Platelet Functions During Extracorporeal Membrane Oxygenation. Platelet–Leukocyte Aggregates Analyzed by Flow Cytometry as a Promising Tool to Monitor Platelet Activation

 , , and
, , and
Abstract
1. Introduction
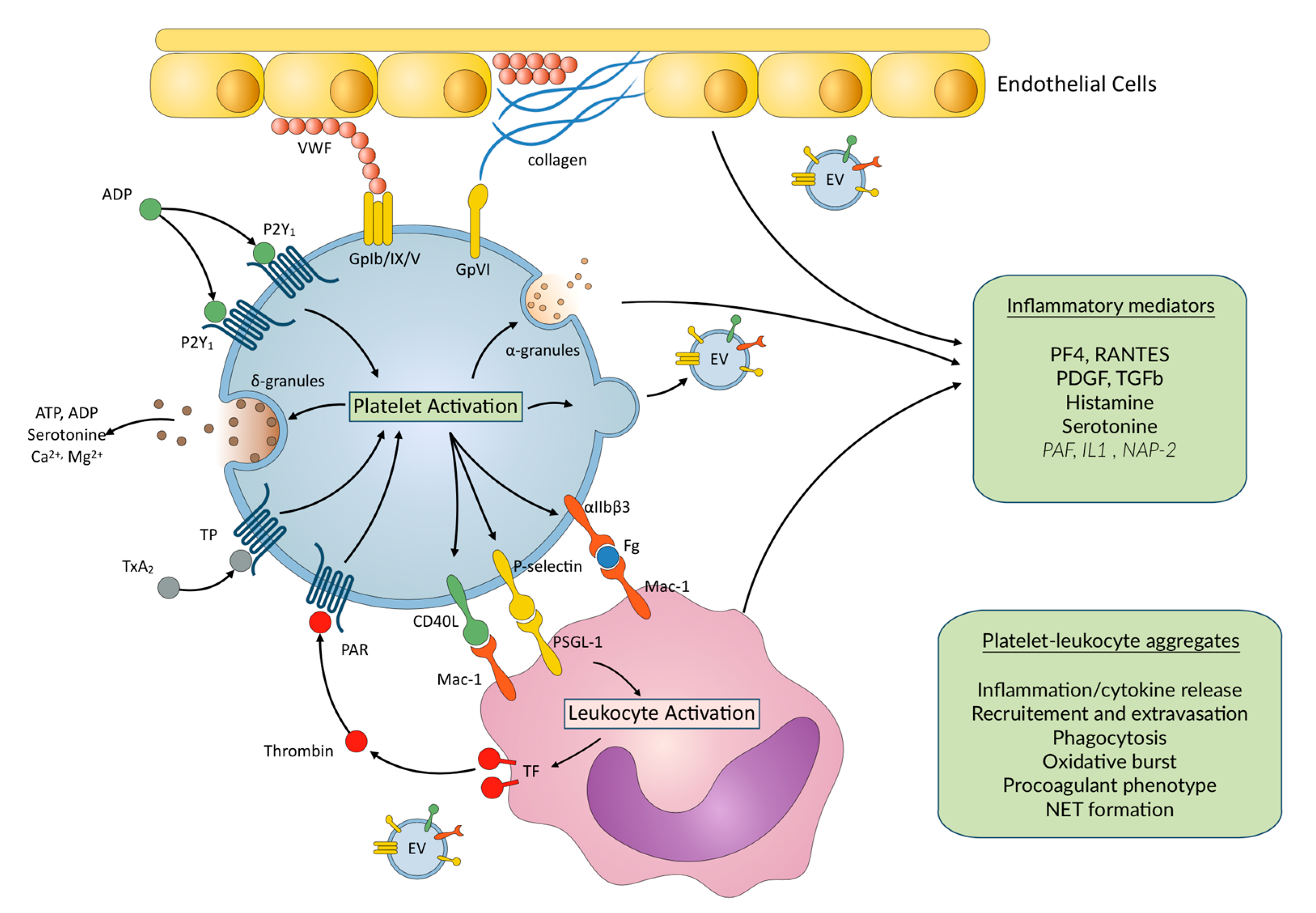
2. Platelet Activation, Aggregation, and ECMO
3. Inflammation in ECMO Patients
4. Inflammation and Platelet–Leukocyte Interactions
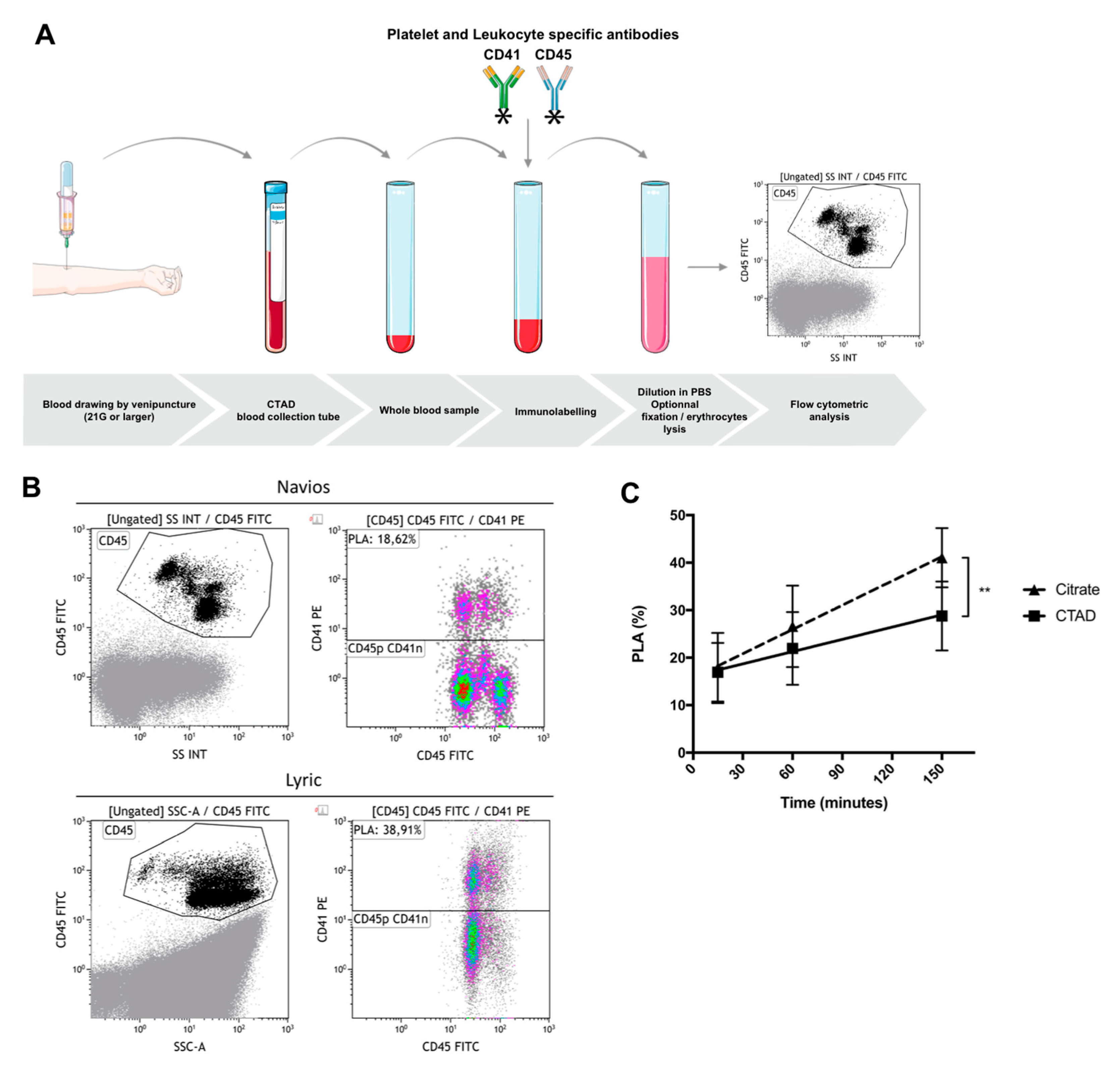
5. Flow Cytometric Whole Blood Measurement of Platelet–Leukocyte Aggregates
5.1. General Considerations
5.2. Pre-Analytical Requirements—Choice of Anticoagulant, Delay between Sampling and Immunolabeling, Sample Stability after Immunolabeling, Effect of Strong Vortex Agitation prior to Immunolabeling
5.3. Optimal Conditions for Whole Blood Cytometric Measurement of PLAs: A Proposal
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Doyle, A.J.; Hunt, B.J. Current understanding of how extracorporeal membrane oxygenators activate haemostasis and other blood components. Front. Med. 2018, 5, 352. [Google Scholar] [CrossRef] [PubMed]
- Nedelec-Gac, F.; Outier, A.S.; Roisne, A.; Mansour, A.; Jacquet, H.; Frerou, A.; Gueret, P.; Le Pabic, E.; Fest, T.; Verhoye, J.P.; et al. The effect of two extracorporeal membrane oxygenation pumps with different rotational speeds on hemostasis: A pilot study. 2020. in preparation. [Google Scholar]
- Kalbhenn, J.; Schmidt, R.; Nakamura, L.; Schelling, J.; Rosenfelder, S.; Zieger, B. Early diagnosis of acquired von Willebrand Syndrome (AVWS) is elementary for clinical practice in patients treated with ECMO therapy. J. Atheroscler. Thromb. 2015, 22, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Vincent, F.; Rauch, A.; Loobuyck, V.; Robin, E.; Nix, C.; Vincentelli, A.; Smadja, D.M.; Leprince, P.; Amour, J.; Lemesle, G.; et al. Arterial pulsatility and circulating von Willebrand factor in patients on mechanical circulatory support. J. Am. Coll. Cardiol. 2018, 71, 2106–2118. [Google Scholar] [CrossRef] [PubMed]
- Malfertheiner, M.V.; Pimenta, L.P.; von Bahr, V.; Millar, J.E.; Obonyo, N.G.; Suen, J.Y.; Pellegrino, V.; Fraser, J.F. Acquired von Willebrand syndrome in respiratory extracorporeal life support: A systematic review of the literature. Crit. Care Resusc. 2017, 19, 45–52. [Google Scholar]
- Mazzeffi, M.; Tanaka, K. Platelets and ECMO: Should we worry about count, function, or both? Intensive Care Med. 2016, 42, 1199–1200. [Google Scholar] [CrossRef]
- Balle, C.M.; Jeppesen, A.N.; Christensen, S.; Hvas, A.-M. Platelet function during extracorporeal membrane Oxygenation in adult patients: A systematic review. Front. Cardiovasc. Med. 2018, 5, 157. [Google Scholar] [CrossRef]
