Endothelial Dysfunction in Chronic Kidney Disease, from Biology to Clinical Outcomes: A 2020 Update




{kind=link}
{kind=link}
Abstract
1. Introduction
2. Basic Molecular Biochemistry of the NO System
3. Major Factors Modulating Bioavailability of NO
3.1. eNOS, Caveolae, and Ox-LDL
3.2. Age and Estrogens
3.3. Shear Stress
3.4. BH4 and L-Arginine
3.5. Asymmetric Dimethylarginine (ADMA)
3.6. Oxidative Stress
3.7. Adipokines
3.8. cGMP
4. Methods of Assessment of Endothelial Function
4.1. Functional Methods
4.2. Biomarkers of Endothelial Biology
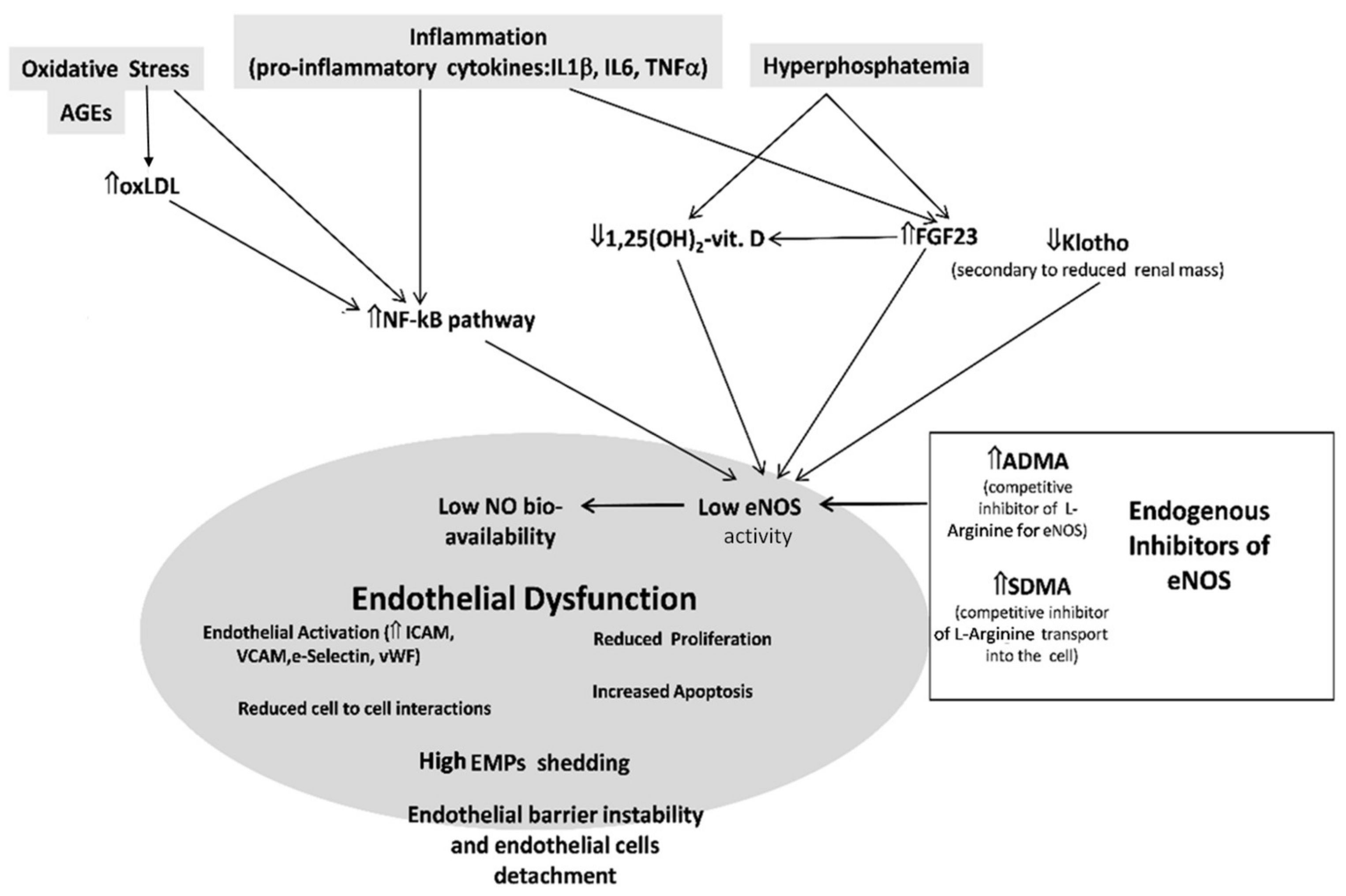
5. The Endothelium in CKD
6. Endogenous Inhibitors of Nitric Oxide Synthase in CKD
7. Protecting Endothelial Health in CKD
7.1. Lipid-Lowering Drugs
7.2. Vitamins C and E
7.3. L-Arginine
7.4. Vitamin D
7.5. Klotho, FGF23, and Phosphate Binders
7.6. Antihypertensive and Antidiabetic Agents
7.7. Renal Transplantation
7.8. Novel Therapeutic Approaches
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.E.; Tang, K.; Yuan, C.-W.; et al. Forecasting life expectancy, years of life lost, and all-Cause and cause-Specific mortality for 250 causes of death: Reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet (Lond. Engl.) 2018, 392, 2052–2090. [Google Scholar] [CrossRef]
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication-Worldwide more than 850 million individuals have kidney diseases. Nephrol. Dial. Transplant. 2019, 34, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.; Matsushita, K.; Abate, K.H.; Al-Aly, Z.; Ärnlöv, J.; Asayama, K.; Atkins, R.; Badawi, A.; Ballew, S.H.; Banerjee, A.; et al. Global Cardiovascular and Renal Outcomes of Reduced GFR. J. Am. Soc. Nephrol. 2017, 28, 2167–2179. [Google Scholar] [CrossRef] [PubMed]
- Weiner, D.E.; Tighiouart, H.; Elsayed, E.F.; Griffith, J.L.; Salem, D.N.; Levey, A.S.; Sarnak, M.J. The Framingham Predictive Instrument in Chronic Kidney Disease. J. Am. Coll. Cardiol. 2007, 50, 217–224. [Google Scholar] [CrossRef]
- Zoccali, C. Traditional and emerging cardiovascular and renal risk factors: An epidemiologic perspective. Kidney Int. 2006, 70, 26–33. [Google Scholar] [CrossRef]
- Zoccali, C.; Vanholder, R.; Massy, Z.A.; Ortiz, A.; Sarafidis, P.; Dekker, F.W.; Fliser, D.; Fouque, D.; Heine, G.H.; Jager, K.J.; et al. The systemic nature of CKD. Nat. Rev. Nephrol. 2017, 13, 344–358. [Google Scholar] [CrossRef]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and clinical impact of organic uremic retention solutes: A comprehensive update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Yilmaz, M.I.M.I.; Saglam, M.; Caglar, K.; Cakir, E.; Sonmez, A.; Ozgurtas, T.; Aydin, A.; Eyileten, T.; Ozcan, O.; Acikel, C.; et al. The determinants of endothelial dysfunction in CKD: Oxidative stress and asymmetric dimethylarginine. Am. J. Kidney Dis. 2006, 47, 42–50. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Zhao, Y.; Xu, A.; Leung, S.W.S. Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circ. Res. 2016, 119, 375–396. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H.C. Endothelial dysfunction and vascular disease—A 30th anniversary update. Acta Physiol. 2017, 219, 22–96. [Google Scholar] [CrossRef] [PubMed]
