Maternal Immunity in Autism Spectrum Disorders: Questions of Causality, Validity, and Specificity

Abstract
1. Introduction
2. Search Strategy
3. Animal Models of Maternal Immune Activation (MIA)
4. MIA and Disease Specificity
5. Disease Specificity Arising from Immune Challenge

{kind=link}
{kind=link}
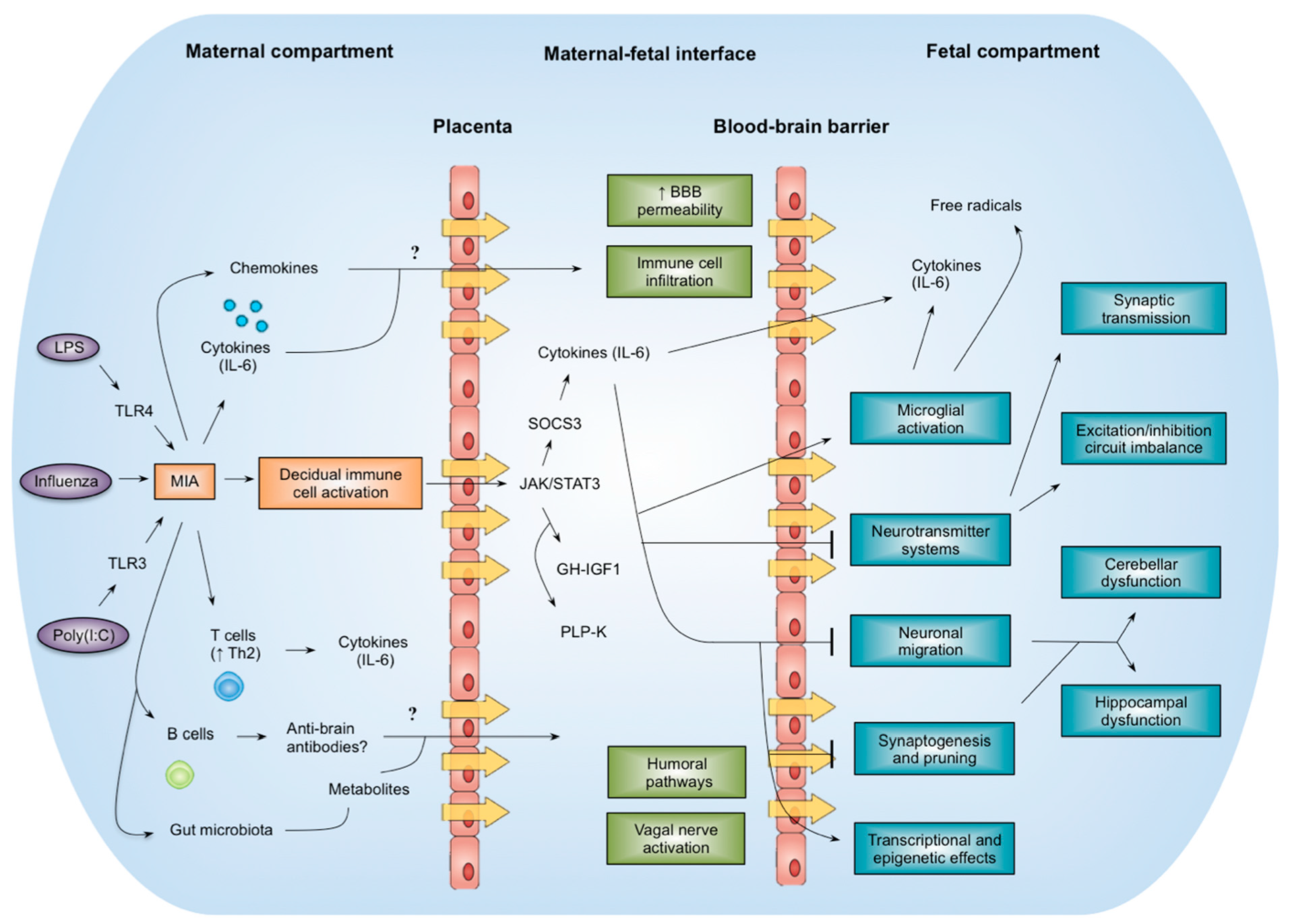{kind=link}
| Reference | Treatment and Species | Timing of Challenge | Behavioural Findings | Neuropathological Findings |
|---|---|---|---|---|
| Fatemi et al. (1999) [18] | Influenza (IN) Mouse | E9 | ND | ↓ Reelin expression in cortical layer I and hippocampus ↓ Ventricular size ↑ Brain size Defective corticogenesis, pyramidal cell atrophy |
| Shi et al. (2003) [19] | Influenza (IN) Mouse | E9.5 | ↓ PPI ↓ Exploratory behaviour ↓ Social interaction | ND |
| Shi et al. (2009) [27] | Influenza (IN) Poly(I:C) (20 mg/kg, IP) Mouse | E9.5 (influenza) E12.5 (Poly(I:C)) | ND | ↓ Purkinje cells in lobule VII of cerebellum Delayed migration of granule cells |
| Kirsten et al. (2012) [48] | LPS (100 μg/kg, IP) Rat | E9.5 | ↓ Play behaviour ↓ Social interaction ↓ USVs ↓ Exploratory behaviour ↑ Repetitive behaviour No change in anxiety-like behaviour | ND |
| Xuan et al. (2014) [49] | LPS (75 μg/kg, IP) Poly(I:C) (20 mg/kg, IP) Mouse | E11.5, 12 (LPS) E12.5 (Poly(I:C)) | ↓ Locomotor activity * ↓ Social approach * ↑ Repetitive behaviour * | ND |
| Sharova et al. (2014) [50] | LPS (45 μg/kg, IP) Mouse | E11.5 | ND | ↓ Neuron number in forebrain area ↑ Neuron number in nasal and olfactory bulb Altered gonadotropin-releasing hormone neuron migration |
| Foley et al. (2015) [51] | LPS (50 μg/kg, SC) Rat | E15, 16 | ↑ Acoustic startle * No change in PPI * | ND |
| Wischhof et al. (2015) [52] | LPS (100 μg/kg, IP) Rat | E15, 16 | ↓ PPI * ↓ Recognition memory | ↓ Myelination in cortex and limbic regions ↓ PARV-expressing interneurons in medial prefrontal cortex, entorhinal cortex, and hippocampus |
| Batinić et al. (2016) [53] | LPS (100 μg/kg, IP) Rat | E15, 16 | ↓ Locomotion * ↓ Response to amphetamines * ↓ Spatial learning and memory * | ND |
| Fernández de Cossío et al. (2017) [28] | LPS (100 μg/kg, IP) Mouse | E15 | ↓ USV duration ↓ Social behaviour ↑ Stereotypical behaviour ↑ Repetitive behaviour * | Increased spine density in granule cells * No effect on pyramidal cells |
| Kirsten et al. (2017) [48] | LPS (100 μg/kg, IP) Rats | E9.5 | ↑ Repetitive behaviour | Reduced dopaminergic activity in the hypothalamus |
| Wu et al. (2018) [54] | LPS (75 μg/kg, IP) Mouse | E14.5 | ↑ Anxiety-like behaviour ↓ Social interaction ↑ Depression-like behaviour | Aberrant cytoarchitecture and lamination in neocortex ↓ Intermediate progenitor cells and astrocytes in neocortex |
| Meyer et al. (2006, 2008) [26,55] Li et al. (2009) [46] | Poly(I:C) (5 mg/kg, IV) Mouse | E9, 17 | ↓ Exploratory behaviour * ↓ Reversal learning ↓ PPI * ↑ Repetitive behaviour * ↑ Locomotor activity | ↓ Hippocampal reelin expression ↓ Ventricular volume * ↑ Dopamine-relater marker expression Altered glutamate-related marker expression |
| Abazyan et al. (2010) [56] Lipina et al. (2013) [57] | Poly(I:C) (5 mg/kg, IP) Mouse | E9 | ↓ PPI ↓ LI ↓ Exploratory behaviour Hyperactivity | ND |
| Hsiao et al. (2012) [44] | Poly(I:C) (20 mg/kg, IP) IL-6 (5 μg, IP) Mouse | E12.5 | ↓ PPI ↓ LI ↓ Exploratory behaviour | ND |
| Malkova et al. (2012) [21] | Poly(I:C) (5 mg/kg, IP) Mouse | E10.5, 12.5, 14.5 | ↓ USVs ↓ Social interaction ↓ Female-induced scent marking ↑ Repetitive behaviour | ND |
