification of metabolic and ultrastructural changes to the kidney following ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2018, 314, F154–F166. [Google Scholar] [CrossRef] [PubMed]
- Bon, D.; Billault, C.; Claire, B.; Thuillier, R.; Hebrard, W.; Boildieu, N.; Celhay, O.; Irani, J.; Seguin, F.; Hauet, T. Analysis of perfusates during hypothermic machine perfusion by NMR spectroscopy: A potential tool for predicting kidney graft outcome. Transplantation 2014, 97, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Guy, A.J.; Nath, J.; Cobbold, M.; Ludwig, C.; Tennant, D.A.; Inston, N.G.; Ready, A.R. Metabolomic analysis of perfusate during hypothermic machine perfusion of human cadaveric kidneys. Transplantation 2015, 99, 754–759. [Google Scholar] [CrossRef]
- Stryjak, I.; Warmuzińska, N.; Bogusiewicz, J.; Łuczykowski, K.; Bojko, B. Monitoring of the influence of long-term oxidative stress and ischemia on the condition of kidney using solid phase microextraction chemical biopsy coupled with liquid chromatography high resolution mass spectrometry. J. Sep. Sci. 2020, 43, 1867–1878. [Google Scholar] [CrossRef]
- Nath, J.; Smith, T.B.; Patel, K.; Ebbs, S.R.; Hollis, A.; Tennant, D.A.; Ludwig, C.; Ready, A.R. Metabolic differences between cold stored and machine perfused porcine kidneys: A 1H NMR based study. Cryobiology 2017, 74, 115–120. [Google Scholar] [CrossRef] [PubMed]
- DiRito, J.R.; Hosgood, S.A.; Tietjen, G.T.; Nicholson, M.L. The future of marginal kidney repair in the context of normothermic machine perfusion. Am. J. Transplant. 2018, 18, 2400–2408. [Google Scholar] [CrossRef]
- Weissenbacher, A.; Vrakas, G.; Nasralla, D.; Ceresa, C.D.L. The future of organ perfusion and re-conditioning. Transpl. Int. 2019, 32, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Woroniecki, R.; Ferdinand, J.R.; Morrow, J.S.; Devarajan, P. Dissociation of spectrin-ankyrin complex as a basis for loss of Na-K-ATPase polarity after ischemia. Am. J. Physiol. Ren. Physiol. 2003, 284, F358–F364. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Llama, P.; Andrews, P.; Turner, R.; Saggi, S.; Dimari, J.; Kwon, T.H.; Nielsen, S.; Safirstein, R.; Knepper, M.A. Decreased abundance of collecting duct aquaporins in post-ischemic renal failure in rats. J. Am. Soc. Nephrol. 1999, 10, 1658–1668. [Google Scholar]
- Miltényi, M.; Tulassay, T.; Körner, A.; Szabó, A.; Dobos, M. Tubular dysfunction in metabolic acidosis. First step to acute renal failure. Contrib. Nephrol. 1988, 67, 58–66. [Google Scholar]
- Liu, J.; Litt, L.; Segal, M.R.; Kelly, M.J.S.; Pelton, J.G.; Kim, M. Metabolomics of oxidative stress in recent studies of endogenous and exogenously administered intermediate metabolites. Int. J. Mol. Sci. 2011, 12, 6469–6501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliwell, B.; Cheah, I.K.; Drum, C.L. Ergothioneine, an adaptive antioxidant for the protection of injured tissues? A hypothesis. Biochem. Biophys. Res. Commun. 2016, 470, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Reddy, I.J.; Gupta, P.S.P.; Mondal, S. L-carnitine Mediated Reduction in Oxidative Stress and Alteration in Transcript Level of Antioxidant Enzymes in Sheep Embryos Produced In Vitro. Reprod. Domest. Anim. 2016, 51, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Ribas, G.S.; Vargas, C.R.; Wajner, M. L-carnitine supplementation as a potential antioxidant therapy for inherited neurometabolic disorders. Gene 2014, 533, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yan, S.; Ji, C.; Dai, W.; Hu, W.; Zhang, W.; Mei, C. Metabolomic changes and protective effect of (L)-carnitine in rat kidney ischemia/reperfusion injury. Kidney Blood Press. Res. 2012, 35, 373–381. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinopoulos, C. Which way does the citric acid cycle turn during hypoxia? The critical role of α-ketoglutarate dehydrogenase complex. J. Neurosci. Res. 2013, 91, 1030–1043. [Google Scholar] [CrossRef] [Green Version]