- Renhao, L. The Glycoprotein Ib-IX-V complex. In Platelets; Michelson, A.D., Ed.; Elsevier: London, UK, 2019; pp. 193–211. [Google Scholar]
- Gardiner, E.E. Proteolytic processing of platelet receptors. Res. Pract. Thromb. Haemost. 2018, 2, 240–250. [Google Scholar] [CrossRef]
- Lukito, P.; Wong, A.; Jing, J.; Arthur, J.F.; Marasco, S.F.; Murphy, D.A.; Bergin, P.J.; Shaw, J.A.; Collecutt, M.; Andrews, R.K.; et al. Mechanical circulatory support is associated with loss of platelet receptors glycoprotein Ibα and glycoprotein VI. J. Thromb. Haemost. 2016, 14, 2253–2260. [Google Scholar] [CrossRef]
- Mazzeffi, M.; Hasan, S.; Abuelkasem, E.; Meyer, M.; Deatrick, K.; Taylor, B.; Kon, Z.; Herr, D.; Tanaka, K. Von Willebrand Factor-GP1bα interactions in venoarterial extracorporeal membrane Oxygenation patients. J. Cardiothorac. Vasc. Anesth. 2019, 33, 2125–2132. [Google Scholar] [CrossRef]
- Nair, P.; Hoechter, D.J.; Buscher, H.; Venkatesh, K.; Whittam, S.; Joseph, J.; Jansz, P. Prospective observational study of hemostatic alterations during adult extracorporeal membrane oxygenation (ECMO) using point-of-care thromboelastometry and platelet aggregometry. J. Cardiothorac. Vasc. Anesth. 2015, 29, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Tauber, H.; Streif, W.; Fritz, J.; Ott, H.; Weigel, G.; Loacker, L.; Heinz, A.; Velik-Salchner, C. Predicting Transfusion Requirements During Extracorporeal Membrane Oxygenation. J. Cardiothorac. Vasc. Anesth. 2016, 30, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Wand, S.; Huber-Petersen, J.F.; Schaeper, J.; Binder, C.; Moerer, O. Platelet function disturbance during Veno-Venous ECMO in ARDS patients assessed by multiple electrode aggregometry—A prospective, observational cohort study. J. Clin. Med. 2019, 8, 1056. [Google Scholar] [CrossRef] [PubMed]
- Balle, C.M.; Jeppesen, A.N.; Christensen, S.; Hvas, A.-M. Platelet function during extracorporeal membrane oxygenation in adult patients. Front. Cardiovasc. Med. 2019, 6, 114. [Google Scholar] [CrossRef] [PubMed]
- Flaujac, C.; Pouard, P.; Boutouyrie, P.; Emmerich, J.; Bachelot-Loza, C.; Lasne, D. Platelet dysfunction after normothermic cardiopulmonary bypass in children: Effect of high-dose aprotinin. Thromb. Haemost. 2007, 98, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Kalbhenn, J.; Schlagenhauf, A.; Rosenfelder, S.; Schmutz, A.; Zieger, B. Acquired von Willebrand syndrome and impaired platelet function during venovenous extracorporeal membrane oxygenation: Rapid onset and fast recovery. J. Heart Lung Transplant. 2018, 37, 985–991. [Google Scholar] [CrossRef]