- Heiss, E.; Dirsch, V. Regulation of eNOS Enzyme Activity by Posttranslational Modification. Curr. Pharm. Des. 2014, 20, 3503–3513. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.M.; Morishima, Y.; Pratt, W.B.; Osawa, Y. Modulation of heme/substrate binding cleft of neuronal nitric-Oxide synthase (nNOS) regulates binding of Hsp90 and Hsp70 proteins and nNOS ubiquitination. J. Biol. Chem. 2012, 287, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Blair, A.; Shaul, P.W.; Yuhanna, I.S.; Conrad, P.A.; Smart, E.J. Oxidized low density lipoprotein displaces endothelial nitric-Oxide synthase (eNOS) from plasmalemmal caveolae and impairs eNOS activation. J. Biol. Chem. 1999, 274, 32512–32519. [Google Scholar] [CrossRef] [PubMed]
- Gharavi, N.M.; Baker, N.A.; Mouillesseaux, K.P.; Yeung, W.; Honda, H.M.; Hsieh, X.; Yeh, M.; Smart, E.J.; Berliner, J.A. Role of endothelial nitric oxide synthase in the regulation of SREBP activation by oxidized phospholipids. Circ. Res. 2006, 98, 768–776. [Google Scholar] [CrossRef]
- Puccetti, L.; Sawamura, T.; Pasqui, A.L.; Pastorelli, H.; Auteri, A.; Bruni, F. Atorvastatin reduces platelet-oxidized-LDL receptor expression in hypercholesterolaemic patients. Eur. J. Clin. Investig. 2005, 35, 47–51. [Google Scholar] [CrossRef]
- Kinlay, S.; Libby, P.; Ganz, P. Endothelial function and coronary artery disease. Curr. Opin. Lipidol. 2001, 12, 383–389. [Google Scholar] [CrossRef]
- Nawrot, T.S.; Staessen, J.A.; Holvoet, P.; Struijker-Boudier, H.A.; Schiffers, P.; Van Bortel, L.M.; Fagard, R.H.; Gardner, J.P.; Kimura, M.; Aviv, A. Telomere length and its associations with oxidized-LDL, carotid artery distensibility and smoking. Front. Biosci. Elit. 2010, 2, 1164–1168. [Google Scholar]
- van der Zwan, L.P.; Teerlink, T.; Dekker, J.M.; Henry, R.M.A.; Stehouwer, C.D.A.; Jakobs, C.; Heine, R.J.; Scheffer, P.G. Circulating oxidized LDL: Determinants and association with brachial flow-Mediated dilation. J. Lipid Res. 2009, 50, 342–349. [Google Scholar] [CrossRef]
- Demir, M.; Kucuk, A.; Sezer, M.T.; Altuntas, A.; Kaya, S. Malnutrition-inflammation score and endothelial dysfunction in hemodialysis patients. J. Ren. Nutr. 2010, 20, 377–383. [Google Scholar] [CrossRef]
- Walker, A.E.; Kaplon, R.E.; Lucking, S.M.S.; Russell-Nowlan, M.J.; Eckel, R.H.; Seals, D.R. Fenofibrate improves vascular endothelial function by reducing oxidative stress while increasing endothelial nitric oxide synthase in healthy normolipidemic older adults. Hypertension 2012, 60, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Yubero-Serrano, E.M.; Delgado-Casado, N.; Delgado-Lista, J.; Perez-Martinez, P.; Tasset-Cuevas, I.; Santos-Gonzalez, M.; Caballero, J.; Garcia-Rios, A.; Marin, C.; Gutierrez-Mariscal, F.M.; et