| Ehninger et al. (2012) [58] | Poly(I:C) (20 mg/kg, IP) Mouse | E10.5, 12.5, 14.5 | No change in social approach, exploratory behaviour, or olfactory function | ND |
| Hsiao et al. (2013) [32] Schwartzer et al. (2013) [25] Wu et al. (2015) [29] | Poly(I:C) (20 mg/kg, IP) Mouse | E12.5 | ↓ PPI ↓ USVs ↓ LI ↓ Social approach ↑ Repetitive behaviour ↑ Anxiety-like behaviour ↑ Depressive-like behaviour | ↓ Neuronal proliferation, maturation and survival in dentate gyrus ↓ VEGFA-VEGFR2 hippocampal expression Impaired hippocampal long-term potentiation |
| Giovanoli et al. (2013) [59] | Poly(I:C) (1 mg/kg, IP) Mouse | E9 | ↓ LI No change in PPI or anxiety-like behaviour | ↑ Reelin expression No change in microglia activation or GABAergic interneurons in CA regions |
| Missault et al. (2014) [43] | Poly(I:C) (4 mg/kg, IP, IV) Rat | E15 | ↓ PPI ↑ Depressive-like behaviour No change in locomotor activity | No change in microglial number |
| Giovanoli et al. (2016) [60] | Poly(I:C) (5 mg/kg, IV) Mouse | E9 | ND | Presynaptic hippocampal deficits No change in microglia or astrocyte density |
| Pendyala et al. (2017) [12] | Poly(I:C) (20 mg/kg, IP) Mouse | E12.5 | ↓ USVs ↑ Repetitive behaviour No change in motor coordination | Proinflammatory state in cerebellum Reduction in cerebellar synaptic organising proteins |
| Li et al. (2018) [61] | Poly(I:C) (10 mg/kg, IV) Rat | E9 | ↑ Locomotion ↓ PPI ↓ LI | Increased prefrontal cortex and hippocampus activity Increased activation of microglia |
| Murray et al. (2019) [9] | Poly(I:C) (10 mg/kg, IP) Rat | E15 | ND | Increased activation of microglia in hippocampus * |
| Lins et al. (2019) [62] | Poly(I:C) (20 mg/kg, IP) Rat | E15 | ↓ Social interaction * No change in startle or PPI | ND |
| Amodeo et al. (2019) [10] | Poly(I:C) (20 mg/kg, IP) Mouse | E12.5 | ↓ Social behaviour ↓Reversal learning | Dysregulation of potassium ion channel activity in frontal cortex |
| Carlezon et al. (2019) [22] | Poly(I:C) (20 mg/kg, IP) LPS (10 mg/kg, SC, postnatal day 9) Mouse | E12.5 | ↓ USVs ↓ Social behaviour * ↑ Anxiety-like behaviour * | Pro-inflammatory state in prefrontal cortex, amygdala, hippocampus and thalamus * |
| Haida et al. (2019) [11] | Poly(I:C) (20 mg/kg, IP) Mouse | E12.5 | ↓ Social behaviour * ↓Motor coordination * | Reduced number of Purkinje cells in cerebellum * Reduced number of neurons in motor cortex* |
| Samuelsson et al. (2006) [63] | IL-6 (9 μg/kg, IP) Rat | E10, 12, 16 or E16, 18, 20 | ↓ Spatial learning | ↓ Neuron number in CA regions ↑ Astrogliosis ↑ Apoptosis ↑ GFAP expression |
| Smith et al. (2007) [64] | IL-6 (5 μg, IP) Poly(I:C) (20 mg/kg, IP) Mouse | E12.5 | ↓ PPI ↓ LI ↓ Exploratory behaviour ↓ Social interaction | ND |
| Reference | Treatment and Species | Timing of Challenge | Behavioural Findings | Neuropathological Findings |
|---|---|---|---|---|
| Short et al. (2010) [20] | Influenza (IN) Rhesus macaque | Early 3rd trimester | Altered mother-infant interaction | ↓ Total and cortical grey matter |
| Willette et al. (2011) [37] | LPS (2 or 4 ng/kg, IV) Rhesus macaque | Early 3rd trimester | ↓ PPI ↑ Emotionality No change in social interaction | ↓ Medial temporal lobe grey matter ↑ Total white matter volume |
| Bauman et al. (2014) [33] | Modified poly(I:C) (0.25 mg/kg, IV) Rhesus macaque | Late 1st and 2nd trimester | ↓ Affiliative vocalisations ↓ Social approach ↑ Motor stereotypy Abnormal social behaviour Abnormal attachment to mother | ND |
| Machado et al. (2015) [65] | Modified poly(I:C) (0.25 mg/kg, IV) Rhesus macaque | Late 1st trimester | ↓ Social attention ↓ Visual fixation | ND |