- Millar, J.E.; Fanning, J.P.; McDonald, C.I.; McAuley, D.F.; Fraser, J.F. The inflammatory response to extracorporeal membrane oxygenation (ECMO): A review of the pathophysiology. Crit. Care 2016, 20, 387. [Google Scholar] [CrossRef]
- Al-Fares, A.; Pettenuzzo, T.; Del Sorbo, L. Extracorporeal life support and systemic inflammation. Intensive Care Med. Exp. 2019, 7, 46. [Google Scholar] [CrossRef]
- Rondina, M.T.; Zimmerman, G.A. 28-the role of platelets in inflammation. In Platelets, 4th ed.; Michelson, A.D., Ed.; Elsevier: London, UK, 2019; pp. 505–522. ISBN 978-0-12-813456-6. [Google Scholar]
- Cognasse, F.; Laradi, S.; Berthelot, P.; Bourlet, T.; Marotte, H.; Mismetti, P.; Garraud, O.; Hamzeh-Cognasse, H. Platelet inflammatory response to stress. Front. Immunol. 2019, 10, 1478. [Google Scholar] [CrossRef]
- Rossaint, J.; Margraf, A.; Zarbock, A. Role of platelets in leukocyte recruitment and resolution of inflammation. Front. Immunol. 2018, 9, 2712. [Google Scholar] [CrossRef]
- Schrottmaier, W.C.; Kral, J.B.; Badrnya, S.; Assinger, A. Aspirin and P2Y12 Inhibitors in platelet-mediated activation of neutrophils and monocytes. Thromb. Haemost. 2015, 114, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Loguinova, M.; Pinegina, N.; Kogan, V.; Vagida, M.; Arakelyan, A.; Shpektor, A.; Margolis, L.; Vasilieva, E. Monocytes of different subsets in complexes with platelets in patients with myocardial infarction. Thromb. Haemost. 2018, 118, 1969–1981. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Smyth, S. Interactions between platelets, leukocytes, and the endothelium. In Platelets; Michelson, A.D., Ed.; Elsevier: London, UK, 2019; pp. 295–305. [Google Scholar]
- Graham, S.M.; Liles, W.C. Platelets in sepsis: Beyond hemostasis. Blood 2016, 127, 2947–2949. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, V.; Totani, L.; Manfredi, A.A.; Maugeri, N. Platelet–leukocyte interactions. In Platelets in Thrombotic and Non-Thrombotic Disorders; Gresele, P., Kleiman, N.S., Lopez, J.A., Page, C.P., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 407–433. ISBN 978-3-319-47460-1. [Google Scholar]
