The Role of Secondary Brain Insults in Status Epilepticus: A Systematic Review

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Section
2.1. Definitions
2.2. Eligibility Criteria
2.3. Search Strategy
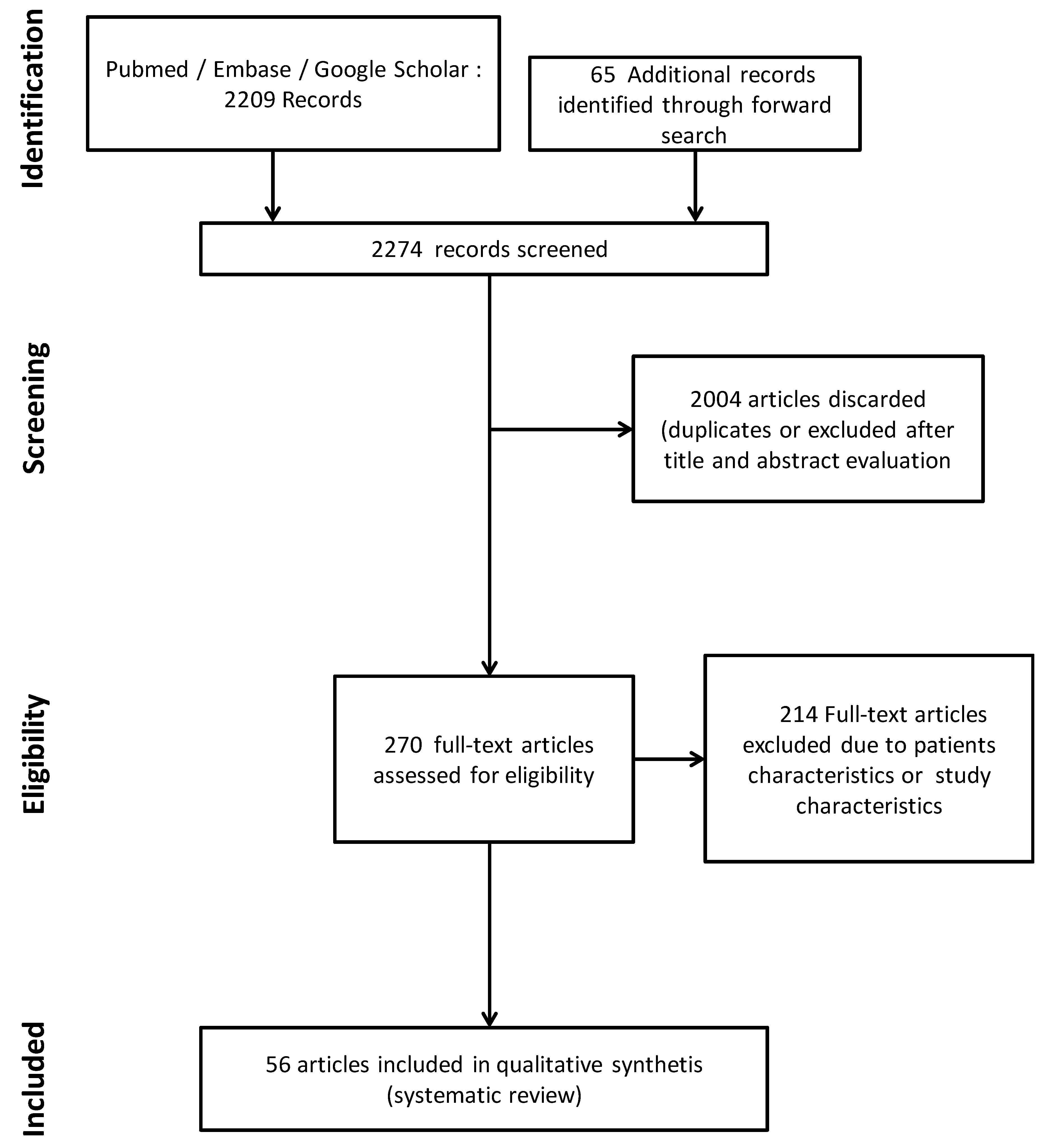
2.4. Study Selection and Data Extraction
3. Results
3.1. Experimental Data
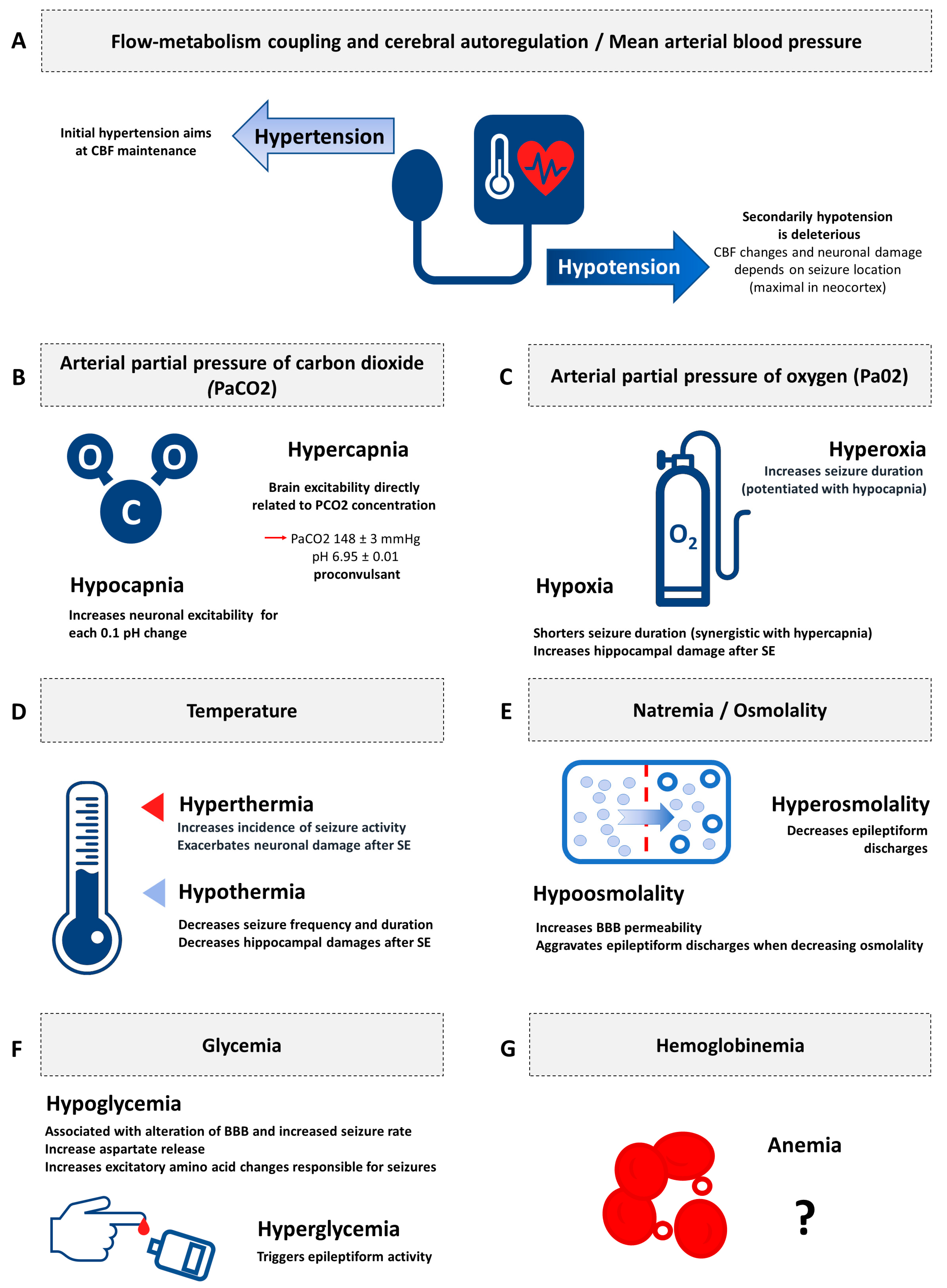
3.1.1. Flow-Metabolism Coupling and Cerebral Autoregulation/Mean Arterial Blood Pressure
3.1.2. Arterial Partial Pressure of Carbon Dioxide
3.1.3. Arterial Partial Pressure of Oxygen
3.1.4. Temperature
3.1.5. Natremia/Osmolality
3.1.6. Glycemia
3.1.7. Hemoglobinemia
3.2. Clinical Applications of Secondary Brain Insults in the Management of Status Epilepticus
4. Perspectives
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holleville, M.; Jacq, G.; Perier, F.; Fontaine, C.; Legriel, S. Epileptic Seizures in Critically Ill Patients: Diagnosis, Management, and Outcomes. J. Clin. Med. 2020, 9, 2218. [Google Scholar] [CrossRef] [PubMed]
- Legriel, S.; Mourvillier, B.; Bele, N.; Amaro, J.; Fouet, P.; Manet, P.; Hilpert, F. Outcomes in 140 critically ill patients with status epilepticus. Intensive Care Med. 2008, 34, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Legriel, S.; Azoulay, E.; Resche-Rigon, M.; Lemiale, V.; Mourvillier, B.; Kouatchet, A.; Troche, G.; Wolf, M.; Galliot, R.; Dessertaine, G.; et al. Functional outcome after convulsive status epilepticus. Crit. Care Med. 2010, 38, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.; Engelhard, K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Siesjo, B.K.; Siesjo, P. Mechanisms of secondary brain injury. Eur. J. Anaesthesiol. 1996, 13, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, C.J.; Delanty, N. Hypertensive emergencies. Lancet 2000, 356, 411–417. [Google Scholar] [CrossRef]
- Geeraerts, T.; Velly, L.; Abdennour, L.; Asehnoune, K.; Audibert, G.; Bouzat, P.; Bruder, N.; Carrillon, R.; Cottenceau, V.; Cotton, F.; et al. Management of severe traumatic brain injury (first 24 hours). Anaesth. Crit. Care Pain Med. 2018, 37, 171–186. [Google Scholar] [CrossRef]
- Chesnut, R.M.; Marshall, L.F.; Klauber, M.R.; Blunt, B.A.; Baldwin, N.; Eisenberg, H.M.; Jane, J.A.; Marmarou, A.; Foulkes, M.A. The role of secondary brain injury in determining outcome from severe head injury. J. Trauma 1993, 34, 216–222. [Google Scholar] [CrossRef]