| Weir et al. (2015) [66] | Modified poly(I:C) (0.25, 0.5, or 1 mg/kg, IV) Rhesus macaque | Late 1st trimester | ND | ↓ Apical dendrite size in prefrontal cortex ↑ Number of oblique dendrites |
| Rose et al. (2017) [67] | Modified poly(I:C) (0.25 mg/kg, IV) | Late 1st or 2nd trimester | ↑ Stereotyped behaviours | ND |
| Bauman et al. (2019) [68] | Modified poly(I:C) (0.25 mg/kg, IV) | Late 1st or 2nd trimester | ND | ↑ Striatal dopamine in late adolescence |
| Reference | Treatment and Species | Timing of Challenge | Immune Findings |
|---|---|---|---|
| Meyer et al. (2006, 2008) [26,55] | Poly(I:C) (5 mg/kg, IV) Mouse | E9, 17 | ↑ TNFα, IL-1α, IL-6, IL-10 |
| Abazyan et al. (2010) [56] | Poly(I:C) (5 mg/kg, IP) Mouse | E9 | ↑ IL-1α, IL-4, IL-5 |
| Arrode-Brusés et al. (2012) [69] | Poly(I:C) (20 mg/kg, IP) Mouse | E16 | ↑ TNFα, IL-1α, IL-7, IL-13, MCP-1, MIP-1α |
| Lipina et al. (2013) [57] | Poly(I:C) (2.5 or 5 mg/kg, IV) Mouse | E9 | ↑ IL-6 |
| Wu et al. (2015) [29] | Poly(I:C) (20 mg/kg, IP) Mouse | E12.5 | ↑ IL-6 |
| Giovanoli et al. (2016) [60] | Poly(I:C) (5 mg/kg, IV) Mouse | E9 | ↑ IL-1 |
| Pendyala et al. (2017) [12] | Poly(I:C) (20 mg/kg, IP) Mouse | E12.5 | ↑ IL-2, IL-3, IL-6, TNFRI, TNF-α, FasL |
| Rose et al. (2017) [67] | Modified poly(I:C) (0.25 mg/kg, IV) Rhesus macaque | 1st or 2nd trimester | ↑ IL-1α, IL-4, IL-13 |
6. Disease Specificity Arising from Genetic and Environmental Factors
7. Mechanistic Insights
8. Conclusions
Funding
Conflicts of Interest
References
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Taylor, M.J.; Rosenqvist, M.A.; Larsson, H.; Gillberg, C.; D’Onofrio, B.M.; Lichtenstein, P.; Lundstrom, S. Etiology of Autism Spectrum Disorders and Autistic Traits Over Time. JAMA Psychiatry 2020. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Hultman, C.; Larsson, H.; Reichenberg, A. The Heritability of Autism Spectrum Disorder. JAMA 2017, 318, 1182–1184. [Google Scholar] [CrossRef] [PubMed]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child. Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-Y.; Xu, L.-L.; Shao, L.; Xia, R.-M.; Yu, Z.-H.; Ling, Z.-X.; Yang, F.; Deng, M.; Ruan, B. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain Behav. Immun. 2016, 58, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.K.; Magnusson, C.; Gardner, R.M.; Blomstrom, A.; Newschaffer, C.J.; Burstyn, I.; Karlsson, H.; Dalman, C. Maternal hospitalization with infection during pregnancy and risk of autism spectrum disorders. Brain Behav. Immun. 2015, 44, 100–105. [Google Scholar] [CrossRef]
- Atladóttir, H.O.; Thorsen, P.; Schendel, D.E.; Østergaard, L.; Lemcke, S.; Partner, E.T. Association of hospitalization for infection in childhood with diagnosis of autism spectrum disorders: A Danish cohort study. Arch. Pediatr. Adolesc. Med. 2010, 164, 470–477. [Google Scholar] [CrossRef]
- Murray, K.N.; Edye, M.E.; Manca, M.; Vernon, A.C.; Oladipo, J.M.; Fasolina, V.; Harte, M.K.; Mason, V.; Grayson, B.; McHugh, P.C. Evolution of a maternal immune activation (mIA) model in rats: Early developmental effects. Brain Behav. Immun. 2019, 75, 48–59. [Google Scholar] [CrossRef]
- Amodeo, D.A.; Lai, C.-Y.; Hassan, O.; Mukamel, E.A.; Behrens, M.M.; Powell, S.B. Maternal immune activation impairs cognitive flexibility and alters transcription in frontal cortex. Neurobiol. Dis. 2019, 125, 211–218. [Google Scholar] [CrossRef]