- Thomas, M.R.; Storey, R.F. The role of platelets in inflammation. Thromb. Haemost. 2015, 114, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Ivert, T.; Dalén, M.; Ander, C.; Stålesen, R.; Lordkipanidzé, M.; Hjemdahl, P. Increased platelet reactivity and platelet-leukocyte aggregation after elective coronary bypass surgery. Platelets 2019, 30, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Finsterbusch, M.; Schrottmaier, W.C.; Kral-Pointner, J.B.; Salzmann, M.; Assinger, A. Measuring and interpreting platelet-leukocyte aggregates. Platelets 2018, 29, 677–685. [Google Scholar] [CrossRef]
- Rossaint, J.; Herter, J.M.; van Aken, H.; Napirei, M.; Döring, Y.; Weber, C.; Soehnlein, O.; Zarbock, A. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood 2014, 123, 2573–2584. [Google Scholar] [CrossRef]
- Grommes, J.; Alard, J.-E.; Drechsler, M.; Wantha, S.; Mörgelin, M.; Kuebler, W.M.; Jacobs, M.; von Hundelshausen, P.; Markart, P.; Wygrecka, M.; et al. Disruption of platelet-derived chemokine heteromers prevents neutrophil extravasation in acute lung injury. Am. J. Respir. Crit. Care Med. 2012, 185, 628–636. [Google Scholar] [CrossRef]
- Freynhofer, M.K.; Gruber, S.C.; Grove, E.L.; Weiss, T.W.; Wojta, J.; Huber, K. Antiplatelet drugs in patients with enhanced platelet turnover: Biomarkers versus platelet function testing. Thromb. Haemost. 2015, 114, 459–468. [Google Scholar] [CrossRef]
- Liverani, E.; Kilpatrick, L.E.; Tsygankov, A.Y.; Kunapuli, S.P. The role of P2Y12 receptor and activated platelets during inflammation. Curr. Drug. Targets 2014, 15, 720–728. [Google Scholar] [CrossRef]
- Liverani, E.; Rico, M.C.; Tsygankov, A.Y.; Kilpatrick, L.E.; Kunapuli, S.P. P2Y12 receptor modulates sepsis-induced inflammation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Storey, R.F.; James, S.K.; Siegbahn, A.; Varenhorst, C.; Held, C.; Ycas, J.; Husted, S.E.; Cannon, C.P.; Becker, R.C.; Steg, P.G.; et al. Lower mortality following pulmonary adverse events and sepsis with ticagrelor compared to clopidogrel in the PLATO study. Platelets 2014, 25, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Nylander, S.; Schulz, R. Effects of P2Y 12 receptor antagonists beyond platelet inhibition-comparison of ticagrelor with thienopyridines: P2Y 12 receptors beyond platelet inhibition. Br. J. Pharmacol. 2016, 173, 1163–1178. [Google Scholar] [CrossRef] [PubMed]
- Lancellotti, P.; Musumeci, L.; Jacques, N.; Servais, L.; Goffin, E.; Pirotte, B.; Oury, C. Antibacterial activity of ticagrelor in conventional antiplatelet dosages against antibiotic-resistant gram-positive bacteria. JAMA Cardiol. 2019, 4, 596–599. [Google Scholar] [CrossRef]
- Rinder, H.M.; Bonan, J.L.; Rinder, C.S.; Ault, K.A.; Smith, B.R. Activated and unactivated platelet adhesion to monocytes and neutrophils. Blood 1991, 78, 1760–1769. [Google Scholar] [CrossRef]