- Tajima, T.; Yoshifuji, A.; Matsui, A.; Itoh, T.; Uchiyama, K.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. β-hydroxybutyrate attenuates renal ischemia-reperfusion injury through its anti-pyroptotic effects. Kidney Int. 2019, 95, 1120–1137. [Google Scholar] [CrossRef] [Green Version]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Andrade, L.; Rodrigues, C.E.; Gomes, S.A.; Noronha, I.L. Acute Kidney Injury as a Condition of Renal Senescence. Cell Transplant. 2018, 27, 739–753. [Google Scholar] [CrossRef]
- Qi, R.; Yang, C. Renal tubular epithelial cells: The neglected mediator of tubulointerstitial fibrosis after injury. Cell Death Dis. 2018, 9, 1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huls, M.; Russel, F.G.M.; Masereeuw, R. The Role of ATP Binding Cassette Transporters in Tissue Defense and Organ Regeneration. J. Pharm. Exp. Ther. 2009, 328, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphreys, B.D.; Czerniak, S.; DiRocco, D.P.; Hasnain, W.; Cheema, R.; Bonventre, J.V. Repair of injured proximal tubule does not involve specialized progenitors. Proc. Natl. Acad. Sci. USA 2011, 108, 9226–9231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, K.; Moeller, M.J. Mechanisms of epithelial repair and regeneration after acute kidney injury. Semin. Nephrol. 2014, 34, 394–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.-Y.; Nigam, S.K. Toward a systems level understanding of organic anion and other multispecific drug transporters: A remote sensing and signaling hypothesis. Mol. Pharmacol. Exp. 2009, 76, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, S.B.; Bush, K.T.; Nigam, S.K. A Network of SLC and ABC Transporter and DME Genes Involved in Remote Sensing and Signaling in the Gut-Liver-Kidney Axis. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Nigam, S.K.; Bush, K.T. Uraemic syndrome of chronic kidney disease: Altered remote sensing and signalling. Nat. Rev. Nephrol. 2019, 15, 301–316. [Google Scholar] [CrossRef]
- Chen, C.; Slitt, A.L.; Dieter, M.Z.; Tanaka, Y.; Scheffer, G.L.; Klaassen, C.D. Up-regulation of Mrp4 expression in kidney of Mrp2-deficient TR- rats. Biochem. Pharmacol. 2005, 70, 1088–1095. [Google Scholar] [CrossRef]
- Shiao, C.-C.; Wu, P.-C.; Huang, T.-M.; Lai, T.-S.; Yang, W.-S.; Wu, C.-H.; Lai, C.-F.; Wu, V.-C.; Chu, T.-S.; Wu, K.-D.; et al. Long-term remote organ consequences following acute kidney injury. Crit. Care 2015, 19, 438. [Google Scholar] [CrossRef] [Green Version]
- Dépret, F.; Prud’homme, M.; Legrand, M. A Role of Remote Organs Effect in Acute Kidney Injury Outcome. Nephron 2017, 137, 273–276. [Google Scholar] [CrossRef]
- Kao, C.-C.; Yang, W.-S.; Fang, J.-T.; Liu, K.D.; Wu, V.-C. Remote organ failure in acute kidney injury. J. Formos. Med. Assoc. 2019, 118, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Serteser, M.; Koken, T.; Kahraman, A.; Yilmaz, K.; Akbulut, G.; Dilek, O.N. Changes in hepatic TNF-alpha levels, antioxidant status, and oxidation products after renal ischemia/reperfusion injury in mice. J. Surg. Res. 2002, 107, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Vaghasiya, J.D.; Sheth, N.R.; Bhalodia, Y.S.; Jivani, N.P. Exaggerated Liver Injury Induced by Renal Ischemia Reperfusion in Diabetes: Effect of Exenatide. Saudi J. Gastroenterol. 2010, 16, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Huang, M.; Wei, R.; Wang, Y.; Su, T.; Li, P.; Chen, X. The uremic toxin hippurate promotes endothelial dysfunction via the activation of Drp1-mediated mitochondrial fission. Redox Biol. 2018, 16, 303–313. [Google Scholar] [CrossRef]