- Haida, O.; Al Sagheer, T.; Balbous, A.; Francheteau, M.; Matas, E.; Soria, F.; Fernagut, P.O.; Jaber, M. Sex-dependent behavioral deficits and neuropathology in a maternal immune activation model of autism. Transl. Psychiatry 2019, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, G.; Chou, S.; Jung, Y.; Coiro, P.; Spartz, E.; Padmashri, R.; Li, M.; Dunaevsky, A. Maternal Immune Activation Causes Behavioral Impairments and Altered Cerebellar Cytokine and Synaptic Protein Expression. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2017, 42, 1435–1446. [Google Scholar] [CrossRef]
- Kirsten, T.B.; Bernardi, M.M. Prenatal lipopolysaccharide induces hypothalamic dopaminergic hypoactivity and autistic-like behaviors: Repetitive self-grooming and stereotypies. Behav. Brain Res. 2017, 331, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; van Bakel, H.; Varghese, M.; Wang, Y.; et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed]
- Eftekharian, M.M.; Ghafouri-Fard, S.; Noroozi, R.; Omrani, M.D.; Arsang-jang, S.; Ganji, M.; Gharzi, V.; Noorozi, H.; Komaki, A.; Mazdeh, M.; et al. Cytokine profile in autistic patients. Cytokine 2018, 108, 120–126. [Google Scholar] [CrossRef]
- Tylee, D.S.; Hess, J.L.; Quinn, T.P.; Barve, R.; Huang, H.; Zhang-James, Y.; Chang, J.; Stamova, B.S.; Sharp, F.R.; Hertz-Picciotto, I. Blood transcriptomic comparison of individuals with and without autism spectrum disorder: A combined-samples mega-analysis. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017, 174, 181–201. [Google Scholar] [CrossRef]
- Gupta, S.; Ellis, S.E.; Ashar, F.N.; Moes, A.; Bader, J.S.; Zhan, J.; West, A.B.; Arking, D.E. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat. Commun. 2014, 5, 5748. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Emamian, E.S.; Kist, D.; Sidwell, R.W.; Nakajima, K.; Akhter, P.; Shier, A.; Sheikh, S.; Bailey, K. Defective corticogenesis and reduction in Reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Mol. Psychiatry 1999, 4, 145–154. [Google Scholar] [CrossRef]
- Shi, L.; Fatemi, S.H.; Sidwell, R.W.; Patterson, P.H. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 297–302. [Google Scholar] [CrossRef]
- Short, S.J.; Lubach, G.R.; Karasin, A.I.; Olsen, K.W.; Styner, M.; Knickmeyer, R.C.; Gilmore, J.H.; Coe, C.L. Maternal influenza infection during pregnancy impacts postnatal brain development in the rhesus monkey. Biol. Psychiatry 2010, 67, 965–973. [Google Scholar] [CrossRef]
- Malkova, N.V.; Yu, C.Z.; Hsiao, E.Y.; Moore, M.J.; Patterson, P.H. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav. Immun. 2012, 26, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Carlezon, W.A.; Kim, W.; Missig, G.; Finger, B.C.; Landino, S.M.; Alexander, A.J.; Mokler, E.L.; Robbins, J.O.; Li, Y.; Bolshakov, V.Y. Maternal and early postnatal immune activation produce sex-specific effects on autism-like behaviors and neuroimmune function in mice. Sci. Rep. 2019, 9, 16928. [Google Scholar] [CrossRef] [PubMed]
- Glass, R.; Norton, S.; Fox, N.; Kusnecov, A.W. Maternal immune activation with staphylococcal enterotoxin A produces unique behavioral changes in C57BL/6 mouse offspring. Brain Behav. Immun. 2019, 75, 12–25. [Google Scholar] [CrossRef]
- Missig, G.; Robbins, J.O.; Mokler, E.L.; Mccullough, K.; Bilbo, S.D.; McDougle, C.J.; Carlezon, W.A. Sex-dependent neurobiological features of prenatal immune activation via TLR7. Mol. Psychiatry 2019. [Google Scholar] [CrossRef] [PubMed]
- Schwartzer, J.J.; Careaga, M.; Chang, C.; Onore, C.E.; Ashwood, P. Allergic fetal priming leads to developmental, behavioral and neurobiological changes in mice. Transl. Psychiatry 2015, 5, e543. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.; Nyffeler, M.; Schwendener, S.; Knuesel, I.; Yee, B.K.; Feldon, J. Relative prenatal and postnatal maternal contributions to schizophrenia-related neurochemical dysfunction after in utero immune challenge. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2008, 33, 441–456. [Google Scholar] [CrossRef]