- Steiger, T.; Foltan, M.; Philipp, A.; Mueller, T.; Gruber, M.; Bredthauer, A.; Krenkel, L.; Birkenmaier, C.; Lehle, K. Accumulations of von Willebrand factor within ECMO oxygenators: Potential indicator of coagulation abnormalities in critically ill patients? Artif. Organs 2019, 43, 1065–1076. [Google Scholar] [CrossRef]
- Li, N.; Goodall, A.H.; Hjemdahl, P. A sensitive flow cytometric assay for circulating platelet-leucocyte aggregates. Br. J. Haematol. 1997, 99, 808–816. [Google Scholar] [CrossRef]
- Carubbi, C.; Masselli, E.; Vitale, M. Flow Cytometry. In Platelets in Thrombotic and Non-Thrombotic Disorders: Pathophysiology, Pharmacology and Therapeutics: An Update; Gresele, P., Kleiman, N.S., Lopez, J.A., Page, C.P., Eds.; Springer International Publishing: Cham, Switwerland, 2017; pp. 589–617. ISBN 978-3-319-47462-5. [Google Scholar]
- Nagasawa, A.; Matsuno, K.; Tamura, S.; Hayasaka, K.; Shimizu, C.; Moriyama, T. The basis examination of leukocyte-platelet aggregates with CD45 gating as a novel platelet activation marker. Int. J. Lab. Hematol. 2013, 35, 534–541. [Google Scholar] [CrossRef]
- Gerrits, A.J.; Frelinger, A.L.; Michelson, A.D. Whole blood analysis of leukocyte-platelet aggregates. Curr. Protoc. Cytom. 2016, 78, 6–15. [Google Scholar] [CrossRef]
- Simon, D.I.; Chen, Z.; Xu, H.; Li, C.Q.; Dong, J.F.; McIntire, L.V.; Ballantyne, C.M.; Zhang, L.; Furman, M.I.; Berndt, M.C.; et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J. Exp. Med. 2000, 192, 193–204. [Google Scholar] [CrossRef]
- Harding, S.A.; Din, J.N.; Sarma, J.; Jessop, A.; Weatherall, M.; Fox, K.A.A.; Newby, D.E. Flow cytometric analysis of circulating platelet-monocyte aggregates in whole blood: Methodological considerations. Thromb. Haemost. 2007, 98, 451–456. [Google Scholar] [PubMed]
- Gachet, C.; Hanau, D.; Spehner, D.; Brisson, C.; Garaud, J.C.; Schmitt, D.A.; Ohlmann, P.; Cazenave, J.P. Alpha IIb beta 3 integrin dissociation induced by EDTA results in morphological changes of the platelet surface-connected canalicular system with differential location of the two separate subunits. J. Cell Biol. 1993, 120, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Bihari, P.; Fent, J.; Hamar, J.; Furész, J.; Lakatos, S. An easy-to-use practical method to measure coincidence in the flow cytometer--the case of platelet-granulocyte complex determination. J. Biochem. Biophys. Methods 2008, 70, 1080–1085. [Google Scholar] [CrossRef] [PubMed]