- Carney, N.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Hawryluk, G.W.; Bell, M.J.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; et al. Guidelines for the Management of Severe Traumatic Brain Injury, Fourth Edition. Neurosurgery 2017, 80, 6–15. [Google Scholar] [CrossRef]
- Cariou, A.; Payen, J.F.; Asehnoune, K.; Audibert, G.; Botte, A.; Brissaud, O.; Debaty, G.; Deltour, S.; Deye, N.; Engrand, N.; et al. Targeted temperature management in the ICU: Guidelines from a French expert panel. Ann. Intensive Care 2017, 7, 70. [Google Scholar] [CrossRef]
- Ntaios, G.; Dziedzic, T.; Michel, P.; Papavasileiou, V.; Petersson, J.; Staykov, D.; Thomas, B.; Steiner, T.; European Stroke, O. European Stroke Organisation (ESO) guidelines for the management of temperature in patients with acute ischemic stroke. Int. J. Stroke 2015, 10, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Bederson, J.B.; Connolly, E.S., Jr.; Batjer, H.H.; Dacey, R.G.; Dion, J.E.; Diringer, M.N.; Duldner, J.E., Jr.; Harbaugh, R.E.; Patel, A.B.; Rosenwasser, R.H.; et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: A statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke 2009, 40, 994–1025. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, B.S.; Horton, R.W. Physiology of status epilepticus in primates. Arch. Neurol. 1973, 28, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Walton, N.Y. Systemic effects of generalized convulsive status epilepticus. Epilepsia 1993, 34 (Suppl. 1), S54–S58. [Google Scholar] [CrossRef]
- Wasterlain, C.G.; Fujikawa, D.G.; Penix, L.; Sankar, R. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia 1993, 34 (Suppl. 1), S37–S53. [Google Scholar] [CrossRef]
- Trinka, E.; Cock, H.; Hesdorffer, D.; Rossetti, A.O.; Scheffer, I.E.; Shinnar, S.; Shorvon, S.; Lowenstein, D.H. A definition and classification of status epilepticus--Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 2015, 56, 1515–1523. [Google Scholar] [CrossRef]
- Aminoff, M.J.; Simon, R.P. Status epilepticus. Causes, clinical features and consequences in 98 patients. Am. J. Med. 1980, 69, 657–666. [Google Scholar] [CrossRef]
- Simon, R.P. Physiologic consequences of status epilepticus. Epilepsia 1985, 26 (Suppl. 1), S58–S66. [Google Scholar] [CrossRef]
- Posner, J.B.; Plum, F. Cerebral metabolism during electrically-induced seizures in man. Trans. Am. Neurol. Assoc. 1968, 93, 84–88. [Google Scholar] [CrossRef]