- Shi, L.; Smith, S.E.P.; Malkova, N.; Tse, D.; Su, Y.; Patterson, P.H. Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav. Immun. 2009, 23, 116–123. [Google Scholar] [CrossRef]
- Fernández de Cossío, L.; Guzmán, A.; van der Veldt, S.; Luheshi, G.N. Prenatal infection leads to ASD-like behavior and altered synaptic pruning in the mouse offspring. Brain Behav. Immun. 2017, 63, 88–98. [Google Scholar] [CrossRef]
- Wu, W.-L.; Adams, C.E.; Stevens, K.E.; Chow, K.-H.; Freedman, R.; Patterson, P.H. The interaction between maternal immune activation and alpha 7 nicotinic acetylcholine receptor in regulating behaviors in the offspring. Brain Behav. Immun. 2015, 46, 192–202. [Google Scholar] [CrossRef]
- DiLalla, L.F.; McCrary, M.; Diaz, E. A review of endophenotypes in schizophrenia and autism: The next phase for understanding genetic etiologies. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 354–361. [Google Scholar] [CrossRef]
- Bjorklund, G.; Pivina, L.; Dadar, M.; Meguid, N.A.; Semenova, Y.; Anwar, M.; Chirumbolo, S. Gastrointestinal Alterations in Autism Spectrum Disorder: What Do We Know? Neurosci. Biobehav. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Bauman, M.D.; Iosif, A.-M.; Smith, S.E.P.; Bregere, C.; Amaral, D.G.; Patterson, P.H. Activation of the maternal immune system during pregnancy alters behavioral development of rhesus monkey offspring. Biol. Psychiatry 2014, 75, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Gumusoglu, S.B.; Stevens, H.E. Maternal Inflammation and Neurodevelopmental Programming: A Review of Preclinical Outcomes and Implications for Translational Psychiatry. Biol. Psychiatry 2019, 85, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Meyer, U. Maternal Immune Activation and Neuropsychiatric Illness: A Translational Research Perspective. Am. J. Psychiatry 2018, 175, 1073–1083. [Google Scholar] [CrossRef]
- Estes, M.L.; McAllister, A.K. Maternal immune activation: Implications for neuropsychiatric disorders. Science 2016, 353, 772–777. [Google Scholar] [CrossRef]
- Willette, A.A.; Lubach, G.R.; Knickmeyer, R.C.; Short, S.J.; Styner, M.; Gilmore, J.H.; Coe, C.L. Brain enlargement and increased behavioral and cytokine reactivity in infant monkeys following acute prenatal endotoxemia. Behav. Brain Res. 2011, 219, 108–115. [Google Scholar] [CrossRef]
- Oshiumi, H.; Matsumoto, M.; Funami, K.; Akazawa, T.; Seya, T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 2003, 4, 161–167. [Google Scholar] [CrossRef]
- Goines, P.E.; Croen, L.A.; Braunschweig, D.; Yoshida, C.K.; Grether, J.; Hansen, R.; Kharrazi, M.; Ashwood, P.; Van de Water, J. Increased midgestational IFN-γ, IL-4 and IL-5 in women bearing a child with autism: A case-control study. Mol. Autism 2011, 2, 13. [Google Scholar] [CrossRef]
- Xie, J.; Huang, L.; Li, X.; Li, H.; Zhou, Y.; Zhu, H.; Pan, T.; Kendrick, K.M.; Xu, W. Immunological cytokine profiling identifies TNF-α as a key molecule dysregulated in autistic children. Oncotarget 2017, 8, 82390–82398. [Google Scholar] [CrossRef]
- Ben-Yehuda, H.; Matcovitch-Natan, O.; Kertser, A.; Spinrad, A.; Prinz, M.; Amit, I.; Schwartz, M. Maternal Type-I interferon signaling adversely affects the microglia and the behavior of the offspring accompanied by increased sensitivity to stress. Mol. Psychiatry 2020, 25, 1050–1067. [Google Scholar] [CrossRef] [PubMed]