- Brandoni, A.; Torres, A.M. Altered Renal Expression of Relevant Clinical Drug Transporters in Different Models of Acute Uremia in Rats. Role of Urea Levels. Cell. Physiol. Biochem. 2015, 36, 907–916. [Google Scholar] [CrossRef]
- Knoflach, A.; Binswanger, U. Serum hippuric acid concentration in renal allograft rejection, ureter obstruction, and tubular necrosis. Transpl. Int. 1994, 7, 17–21. [Google Scholar] [CrossRef]
- Satoh, M.; Hayashi, H.; Watanabe, M.; Ueda, K.; Yamato, H.; Yoshioka, T.; Motojima, M. Uremic toxins overload accelerates renal damage in a rat model of chronic renal failure. Nephron Exp. Nephrol. 2003, 95, e111–e118. [Google Scholar] [CrossRef]
- Oliva-Damaso, E.; Oliva-Damaso, N.; Rodriguez-Esparragon, F.; Payan, J.; Baamonde-Laborda, E.; Gonzalez-Cabrera, F.; Santana-Estupiñan, R.; Rodriguez-Perez, J.C. Asymmetric (ADMA) and Symmetric (SDMA) Dimethylarginines in Chronic Kidney Disease: A Clinical Approach. Int. J. Mol. Sci. 2019, 20, 3668. [Google Scholar] [CrossRef] [Green Version]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreto, F.C.; Barreto, D.V.; Canziani, M.E.F. Uremia Retention Molecules and Clinical Outcomes. Contrib Nephrol. 2017, 191, 18–31. [Google Scholar] [PubMed]
- Liabeuf, S.; Cheddani, L.; Massy, Z.A. Uremic Toxins and Clinical Outcomes: The Impact of Kidney Transplantation. Toxins (Basel) 2018, 10, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massy, Z.A.; Liabeuf, S. Middle-Molecule Uremic Toxins and Outcomes in Chronic Kidney Disease. Contrib. Nephrol. 2017, 191, 8–17. [Google Scholar] [PubMed]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport-an update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef] [Green Version]
- Patel, C.G.; Ogasawara, K.; Akhlaghi, F. Mycophenolic acid glucuronide is transported by multidrug resistance-associated protein 2 and this transport is not inhibited by cyclosporine, tacrolimus or sirolimus. Xenobiotica 2013, 43, 229–235. [Google Scholar] [CrossRef]
- Naesens, M.; Lerut, E.; de Jonge, H.; Van Damme, B.; Vanrenterghem, Y.; Kuypers, D.R.J. Donor age and renal P-glycoprotein expression associate with chronic histological damage in renal allografts. J. Am. Soc. Nephrol. 2009, 20, 2468–2480. [Google Scholar] [CrossRef] [Green Version]
- Hauser, I.A.; Schaeffeler, E.; Gauer, S.; Scheuermann, E.H.; Wegner, B.; Gossmann, J.; Ackermann, H.; Seidl, C.; Hocher, B.; Zanger, U.M.; et al. ABCB1 genotype of the donor but not of the recipient is a major risk factor for cyclosporine-related nephrotoxicity after renal transplantation. J. Am. Soc. Nephrol. 2005, 16, 1501–1511. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.; McKnight, A.J.; Döhler, B.; Simmonds, M.J.; Courtney, A.E.; Brand, O.J.; Briggs, D.; Ball, S.; Cockwell, P.; Patterson, C.C.; et al. Donor ABCB1 variant associates with increased risk for kidney allograft failure. J. Am. Soc. Nephrol. 2012, 23, 1891–1899. [Google Scholar] [CrossRef] [Green Version]
- Woillard, J.-B.; Gatault, P.; Picard, N.; Arnion, H.; Anglicheau, D.; Marquet, P. A donor and recipient candidate gene association study of allograft loss in renal transplant recipients receiving a tacrolimus-based regimen. Am. J. Transplant. 2018, 18, 2905–2913. [Google Scholar] [CrossRef] [Green Version]