- Meldrum, B.S.; Nilsson, B. Cerebral blood flow and metabolic rate early and late in prolonged epileptic seizures induced in rats by bicuculline. Brain 1976, 99, 523–542. [Google Scholar] [CrossRef]
- Ingvar, M.; Siesjo, B.K. Local blood flow and glucose consumption in the rat brain during sustained bicuculline-induced seizures. Acta Neurol. Scand. 1983, 68, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Blennow, G.; Brierley, J.B.; Meldrum, B.S.; Siesjo, B.K. Epileptic brain damage: The role of systemic factors that modify cerebral energy metabolism. Brain 1978, 101, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Laffey, J.G.; Kavanagh, B.P. Hypocapnia. N. Engl. J. Med. 2002, 347, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.D.; Dewaraja, A.S.; Vanhatalo, S. Does hyperventilation elicit epileptic seizures? Epilepsia 2004, 45, 618–620. [Google Scholar] [CrossRef] [PubMed]
- Bergsholm, P.; Gran, L.; Bleie, H. Seizure duration in unilateral electroconvulsive therapy. The effect of hypocapnia induced by hyperventilation and the effect of ventilation with oxygen. Acta Psychiatr. Scand. 1984, 69, 121–128. [Google Scholar] [CrossRef]
- Marrosu, F.; Puligheddu, M.; Giagheddu, M.; Cossu, G.; Piga, M. Correlation between cerebral perfusion and hyperventilation enhanced focal spiking activity. Epilepsy Res. 2000, 40, 79–86. [Google Scholar] [CrossRef]
- Sawayama, E.; Takahashi, M.; Inoue, A.; Nakajima, K.; Kano, A.; Sawayama, T.; Okutomi, T.; Miyaoka, H. Moderate hyperventilation prolongs electroencephalogram seizure duration of the first electroconvulsive therapy. J. ECT 2008, 24, 195–198. [Google Scholar] [CrossRef]
- Jonas, J.; Vignal, J.P.; Baumann, C.; Anxionnat, J.F.; Muresan, M.; Vespignani, H.; Maillard, L. Effect of hyperventilation on seizure activation: Potentiation by antiepileptic drug tapering. J. Neurol. Neurosurg. Psychiatry 2011, 82, 928–930. [Google Scholar] [CrossRef]
- Balestrino, M.; Somjen, G.G. Concentration of carbon dioxide, interstitial pH and synaptic transmission in hippocampal formation of the rat. J. Physiol. 1988, 396, 247–266. [Google Scholar] [CrossRef]
- Crawford, C.D.; Butler, P.; Froese, A. Arterial PaO2 and PaCO2 influence seizure duration in dogs receiving electroconvulsive therapy. Can. J. Anaesth. 1987, 34, 437–441. [Google Scholar]