- Dulken, B.W.; Buckley, M.T.; Navarro Negredo, P.; Saligrama, N.; Cayrol, R.; Leeman, D.S.; George, B.M.; Boutet, S.C.; Hebestreit, K.; Pluvinage, J.V.; et al. Single-cell analysis reveals T cell infiltration in old neurogenic niches. Nature 2019, 571, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Missault, S.; Van den Eynde, K.; Vanden Berghe, W.; Fransen, E.; Weeren, A.; Timmermans, J.P.; Kuman-Singh, S.; Dedeurwaedere, S. The risk for behavioural deficits is determined by the maternal immune response to prenatal immune challenge in a neurodevelopmental model. Brain Behav. Immun. 2014, 42, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Chow, J.; Mazmanian, S.K.; Patterson, P.H. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc. Natl. Acad. Sci. USA 2012, 109, 12776–12781. [Google Scholar] [CrossRef] [PubMed]
- Pineda, E.; Shin, D.; You, S.J.; Auvin, S.; Sankar, R.; Mazarati, A. Maternal immune activation promotes hippocampal kindling epileptogenesis in mice. Ann. Neurol. 2013, 74, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheung, C.; Wei, R.; Hui, E.S.; Feldon, J.; Meyer, U.; Chung, S.; Chue, S.E.; Sham, P.C.; Wu, E.X.; et al. Prenatal immune challenge is an environmental risk factor for brain and behavior change relevant to schizophrenia: Evidence from MRI in a mouse model. PLoS ONE 2009, 4, e6354. [Google Scholar] [CrossRef]
- Clancy, B.; Darlington, R.B.; Finlay, B.L. Translating developmental time across mammalian species. Neuroscience 2001, 105, 7–17. [Google Scholar] [CrossRef]
- Kirsten, T.B.; Chaves-Kirsten, G.P.; Chaible, L.M.; Silva, A.C.; Martins, D.O.; Britto, L.R.G.; Dagli, M.L.Z.; Torrao, A.S.; Palermo-Neto, J.; Bernardi, M.M. Hypoactivity of the central dopaminergic system and autistic-like behavior induced by a single early prenatal exposure to lipopolysaccharide. J. Neurosci. Res. 2012, 90, 1903–1912. [Google Scholar] [CrossRef]
- Xuan, I.C.Y.; Hampson, D.R. Gender-dependent effects of maternal immune activation on the behavior of mouse offspring. PLoS ONE 2014, 9, e104433. [Google Scholar] [CrossRef]
- Sharova, V.S.; Izvolskaia, M.S.; Zakharova, L.A. Lipopolysaccharide-induced maternal inflammation affects the gonadotropin-releasing hormone neuron development in fetal mice. Neuroimmunomodulation 2015, 22, 222–232. [Google Scholar] [CrossRef]
- Warrier, V.; Chee, V.; Smith, P.; Chakrabarti, B.; Baron-Cohen, S. A comprehensive meta-analysis of common genetic variants in autism spectrum conditions. Mol. Autism 2015, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Wischhof, L.; Irrsack, E.; Osorio, C.; Koch, M. Prenatal LPS-exposure—A neurodevelopmental rat model of schizophrenia—Differentially affects cognitive functions, myelination and parvalbumin expression in male and female offspring. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 57, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Batinić, B.; Santrač, A.; Divović, B.; Timic, T.; Stankovic, T.; Obradovic, A.L.; Joksimovic, S.; Savic, M.M. Lipopolysaccharide exposure during late embryogenesis results in diminished locomotor activity and amphetamine response in females and spatial cognition impairment in males in adult, but not adolescent rat offspring. Behav. Brain Res. 2016, 299, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Qi, F.; Song, D.; He, Z.; Zuo, Z.; Yang, Y.; Liu, Q.; Hu, S.; Wang, X.; Zheng, X.; et al. Prenatal influenza vaccination rescues impairments of social behavior and lamination in a mouse model of autism. J. Neuroinflamm. 2018, 15, 228. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.; Nyffeler, M.; Engler, A.; Urwyler, A.; Schedlowski, M.; Knuesel, I.; Yee, B.K.; Feldon, J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 4752–4762. [Google Scholar] [CrossRef] [PubMed]