- Fent, J.; Bihari, P.; Furész, J.; Hamar, J.; Lakatos, S. Impact of coincidence on granulocyte-platelet complex determination by flow cytometry is evaluated by a novel computer simulation model of coincidence. J. Biochem. Biophys. Methods 2008, 70, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Poncelet, P.; Robert, S.; Bailly, N.; Garnache-Ottou, F.; Bouriche, T.; Devalet, B.; Segatchian, J.H.; Saas, P.; Mullier, F. Tips and tricks for flow cytometry-based analysis and counting of microparticles. Transfus. Apher. Sci. 2015, 53, 110–126. [Google Scholar] [CrossRef] [PubMed]
- Majumder, B.; North, J.; Mavroudis, C.; Rakhit, R.; Lowdell, M.W. Improved accuracy and reproducibility of enumeration of platelet-monocyte complexes through use of doublet-discriminator strategy. Cytom. B Clin. Cytom. 2012, 82, 353–359. [Google Scholar] [CrossRef]
- Hui, H.; Fuller, K.A.; Chuah, H.; Liang, J.; Sidiqi, H.; Radeski, D.; Erber, W.N. Imaging flow cytometry to assess chromosomal abnormalities in chronic lymphocytic leukaemia. Methods 2018, 134, 32–40. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type of ECMO and PumpNumber of Patients Study Reference | Blood Collection | Results |
|---|---|---|
| Centrifugal pump Jostra pumphead, Maquet® VA n = 7; VV n = 3 Nair et al. [12] | During ECMO. Citrated whole blood. | Patients with platelet count > 100 × 109/L 50% to 72% of patients within low range of ADP, TRAP, collagen aggregation, and ristocetin agglutination |
| VA n = 26; VV n = 12 Tauber et al. [13] | Before, after 24 h and 48 h initiation of ECMO therapy and 24 h after ECMO termination. Hirudin anticoagulated whole blood. | 30% to 40% decrease in aggregation with ADP, TRAP, AA after 24 h/baseline. After 48 h, aggregation with TRAP = baseline, ADP and AA lower/baseline Return to baseline after 24 h |
| ROTAFLOW® or CARDIOHELP® centrifugal pumps. VV n = 20 Wand et al. [14] | Before, 6 h, 1, 2, 3 and 7 days after the start of ECMO therapy. Whole blood sample on heparin-anticoagulated and calcium-balanced tubes. | Patients with platelet count > 70 × 109/L. Reduced platelet aggregation with ADP, TRAP, AA 6 h after ECMO initiation/before, spontaneous recovery on day 2 with values exceeding baseline afterwards |
| VA n = 23; VV n = 10 Balle et al. [15] | Every day from day 1 to day 7 in 33 patients. Hirudin anticoagulated whole blood. | Platelet aggregation with ADP, TRAP, AA: lower compared to healthy volunteers from day 1 up to day 7 but similar when analyzed relative to platelet count |
| Sample Handling and Preparation | |
|---|---|
| Anticoagulant in blood sampling tube | Preferably CTAD (other option: sodium citrate) |