- Dulla, C.G.; Dobelis, P.; Pearson, T.; Frenguelli, B.G.; Staley, K.J.; Masino, S.A. Adenosine and ATP link PCO2 to cortical excitability via pH. Neuron 2005, 48, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Ziemann, A.E.; Schnizler, M.K.; Albert, G.W.; Severson, M.A.; Howard Iii, M.A.; Welsh, M.J.; Wemmie, J.A. Seizure termination by acidosis depends on ASIC1a. Nat. Neurosci. 2008, 11, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Legriel, S.; Mentec, H. Status epilepticus during acute hypercapnia: A case report. Intensive Care Med. 2005, 31, 314. [Google Scholar] [CrossRef] [PubMed]
- Brodie, D.A.; Woodbury, D.M. Acid-base changes in brain and blood of rats exposed to high concentrations of carbon dioxide. Am. J. Physiol. 1958, 192, 91–94. [Google Scholar] [CrossRef]
- Withrow, C.D.; Nord, N.M.; Turner, L.M., Jr.; Woodbury, D.M. Carbon dioxide seizures in immature rats. Proc. Soc. Exp. Biol. Med. 1967, 125, 288–291. [Google Scholar] [CrossRef]
- Katsura, K.; Kristian, T.; Smith, M.L.; Siesjo, B.K. Acidosis induced by hypercapnia exaggerates ischemic brain damage. J. Cereb. Blood Flow Metab. 1994, 14, 243–250. [Google Scholar] [CrossRef]
- Mathern, G.W.; Price, G.; Rosales, C.; Pretorius, J.K.; Lozada, A.; Mendoza, D. Anoxia during kainate status epilepticus shortens behavioral convulsions but generates hippocampal neuron loss and supragranular mossy fiber sprouting. Epilepsy Res. 1998, 30, 133–151. [Google Scholar] [CrossRef]
- Aksay, S.S.; Bumb, J.M.; Janke, C.; Hoyer, C.; Kranaster, L.; Sartorius, A. New evidence for seizure quality improvement by hyperoxia and mild hypocapnia. J. ECT 2014, 30, 287–291. [Google Scholar] [CrossRef]
- Wasterlain, C.G. Mortality and morbidity from serial seizures. An experimental study. Epilepsia 1974, 15, 155–176. [Google Scholar] [CrossRef]
- Nevander, G.; Ingvar, M.; Auer, R.; Siesjo, B.K. Status epilepticus in well-oxygenated rats causes neuronal necrosis. Ann. Neurol. 1985, 18, 281–290. [Google Scholar] [CrossRef]
- Amano, S.; Obata, T.; Hazama, F.; Kashiro, N.; Shimada, M. Hypoxia prevents seizures and neuronal damages of the hippocampus induced by kainic acid in rats. Brain Res. 1990, 523, 121–126. [Google Scholar] [CrossRef]
- Emerson, M.R.; Nelson, S.R.; Samson, F.E.; Pazdernik, T.L. Hypoxia preconditioning attenuates brain edema associated with kainic acid-induced status epilepticus in rats. Brain Res. 1999, 825, 189–193. [Google Scholar] [CrossRef]