- Abazyan, B.; Nomura, J.; Kannan, G.; Ishizuka, K.; Tamashiro, K.L.; Nucifora, F.; Pogorelov, V.; Ladenheim, B.; Yang, C.; Krasnova, I.N.; et al. Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol. Psychiatry 2010, 68, 1172–1181. [Google Scholar] [CrossRef]
- Lipina, T.V.; Zai, C.; Hlousek, D.; Order, J.; Wong, A.H.C. Maternal immune activation during gestation interacts with Disc1 point mutation to exacerbate schizophrenia-related behaviors in mice. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 7654–7666. [Google Scholar] [CrossRef]
- Ehninger, D.; Sano, Y.; de Vries, P.J.; Dies, K.; Franz, D.; Geschwind, D.H.; Kaur, M.; Lee, Y.-S.; Li, W.; Lowe, J.K.; et al. Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Mol. Psychiatry 2012, 17, 62–70. [Google Scholar] [CrossRef]
- Giovanoli, S.; Engler, H.; Engler, A.; Richetto, J.; Voget, M.; Willi, R.; Winter, C.; Riva, M.A.; Brobech, P.; Schedlowski, M. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science 2013, 339, 1095–1099. [Google Scholar] [CrossRef]
- Giovanoli, S.; Weber-Stadlbauer, U.; Schedlowski, M.; Meyer, U.; Engler, H. Prenatal immune activation causes hippocampal synaptic deficits in the absence of overt microglia anomalies. Brain Behav. Immun. 2016, 55, 25–38. [Google Scholar] [CrossRef]
- Li, X.; Tian, X.; Lv, L.; Hei, G.; Huang, X.; Fan, X.; Zhang, J.; Zhang, J.; Pang, L.; Song, X. Microglia activation in the offspring of prenatal Poly I: C exposed rats: A PET imaging and immunohistochemistry study. Gen. Psychiatry 2018, 31, e000006. [Google Scholar] [CrossRef] [PubMed]
- Lins, B.R.; Marks, W.N.; Zabder, N.K.; Greba, Q.; Howland, J.G. Maternal Immune Activation during Pregnancy Alters the Behavior Profile of Female Offspring of Sprague Dawley Rats. eNeuro 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.-M.; Jennische, E.; Hansson, H.-A.; Holmäng, A. Prenatal exposure to interleukin-6 results in inflammatory neurodegeneration in hippocampus with NMDA/GABA(A) dysregulation and impaired spatial learning. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1345–R1356. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.P.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef]
- Machado, C.J.; Whitaker, A.M.; Smith, S.E.P.; Patterson, P.H.; Bauman, M.D. Maternal immune activation in nonhuman primates alters social attention in juvenile offspring. Biol. Psychiatry 2015, 77, 823–832. [Google Scholar] [CrossRef]
- Weir, R.K.; Forghany, R.; Smith, S.E.P.; Patterson, P.H.; McAllister, A.K.; Schumann, C.M.; Bauman, M.D. Preliminary evidence of neuropathology in nonhuman primates prenatally exposed to maternal immune activation. Brain Behav. Immun. 2015, 48, 139–146. [Google Scholar] [CrossRef]
- Rose, D.R.; Careaga, M.; Van de Water, J.; McAllister, K.; Bauman, M.D.; Ashwood, P. Long-term altered immune responses following fetal priming in a non-human primate model of maternal immune activation. Brain Behav. Immun. 2017, 63, 60–70. [Google Scholar] [CrossRef]
- Bauman, M.D.; Lesh, T.A.; Rowland, D.J.; Schumann, C.M.; Smucny, J.; Kukis, D.L.; Cherry, S.R.; MacAllister, A.K.; Carter, C.S. Preliminary evidence of increased striatal dopamine in a nonhuman primate model of maternal immune activation. Transl. Psychiatry 2019, 9, 135. [Google Scholar] [CrossRef]
- Arrode-Brusés, G.; Brusés, J.L. Maternal immune activation by poly I:C induces expression of cytokines IL-1β and IL-13, chemokine MCP-1 and colony stimulating factor VEGF in fetal mouse brain. J. Neuroinflamm. 2012, 9, 83. [Google Scholar] [CrossRef]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef]