al. Postprandial antioxidant effect of the Mediterranean diet supplemented with coenzyme Q 10 in elderly men and women. Age (Omaha) 2011, 33, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Merino, J.; Ferré, R.; Girona, J.; Aguas, D.; Cabré, A.; Plana, N.; Vinuesa, A.; Ibarretxe, D.; Basora, J.; Buixadera, C.; et al. Even low physical activity levels improve vascular function in overweight and obese postmenopausal women. Menopause 2013, 20, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, R.E.; Gano, L.B.; Seals, D.R. Vascular endothelial function and oxidative stress are related to dietary niacin intake among healthy middle-Aged and older adults. J. Appl. Physiol. 2014, 116, 156–163. [Google Scholar] [CrossRef]
- Orem, A.; Yucesan, F.B.; Orem, C.; Akcan, B.; Kural, B.V.; Alasalvar, C.; Shahidi, F. Hazelnut-Enriched diet improves cardiovascular risk biomarkers beyond a lipid-Lowering effect in hypercholesterolemic subjects. J. Clin. Lipidol. 2013, 7, 123–131. [Google Scholar] [CrossRef]
- Mineo, C.; Deguchi, H.; Griffin, J.H.; Shaul, P.W. Endothelial and antithrombotic actions of HDL. Circ. Res. 2006, 98, 1352–1364. [Google Scholar] [CrossRef]
- Li, X.P.; Zhao, S.P.; Zhang, X.Y.; Liu, L.; Gao, M.; Zhou, Q.C. Protective effect of high density lipoprotein on endothelium-Dependent vasodilatation. Int. J. Cardiol. 2000, 73, 231–236. [Google Scholar] [CrossRef]
- Kuvin, J.T.; Patel, A.R.; Sidhu, M.; Rand, W.M.; Sliney, K.A.; Pandian, N.G.; Karas, R.H. Relation between high-Density lipoprotein cholesterol and peripheral vasomotor function. Am. J. Cardiol. 2003, 92, 275–279. [Google Scholar] [CrossRef]
- Kuvin, J.T.; Rämet, M.E.; Patel, A.R.; Pandian, N.G.; Mendelsohn, M.E.; Karas, R.H. A novel mechanism for the beneficial vascular effects of high-density lipoprotein cholesterol: Enhanced vasorelaxation and increased endothelial nitric oxide synthase expression. Am. Heart J. 2002, 144, 165–172. [Google Scholar] [CrossRef][Green Version]
- Bisoendial, R.J.; Hovingh, G.K.; Levels, J.H.M.; Lerch, P.G.; Andresen, I.; Hayden, M.R.; Kastelein, J.J.P.; Stroes, E.S.G. Restoration of endothelial function by increasing high-Density lipoprotein in subjects with isolated low high-Density lipoprotein. Circulation 2003, 107, 2944–2948. [Google Scholar] [CrossRef]
- Spieker, L.E.; Sudano, I.; Hürlimann, D.; Lerch, P.G.; Lang, M.G.; Binggeli, C.; Corti, R.; Ruschitzka, F.; Lüscher, T.F.; Noll, G. High-Density lipoprotein restores endothelial function in hypercholesterolemic men. Circulation 2002, 105, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Shaul, P.W.; Anderson, R.G.W. Role of plasmalemmal caveolae in signal transduction. Am. J. Physiol. - Lung Cell. Mol. Physiol. 1998, 275, L843–L851. [Google Scholar] [CrossRef] [PubMed]