| Storage temperature | Room temperature |
| Centrifugation before immunolabeling | None (whole blood protocol) |
| Prefixation before immunolabeling | None |
| Erythrocyte lysis before immunolabeling | None |
| Processing time before immunolabeling | Fixed time to allow comparative analysis As short as possible, preferably < 1 h after blood withdrawal |
| Fixation after immunolabeling | Optional, paraformaldehyde fixation (0.5 to 1%) |
| Final blood dilution | ≥1:40 |
| Erythrocyte lysis after immunolabeling | Optional |
| Processing time after immunolabeling | Without delay if no fixation 24 h at 4 °C, after fixation |
| Cytometric flow rates | Low to medium, determined in each laboratory |
| Immunolabeling | |
| Platelet-specific antibody | Preferably: CD41 Other option: CD61 |
| Leukocyte-specific antibody | CD45 |
| Leukocyte subtype-specific antibody | Neutrophils: CD66b |
| Monocytes: CD14 | |
| T-cells: CD3, CD4 and CD8 | |
| B-cells: CD19 | |
| NK-cells: CD56 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansour, A.; Roussel, M.; Gaussem, P.; Nédelec-Gac, F.; Pontis, A.; Flécher, E.; Bachelot-Loza, C.; Gouin-Thibault, I. Platelet Functions During Extracorporeal Membrane Oxygenation. Platelet–Leukocyte Aggregates Analyzed by Flow Cytometry as a Promising Tool to Monitor Platelet Activation. J. Clin. Med. 2020, 9, 2361. https://doi.org/10.3390/jcm9082361
Mansour A, Roussel M, Gaussem P, Nédelec-Gac F, Pontis A, Flécher E, Bachelot-Loza C, Gouin-Thibault I. Platelet Functions During Extracorporeal Membrane Oxygenation. Platelet–Leukocyte Aggregates Analyzed by Flow Cytometry as a Promising Tool to Monitor Platelet Activation. Journal of Clinical Medicine. 2020; 9(8):2361. https://doi.org/10.3390/jcm9082361
Chicago/Turabian StyleMansour, Alexandre, Mikael Roussel, Pascale Gaussem, Fabienne Nédelec-Gac, Adeline Pontis, Erwan Flécher, Christilla Bachelot-Loza, and Isabelle Gouin-Thibault. 2020. "Platelet Functions During Extracorporeal Membrane Oxygenation. Platelet–Leukocyte Aggregates Analyzed by Flow Cytometry as a Promising Tool to Monitor Platelet Activation" Journal of Clinical Medicine 9, no. 8: 2361. https://doi.org/10.3390/jcm9082361
APA StyleMansour, A., Roussel, M., Gaussem, P., Nédelec-Gac, F., Pontis, A., Flécher, E., Bachelot-Loza, C., & Gouin-Thibault, I. (2020). Platelet Functions During Extracorporeal Membrane Oxygenation. Platelet–Leukocyte Aggregates Analyzed by Flow Cytometry as a Promising Tool to Monitor Platelet Activation. Journal of Clinical Medicine, 9(8), 2361. https://doi.org/10.3390/jcm9082361