- Mazina, V.; Gerdts, J.; Trinh, S.; Ankenman, K.; Ward, T.; Dennis, M.Y.; Girirajan, S.; Eichler, E.E.; Berniner, R. Epigenetics of autism-related impairment: Copy number variation and maternal infection. J. Dev. Behav. Pediatr. JDBP 2015, 36, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Foley, K.A.; MacFabe, D.F.; Kavaliers, M.; Ossenkopp, K.-P. Sexually dimorphic effects of prenatal exposure to lipopolysaccharide, and prenatal and postnatal exposure to propionic acid, on acoustic startle response and prepulse inhibition in adolescent rats: Relevance to autism spectrum disorders. Behav. Brain Res. 2015, 278, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Yuen, R.K.C.; Thiruvahindrapuram, B.; Merico, D.; Walker, S.; Tammimies, K.; Hoang, N.; Crysler, C.; Nalpathamkalam, T.; Pellechia, G.; Liu, Y.; et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 2015, 21, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manna, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Autism Spectrum Disorders Working Group of the Psychiatric Genomics Consortium. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism 2017, 8. [Google Scholar] [CrossRef]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallasen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef]
- Patterson, P.H. Maternal infection and immune involvement in autism. Trends Mol. Med. 2011, 17, 389–394. [Google Scholar] [CrossRef]
- Zaretsky, M.V.; Alexander, J.M.; Byrd, W.; Bawdon, R.E. Transfer of inflammatory cytokines across the placenta. Obstet. Gynecol. 2004, 103, 546–550. [Google Scholar] [CrossRef]
- Aaltonen, R.; Heikkinen, T.; Hakala, K.; Laine, K.; Alanen, A. Transfer of proinflammatory cytokines across term placenta. Obstet. Gynecol. 2005, 106, 802–807. [Google Scholar] [CrossRef]
- PrabhuDas, M.; Bonney, E.; Caron, K.; Dey, S.; Erlebacher, A.; Fazleabas, A.; Fisher, S.; Golos, T.; Matzuk, M.; McCune, J.M.; et al. Immune mechanisms at the maternal-fetal interface: Perspectives and challenges. Nat. Immunol. 2015, 16, 328–334. [Google Scholar] [CrossRef]
- Holme, A.M.; Roland, M.C.P.; Lorentzen, B.; Michelsen, T.M.; Henriksen, T. Placental glucose transfer: A human in vivo study. PLoS ONE 2015, 10, e0117084. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, E.; Menassa, D.A.; Jacobson, L.; West, S.J.; Domingos, J.; Moloney, T.C.; Lang, B.; Harrison, B.J.; Bennett, D.L.H.; Bannerman, D.; et al. Persistent microglial activation and synaptic loss with behavioral abnormalities in mouse offspring exposed to CASPR2-antibodies in utero. Acta Neuropathol. 2017, 134, 567–583. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji-Xu, A.; Vincent, A. Maternal Immunity in Autism Spectrum Disorders: Questions of Causality, Validity, and Specificity. J. Clin. Med. 2020, 9, 2590. https://doi.org/10.3390/jcm9082590
Ji-Xu A, Vincent A. Maternal Immunity in Autism Spectrum Disorders: Questions of Causality, Validity, and Specificity. Journal of Clinical Medicine. 2020; 9(8):2590. https://doi.org/10.3390/jcm9082590
Chicago/Turabian StyleJi-Xu, Antonio, and Angela Vincent. 2020. "Maternal Immunity in Autism Spectrum Disorders: Questions of Causality, Validity, and Specificity" Journal of Clinical Medicine 9, no. 8: 2590. https://doi.org/10.3390/jcm9082590
APA StyleJi-Xu, A., & Vincent, A. (2020). Maternal Immunity in Autism Spectrum Disorders: Questions of Causality, Validity, and Specificity. Journal of Clinical Medicine, 9(8), 2590. https://doi.org/10.3390/jcm9082590