- Kimchi-Sarfaty, C.; Oh, J.M.; Kim, I.-W.; Sauna, Z.E.; Calcagno, A.M.; Ambudkar, S.V.; Gottesman, M.M. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 2007, 315, 525–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sheikh, A.A.K.; Greupink, R.; Wortelboer, H.M.; van den Heuvel, J.J.M.W.; Schreurs, M.; Koenderink, J.B.; Masereeuw, R.; Russel, F.G.M. Interaction of immunosuppressive drugs with human organic anion transporter (OAT) 1 and OAT3, and multidrug resistance-associated protein (MRP) 2 and MRP4. Transl Res. 2013, 162, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Cantz, T.; Brom, M.; Leier, I.; Keppler, D. Expression of the apical conjugate export pump, Mrp2, in the polarized hepatoma cell line, WIF-B. Hepatology 1998, 28, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, J.J.; Harper, D.; Getsy, J.; Jusko, W.J. Pharmacokinetic interactions between sirolimus and microemulsion cyclosporine when orally administered jointly and 4 hours apart in healthy volunteers. J. Clin. Pharm. 2003, 43, 1168–1176. [Google Scholar] [CrossRef]
- Emoto, C.; Vinks, A.A.; Fukuda, T. Risk Assessment of Drug-Drug Interactions of Calcineurin Inhibitors Affecting Sirolimus Pharmacokinetics in Renal Transplant Patients. Ther. Drug Monit. 2016, 38, 607–613. [Google Scholar] [CrossRef]
- Podder, H.; Stepkowski, S.M.; Napoli, K.L.; Clark, J.; Verani, R.R.; Chou, T.C.; Kahan, B.D. Pharmacokinetic interactions augment toxicities of sirolimus/cyclosporine combinations. J. Am. Soc. Nephrol. 2001, 12, 1059–1071. [Google Scholar]
- Kahan, B.D. Efficacy of sirolimus compared with azathioprine for reduction of acute renal allograft rejection: A randomised multicentre study. The Rapamune US Study Group. Lancet 2000, 356, 194–202. [Google Scholar] [CrossRef]
- Anglicheau, D.; Pallet, N.; Rabant, M.; Marquet, P.; Cassinat, B.; Méria, P.; Beaune, P.; Legendre, C.; Thervet, E. Role of P-glycoprotein in cyclosporine cytotoxicity in the cyclosporine-sirolimus interaction. Kidney Int. 2006, 70, 1019–1025. [Google Scholar] [CrossRef] [Green Version]
- Hesselink, D.A.; van Hest, R.M.; Mathot, R.A.A.; Bonthuis, F.; Weimar, W.; de Bruin, R.W.F.; van Gelder, T. Cyclosporine interacts with mycophenolic acid by inhibiting the multidrug resistance-associated protein 2. Am. J. Transplant. 2005, 5, 987–994. [Google Scholar] [CrossRef]
- El-Sheikh, A.A.K.; Koenderink, J.B.; Wouterse, A.C.; van den Broek, P.H.H.; Verweij, V.G.M.; Masereeuw, R.; Russel, F.G.M. Renal glucuronidation and multidrug resistance protein 2-/ multidrug resistance protein 4-mediated efflux of mycophenolic acid: Interaction with cyclosporine and tacrolimus. Transl. Res. 2014, 164, 46–56. [Google Scholar] [CrossRef]
- Nigam, S.K. What do drug transporters really do? Nat. Rev. Drug Discov 2015, 14, 29–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, Y.; Motohashi, H.; Ueo, H.; Masuda, S.; Saito, H.; Okuda, M.; Mori, N.; Matsuura, M.; Doi, T.; Fukatsu, A.; et al. Expression levels of renal organic anion transporters (OATs) and their correlation with anionic drug excretion in patients with renal diseases. Pharm. Res. 2004, 21, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Kito, T.; Ito, S.; Mizuno, T.; Maeda, K.; Kusuhara, H. Investigation of non-linear Mate1-mediated efflux of trimethoprim in the mouse kidney as the mechanism underlying drug-drug interactions between trimethoprim and organic cations in the kidney. Drug Metab. Pharmacol. 2019, 34, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Saat, T.C.; van den Akker, E.K.; IJzermans, J.N.M.; Dor, F.J.M.F.; de Bruin, R.W.F. Improving the outcome of kidney transplantation by ameliorating renal ischemia reperfusion injury: Lost in translation? J. Transl. Med. 2016, 14, 20. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, M.L.; Hosgood, S.A. Renal transplantation after ex vivo normothermic perfusion: The first clinical study. Am. J. Transplant. 2013, 13, 1246–1252. [Google Scholar] [CrossRef]