- Elayan, I.M.; Axley, M.J.; Prasad, P.V.; Ahlers, S.T.; Auker, C.R. Effect of hyperbaric oxygen treatment on nitric oxide and oxygen free radicals in rat brain. J. Neurophysiol. 2000, 83, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Lambertsen, C.; Clark, J.; Gelfand, R.; Pisarello, J.; Cobbs, W. Definition of tolerance to continuous hyperoxia in man-An abstract report of Predictive Studies V. In Proceedings of the 9th International Symposium on Underwater and Hyperbaric Physiology, Sydney, Australia, 1–4 March 1987; pp. 717–735. [Google Scholar]
- Soderfeldt, B.; Blennow, G.; Kalimo, H.; Olsson, Y.; Siesjo, B.K. Influence of systemic factors on experimental epileptic brain injury. Structural changes accompanying bicuculline-induced seizures in rats following manipulations of tissue oxygenation or alpha-tocopherol levels. Acta Neuropathol. 1983, 60, 81–91. [Google Scholar] [PubMed]
- Pomper, J.K.; Hoffmann, U.; Kovacs, R.; Gabriel, S.; Heinemann, U. Hyperoxia is not an essential condition for status epilepticus induced cell death in organotypic hippocampal slice cultures. Epilepsy Res. 2004, 59, 61–65. [Google Scholar] [CrossRef]
- Cungi, P.J.; Holleville, M.; Fontaine, C.; Jacq, G.; Legriel, S. Second-line anticonvulsant for convulsive status epilepticus: The dosage matters! Anaesth. Crit. Care Pain Med. 2020, 39, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Duong, T.M.; de Lanerolle, N.C. The neuropathology of hyperthermic seizures in the rat. Epilepsia 1999, 40, 5–19. [Google Scholar] [CrossRef]
- Liu, Z.; Gatt, A.; Mikati, M.; Holmes, G.L. Effect of temperature on kainic acid-induced seizures. Brain Res. 1993, 631, 51–58. [Google Scholar] [CrossRef]
- Lundgren, J.; Smith, M.L.; Blennow, G.; Siesjo, B.K. Hyperthermia aggravates and hypothermia ameliorates epileptic brain damage. Exp. Brain Res. 1994, 99, 43–55. [Google Scholar] [CrossRef]
- Maeda, T.; Hashizume, K.; Tanaka, T. Effect of hypothermia on kainic acid-induced limbic seizures: An electroencephalographic and 14C-deoxyglucose autoradiographic study. Brain Res. 1999, 818, 228–235. [Google Scholar] [CrossRef]