- Kaths, J.M.; Cen, J.Y.; Chun, Y.M.; Echeverri, J.; Linares, I.; Ganesh, S.; Yip, P.; John, R.; Bagli, D.; Mucsi, I.; et al. Continuous Normothermic Ex Vivo Kidney Perfusion Is Superior to Brief Normothermic Perfusion Following Static Cold Storage in Donation After Circulatory Death Pig Kidney Transplantation. Am. J. Transplant. 2017, 17, 957–969. [Google Scholar] [CrossRef]
- Kataria, A.; Magoon, S.; Makkar, B.; Gundroo, A. Machine perfusion in kidney transplantation. Curr. Opin. Organ Transplant. 2019, 24, 378–384. [Google Scholar] [CrossRef]
- Chatterjee, P.K. Novel pharmacological approaches to the treatment of renal ischemia-reperfusion injury: A comprehensive review. Naunyn Schmiedebergs Arch. Pharmacol. 2007, 376, 1–43. [Google Scholar] [CrossRef]
- Chatauret, N.; Thuillier, R.; Hauet, T. Preservation strategies to reduce ischemic injury in kidney transplantation: Pharmacological and genetic approaches. Curr. Opin. Organ Transplant. 2011, 16, 180–187. [Google Scholar] [CrossRef]
- Gong, H.; Sun, J.; Xue, W.; Tian, P.; Ding, X.; Yan, H.; Li, Y.; Zheng, J. Protective effect of truncated Na+/K+-ATPase β on ischemia/reperfusion-induced renal injury in rats. Exp. Biol. Med. (Maywood) 2014, 239, 677–685. [Google Scholar] [CrossRef]
- Belliard, A.; Gulati, G.K.; Duan, Q.; Alves, R.; Brewer, S.; Madan, N.; Sottejeau, Y.; Wang, X.; Kalisz, J.; Pierre, S.V. Ischemia/reperfusion-induced alterations of enzymatic and signaling functions of the rat cardiac Na+/K+-ATPase: Protection by ouabain preconditioning. Physiol. Rep. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, R.; Meusel, M.; Renker, S.; Bauer, C.; Holzinger, H.; Roeder, M.; Wanner, C.; Gekle, M.; Sauvant, C. Low-dose indomethacin after ischemic acute kidney injury prevents downregulation of Oat1/3 and improves renal outcome. Am. J. Physiol. Ren. Physiol. 2009, 297, F1614–F1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saigo, C.; Nomura, Y.; Yamamoto, Y.; Sagata, M.; Matsunaga, R.; Jono, H.; Nishi, K.; Saito, H. Meclofenamate elicits a nephropreventing effect in a rat model of ischemic acute kidney injury by suppressing indoxyl sulfate production and restoring renal organic anion transporters. Drug Des. Dev. Ther. 2014, 8, 1073–1082. [Google Scholar]
- Verma, R.; Mishra, V.; Sasmal, D.; Raghubir, R. Pharmacological evaluation of glutamate transporter 1 (GLT-1) mediated neuroprotection following cerebral ischemia/reperfusion injury. Eur. J. Pharmacol. 2010, 638, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, J.; Ohkita, M.; Takaoka, M.; Kaneshiro, Y.; Matsuo, T.; Kaneko, K.; Matsumura, Y. Role of Na+/H+ exchanger in the pathogenesis of ischemic acute renal failure in mice. J. Cardiovasc. Pharmacol. 2007, 49, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Hropot, M.; Juretschke, H.P.; Langer, K.H.; Schwark, J.R. S3226, a novel NHE3 inhibitor, attenuates ischemia-induced acute renal failure in rats. Kidney Int. 2001, 60, 2283–2289. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.-H.; Liu, H.-B.; Gao, D.-K.; Ge, G.-Q.; Zhang, P.; Sun, S.-R.; Wang, H.-M.; Liu, S.-B. ABCG2 protects kidney side population cells from hypoxia/reoxygenation injury through activation of the MEK/ERK pathway. Cell Transplant. 2013, 22, 1859–1868. [Google Scholar] [CrossRef]
- Kieran, N.E.; Doran, P.P.; Connolly, S.B.; Greenan, M.-C.; Higgins, D.F.; Leonard, M.; Godson, C.; Taylor, C.T.; Henger, A.; Kretzler, M.; et al. Modification of the transcriptomic response to renal ischemia/reperfusion injury by lipoxin analog. Kidney Int. 2003, 64, 480–492. [Google Scholar] [CrossRef] [Green Version]
- Toyohara, T.; Suzuki, T.; Morimoto, R.; Akiyama, Y.; Souma, T.; Shiwaku, H.O.; Takeuchi, Y.; Mishima, E.; Abe, M.; Tanemoto, M.; et al. SLCO4C1 transporter eliminates uremic toxins and attenuates hypertension and renal inflammation. J. Am. Soc. Nephrol. 2009, 20, 2546–2555. [Google Scholar] [CrossRef] [Green Version]
- Nespoux, J.; Patel, R.; Hudkins, K.L.; Huang, W.; Freeman, B.; Kim, Y.C.; Koepsell, H.; Alpers, C.E.; Vallon, V. Gene deletion of the Na+-glucose cotransporter SGLT1 ameliorates kidney recovery in a murine model of acute kidney injury induced by ischemia-reperfusion. Am. J. Physiol. Ren. Physiol. 2019, 316, F1201–F1210. [Google Scholar] [CrossRef]
- Fuller, T.F.; Kusch, A.; Chaykovska, L.; Catar, R.; Pützer, J.; Haller, M.; Troppmair, J.; Hoff, U.; Dragun, D. Protein kinase C inhibition ameliorates posttransplantation preservation injury in rat renal transplants. Transplantation 2012, 94, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Rosenberger, C.; Heyman, S.N.; Eckardt, K.-U. Regulation of hypoxia-inducible factor in kidney disease. Clin. Exp. Pharmacol. Physiol. 2013, 40, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Wang, Y.; Zheng, M.; Liu, Z.; Cai, J.; Tang, C.; Dong, Z. Hypoxia and Hypoxia-Inducible Factors in Kidney Injury and Repair. Cells 2019, 8, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Part 1—Transporters of Endogenous Metabolites | |||||
|---|---|---|---|---|---|
| Transporter | Gene | Location | Transport Mode | Endogenous Substrates | References |
| Inorganic Ions | |||||
| NaPi-IIa | SLC34A1 | Apical | Na+/phosphate co-transporter | Na+; Pi | [24] |
| NaPi-IIc | SLC34A3 | Apical | Na+/phosphate co-transporter | Na+; Pi | |
| NHE3 | SLC9A3 | Apical | Na+/H+ exchanger | Na+; H+ | [34] |
| NBCe1 | SLC4A4 | Basolateral | Na+/HCO3− co-transporter | Na+; HCO3− | [35] |
| KCC3 | SLC12A6 | Basolateral | K+-Cl− cotransporter | K+; Cl− | [36] |
| KCC4 | SLC12A7 | Basolateral | K+-Cl− cotransporter | K+; Cl− | |
| NaS1 | SLC13A1 | Apical | Na+-SO42−-cotransporter | Na+; SO42− | [37] |
| Sat1 | SLC26A1 | Basolateral | sulfate anion transporter | SO42− | |
| Glucose | |||||
| SGLT1 | SLC5A1 | Apical | Na+/glucose co-transporter | Na+; glucose | [38] |
| SGLT2 | SLC5A2 | Apical | Na+/glucose co-transporter | Na+; glucose | |
| GLUT1 | SLC2A1 | Basolateral | passive transport | glucose | |
| GLUT2 | SLC2A2 | Basolateral | passive transport | glucose | |
| Peptides, amino acids | |||||
| PEPT1 | SLC15A1 | Apical | H+/oligopeptide cotransporters | oligopeptides | [39] |
| PEPT2 | SLC15A2 | Apical | H+/oligopeptide cotransporters | oligopeptides | |
| Neutral amino acids (AA0) | |||||
| B0AT1 | SLC6A19 | Apical | Na+-neutral amino acid co-transporter | AA0 | [40] |
| TauT | SLC6A6 | Apical | Na+-dependent co-transporter | taurine; β-alanine; (GABA) | [40,41] |
| IMINOB | SLC6A20 | Apical | Na+-Cl−-dependent co-transporter | proline; hydroxyproline | [40] |
| PAT2 | SLC36A2 | Apical | H+-dependent symporter | glycine; alanine; proline | [40] |
| LAT2-4F2hc | SLC7A8/SLC3A2 | Basolateral | antiporter | ARO; AA0 | [42,43] |
| TAT1 | SLC6A10 | Basolateral | uniporter | ARO | |
| Cationic amino acids (AA+) | |||||
| rBAT/b0,+AT | SLC3A1/SLC7A9 | Apical | amino acids exchanger | AA+; AA0 | [44,45] |
| 4F2hc/y+LAT1 | SLC3A2/SLC7A7 | Basolateral | Na+-dependent amino acids exchanger | AA+; AA0 | [46] |
| Anionic amino acids (AA−) | |||||