- Schmitt, F.C.; Buchheim, K.; Meierkord, H.; Holtkamp, M. Anticonvulsant properties of hypothermia in experimental status epilepticus. Neurobiol. Dis. 2006, 23, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Kowski, A.B.; Kanaan, H.; Schmitt, F.C.; Holtkamp, M. Deep hypothermia terminates status epilepticus--an experimental study. Brain Res. 2012, 1446, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, P.P.; Li, L.Y.; Zhang, H.M.; Li, T. Hypothermia reduces brain edema, spontaneous recurrent seizure attack, and learning memory deficits in the kainic acid treated rats. CNS Neurosci. Ther. 2011, 17, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhou, Y.; Wang, Y. Effect of mild hypothermia on glutamate receptor expression after status epilepticus. Epilepsy Res. 2012, 101, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Phillips, K.F.; Deshpande, L.S.; DeLorenzo, R.J. Hypothermia reduces calcium entry via the N-methyl-D-aspartate and ryanodine receptors in cultured hippocampal neurons. Eur. J. Pharmacol. 2013, 698, 186–192. [Google Scholar] [CrossRef]
- Dudek, F.E.; Obenaus, A.; Tasker, J.G. Osmolality-induced changes in extracellular volume alter epileptiform bursts independent of chemical synapses in the rat: Importance of non-synaptic mechanisms in hippocampal epileptogenesis. Neurosci. Letters 1990, 120, 267–270. [Google Scholar] [CrossRef]
- Huang, R.; Somjen, G.G. Effects of hypertonia on voltage-gated ion currents in freshly isolated hippocampal neurons, and on synaptic currents in neurons in hippocampal slices. Brain Res. 1997, 748, 157–167. [Google Scholar] [CrossRef]
- Andrew, R.D.; Fagan, M.; Ballyk, B.A.; Rosen, A.S. Seizure susceptibility and the osmotic state. Brain Res. 1989, 498, 175–180. [Google Scholar] [CrossRef]
- Roper, S.N.; Obenaus, A.; Dudek, F.E. Osmolality and nonsynaptic epileptiform bursts in rat CA1 and dentate gyrus. Ann. Neurol. 1992, 31, 81–85. [Google Scholar] [CrossRef]
- Oztas, B.; Kaya, M.; Kucuk, M.; Tugran, N. Influence of hypoosmolality on the blood-brain barrier permeability during epileptic seizures. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2003, 27, 701–704. [Google Scholar] [CrossRef]
- Janigro, D. Blood-brain barrier, ion homeostatis and epilepsy: Possible implications towards the understanding of ketogenic diet mechanisms. Epilepsy Res. 1999, 37, 223–232. [Google Scholar] [CrossRef]
- Oztas, B.; Camurcu, S. Blood-brain barrier permeability after electrically induced seizure in normoglycemic, hypoglycemic, and hyperglycemic rats. Psychiatry Res. 1989, 29, 151–159. [Google Scholar] [CrossRef]