| EAAT3 | SLC1A1 | Basolateral | K+-Na+/H+/amino acids exhanger | l-glutamate; l-aspartate | [40] |
| Tricarboxylic acid (TCA) intermediates | |||||
| NaDC1 | SLC13A2 | Apical | Na+/di-tricarboxylate co-transporter | TCA cycle (e.g., succinate, citrate, α-ketoglutarate, fumarate) | [47] |
| NaDC3 | SLC13A3 | Basolateral | Na+/di-tricarboxylate co-transporter | TCA cycle (e.g., succinate, citrate, α-ketoglutarate, fumarate) | |
| SMCT1 | SLC5A8 | Apical | high-affinity Na+-coupled co-transporter | lactate; pyruvate; nicotinate | [48] |
| SMCT2 | SLC5A12 | Apical | low-affinity Na+-coupled co-transporter | lactate; pyruvate; nicotinate | |
| Part 2–Transporters of Uremic Toxins and Drugs | |||||
| Transporter | Endogenous Substrates | Uremic Toxins | Drugs | ||
| Organic anions (OA−) | |||||
| OAT Family | |||||
| OAT1; OAT2; OAT3; OAT4 | mono-carboxylates (e.g., butyrate, lactate, propionate, pyruvate, 3-hydroxybutyrate, benzoate, 3-hydroxypropionate, 3-hydroxyisobutyrate); di-carboxylates (e.g., α-ketoglutarate, N-acetylaspartate); short-chain fatty acids (e.g., hexanoate, heptanoate and octanoate); prostaglandins (PGE2 and PGF2α); cyclic nucleotides (cAMP and cGMP); folate; nicotinate; tryptophan metabolites (quinolinate and kynurenate); purine metabolites (xanthine and hypoxanthine); adenine, adenosine, cytidine, guanidine, guanosine, inosine; hormones (estrone-3-sulfate, 17β-estradiol-3-sulfate, ß-estradiol-3-sulfate, β-estradiol-3,7-disulfate and DHEAS) [22,49,50] | uremic toxins (indoxyl sulfate, p-cresol, p-cresyl sulfate, indole-acetate, uric acid, creatinine, kynurenic acid, orotic acid, benzoate, trimethylamine N-Oxide (TMAO), 3-carboxy-4-methyl-5-propyl-2-furanpropionate (CMPF)); environmental toxins (mercury conjugate, ochratoxin A and aristolochic acid) [9,27,28,51] | Antivirals (adefovir, tenofovir, aciclovir, ciclofovir, cidofovir, lamivudine, stavudine); Non steroidals anti-inflammatory; Methotrexate; Diuretics (furosemide, bumetanide, hydrochlorothiazide); Angiotensin II receptors blockers (candesartan, valsartan, losartan…); β-lactams (penicillins chepalosporines…); Other antibiotics (tetracycline, ciprofloxacine); H2-antihistaminics (cimetidine, ranitidine, famotidine…); ACE inhibitors (captopril, quinaprine); HMG-coA reductor inhibitors (fluvastatin, pravastatin, simvastatin); Oral antidiabetics (glibenclamide…); mycophenolic acid glucuronide; Uricosuric-drugs (probenecid, benzbromarone) [9,29,30,52] | ||
| OATP Family | |||||
| OATP4C1 | thyroid hormones (T3), cAMP [50] | asymmetric dimethylarginine (ADMA), guanidine succinate (GSA), and trans-aconitate [27] | Digoxin, ouabaine, methotrexate [29] | ||
| MRP Family | |||||
| MRP2; MRP4 | glutathione, conjugated glutathione; bilirubin glucuronides and other metabolites (LTC4, E217βG, reduced glutathione (GSH)); cyclic nucleotides and ADP; prostaglandins (PGE1, PGE2); thromboxane B2 (TBX2); proinflammatory leukotrienes B4 et C4 (LTB4, LTC4); estradiol glucuronide (E217βG); folic acid [53,54] | kynurenic acid, probably indoxyl sulfate and hippuric acid [27] | Antivirals (tenofovir); Non steroidals anti-inflammatory; Methotrexate; Diuretics (furosemide); Angiotensin II receptors blockers; β-lactams [29] | ||
| BCRP Family | |||||
| BCRP | Vitamins (folic acid, B2, K3) estrone-3 sulfate, dehydroepiandrosterone sulfate, E217βG [55] | Urate, kynurenic acid, indoxyl sulfate, hippuric acid, p-cresyl sulfate and p-cresyl glucuronide [27,56,57] | Corticosteroids conjugates; Antineoplasics (mitoxantrone, methotrexate, irinotecan…); Antiretroviral nucleosides (AZT, lamivudine…); Fluoroquinolones; Statins (rosuvastatin); sulfasalazine; ITK (imatinib, gefitinib, nilotinib) [29,55] | ||
| Organic cations (OC+) | |||||
| OCT Family | |||||
| OCT2 | choline, thiamine, l-carnitine [31] | polyamine uremic toxins (e.g., cadaverine, putrescine, spermine and spermidine); guanidino group (e.g., creatinine, guanidine or methylguanidine) [27,58] | Metformin; Cisplatin; H2-antihistaminics (cimetidine, famotidine, ranitidine…); antivirals; β blockers (pindolol…); Calcium channel blockers; Antiarrhythmics (procainamide, dofetilide…); Antimalarials; Varenicline; Amantadine; Memantine [29,31] | ||
| OCTN1; OCTN2 | ergothionein; l-carnitine, choline [22,59] | Verapamil; Quinidine; Gabapentine; β-lactams (cephaloridine, cefepime); Valproic acid [10,29] | |||
| MATE Family | |||||
| MATE1; MATE2 | 6β-hydroxycortisol, E3S, N-methylnicotinamide (NMN), l-Arginine and thiamine [60] | guanidine, creatinine and ADMA [60] | Corticosteroids; Metformin; Cimetidine; Platinum compounds; Antibiotics (cefalexine, cephradine); H2-antihistaminics (cimetidine); Memantine [10,29] | ||
| MDR Family | |||||
| MDR1/P-gp | Anticancer drugs (methotrexate, anthacyclines, campthecins, taxanes…); Digoxin; Cardiovascular agents (valsartan, quinidine, nifedipine, verapamil); Immunosuppressant (ciclosporine, tacrolimus); Corticosteroids (corticosterone, dexamethasone); Antibactrial (erythromycine); Antiretrovirals (ritonavir, saquinavir); Phenobarbital; Phenytoin; Statins (lovastatin, simvastatin); H1-antihistaminic (fexofenadine); Anticoagulants (dabigatran); ITK inhibitors (imatinib, gefitinib); Antiepileptics (Topiramate); Analgesics (morphine) [9,10,29,32] | ||||
| Treatment | Action | Model (Species/Organ) | References |
|---|---|---|---|
| tNKA(β) | Enhances Na+/K+-ATPase activity | Rat/Kidney | [172] |
| Ouabain | Preserves Na+/K+-ATPase activity | Rat/Heart | [173] |
| Indomethacin | Prevents OAT1 and OAT3 downregulation, increases PAH excretion | Rat/Kidney | [174] |
| Meclofenamate | Prevents OAT1 and OAT3 downregulation (induced by Protaglandin E2), Decreases indoxyl-sulfate hepatic production | Rat/Kidney, Liver | [175] |
| Ceftriaxone | Upregulates and increases SGLT1 activity | Rat/Brain | [176] |
| 5-(N-ethyl-N-isopropyl) amiloride | Inhibits NHE | Mice/Kidney | [177] |
| S3226 | Inhibits NHE3 | Rat/Kidney | [178] |
| Side population cells | Cells which constitutively express BCRP | Mice/Kidney | [179] |
| Liopoxin analog | Restores expression of genes differentially expressed by IRI, including OCT1 and aquaporin 1 | Mice/Kidney | [180] |
| Pravastatin | Upregulates OATP4C1 | Rat/Kidney | [181] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faucher, Q.; Alarcan, H.; Marquet, P.; Barin-Le Guellec, C. Effects of Ischemia-Reperfusion on Tubular Cell Membrane Transporters and Consequences in Kidney Transplantation. J. Clin. Med. 2020, 9, 2610. https://doi.org/10.3390/jcm9082610
Faucher Q, Alarcan H, Marquet P, Barin-Le Guellec C. Effects of Ischemia-Reperfusion on Tubular Cell Membrane Transporters and Consequences in Kidney Transplantation. Journal of Clinical Medicine. 2020; 9(8):2610. https://doi.org/10.3390/jcm9082610
Chicago/Turabian StyleFaucher, Quentin, Hugo Alarcan, Pierre Marquet, and Chantal Barin-Le Guellec. 2020. "Effects of Ischemia-Reperfusion on Tubular Cell Membrane Transporters and Consequences in Kidney Transplantation" Journal of Clinical Medicine 9, no. 8: 2610. https://doi.org/10.3390/jcm9082610
APA StyleFaucher, Q., Alarcan, H., Marquet, P., & Barin-Le Guellec, C. (2020). Effects of Ischemia-Reperfusion on Tubular Cell Membrane Transporters and Consequences in Kidney Transplantation. Journal of Clinical Medicine, 9(8), 2610. https://doi.org/10.3390/jcm9082610