- Schwechter, E.M.; Veliskova, J.; Velisek, L. Correlation between extracellular glucose and seizure susceptibility in adult rats. Ann. Neurol. 2003, 53, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Schauwecker, P.E. The effects of glycemic control on seizures and seizure-induced excitotoxic cell death. BMC Neurosci. 2012, 13, 94. [Google Scholar] [CrossRef] [PubMed]
- Auer, R.N. Hypoglycemic brain damage. Forensic Sci. Int. 2004, 146, 105–110. [Google Scholar] [CrossRef]
- Kirchner, A.; Veliskova, J.; Velisek, L. Differential effects of low glucose concentrations on seizures and epileptiform activity in vivo and in vitro. Eur. J. Neurosci. 2006, 23, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Claassen, J.; Albers, D.; Schmidt, J.M.; De Marchis, G.M.; Pugin, D.; Falo, C.M.; Mayer, S.A.; Cremers, S.; Agarwal, S.; Elkind, M.S.; et al. Nonconvulsive seizures in subarachnoid hemorrhage link inflammation and outcome. Ann. Neurol. 2014, 75, 771–781. [Google Scholar] [CrossRef]
- Hawkes, M.A.; Hocker, S.E. Systemic Complications Following Status Epilepticus. Curr. Neurol. Neurosci. Rep. 2018, 18, 7. [Google Scholar] [CrossRef]
- Legriel, S.; Lemiale, V.; Schenck, M.; Chelly, J.; Laurent, V.; Daviaud, F.; Srairi, M.; Hamdi, A.; Geri, G.; Rossignol, T.; et al. Hypothermia for Neuroprotection in Convulsive Status Epilepticus. N. Engl. J. Med. 2016, 375, 2457–2467. [Google Scholar] [CrossRef]
- Hawkes, M.A.; English, S.W.; Mandrekar, J.N.; Rabinstein, A.A.; Hocker, S. Causes of Death in Status Epilepticus. Crit. Care Med. 2019, 47, 1226–1231. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontaine, C.; Jacq, G.; Perier, F.; Holleville, M.; Legriel, S. The Role of Secondary Brain Insults in Status Epilepticus: A Systematic Review. J. Clin. Med. 2020, 9, 2521. https://doi.org/10.3390/jcm9082521
Fontaine C, Jacq G, Perier F, Holleville M, Legriel S. The Role of Secondary Brain Insults in Status Epilepticus: A Systematic Review. Journal of Clinical Medicine. 2020; 9(8):2521. https://doi.org/10.3390/jcm9082521
Chicago/Turabian StyleFontaine, Candice, Gwenaelle Jacq, François Perier, Mathilde Holleville, and Stephane Legriel. 2020. "The Role of Secondary Brain Insults in Status Epilepticus: A Systematic Review" Journal of Clinical Medicine 9, no. 8: 2521. https://doi.org/10.3390/jcm9082521
APA StyleFontaine, C., Jacq, G., Perier, F., Holleville, M., & Legriel, S. (2020). The Role of Secondary Brain Insults in Status Epilepticus: A Systematic Review. Journal of Clinical Medicine, 9(8), 2521. https://doi.org/10.3390/jcm9082521