- Chambliss, K.L.; Yuhanna, I.S.; Mineo, C.; Liu, P.; German, Z.; Sherman, T.S.; Mendelsohn, M.E.; Anderson, R.G.; Shaul, P.W. Estrogen receptor alpha and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae. Circ. Res. 2000, 87, e44–e52. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.J.; Ceravolo, G.S.; Dos Santos, R.A.; De Oliveira, M.A.; Araújo, P.X.; Giaquinto, L.R.; Tostes, R.C.; Akamine, E.H.; Fortes, Z.B.; Dantas, A.P.; et al. Association of testosterone with estrogen abolishes the beneficial effects of estrogen treatment by increasing ROS generation in aorta endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, 723–732. [Google Scholar] [CrossRef]
- Zuloaga, K.L.; Davis, C.M.; Zhang, W.; Alkayed, N.J. Role of aromatase in sex-specific cerebrovascular endothelial function in mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H929–H937. [Google Scholar] [CrossRef]
- Harris, R.A.; Tedjasaputra, V.; Zhao, J.; Richardson, R.S. Premenopausal women exhibit an inherent protection of endothelial function following a high-fat meal. Reprod. Sci. 2012, 19, 221–228. [Google Scholar] [CrossRef]
- Moreau, K.L.; Meditz, A.; Deane, K.D.; Kohrt, W.M. Tetrahydrobiopterin improves endothelial function and decreases arterial stiffness in estrogen-Deficient postmenopausal women. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1211–H1218. [Google Scholar] [CrossRef]
- Yan, C.; Huang, A.; Kaley, G.; Sun, D. Chronic high blood flow potentiates shear stress-Induced release of NO in arteries of aged rats. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3105–H3110. [Google Scholar] [CrossRef]
- Bellien, J.; Iacob, M.; Gutierrez, L.; Isabelle, M.; Lahary, A.; Thuillez, C.; Joannides, R. Crucial role of NO and endothelium-Derived hyperpolarizing factor in human sustained conduit artery flow-Mediated dilatation. Hypertension 2006, 48, 1088–1094. [Google Scholar] [CrossRef]
- Babbitt, D.M.; Kim, J.S.; Forrester, S.J.; Brown, M.D.; Park, J.Y. Effect of interleukin-10 and laminar shear stresson endothelial nitric oxide synthase and nitric oxide in African American human umbilical vein endothelial cells. Ethn. Dis. 2015, 25, 413–418. [Google Scholar] [CrossRef]
- Bevan, H.S.; Slater, S.C.; Clarke, H.; Cahill, P.A.; Mathieson, P.W.; Welsh, G.I.; Satchell, S.C. Acute laminar shear stress reversibly increases human glomerular endothelial cell permeability via activation of endothelial nitric oxide synthase. Am. J. Physiol. Ren. Physiol. 2011, 301, F733–F742. [Google Scholar] [CrossRef] [PubMed]
- Bergaya, S.; Hilgers, R.H.P.; Meneton, P.; Dong, Y.; Bloch-Faure, M.; Inagami, T.; Alhenc-Gelas, F.; Lévy, B.I.; Boulanger, C.M. Flow-Dependent dilation mediated by endogenous kinins requires angiotensin AT2 receptors. Circ. Res. 2004, 94, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Gibbons, G.H.; Dzau, V.J.; Cooke, J.P. Shear stress elevates endothelial cGMP: Role of a potassium channel and G protein coupling. Circulation 1993, 88, 193–197. [Google Scholar] [CrossRef]
- Perez-Aguilar, S.; Torres-Tirado, D.; Martell-Gallegos, G.; Velarde-Salcedo, J.; Barba-de la Rosa, A.P.; Knabb, M.; Rubio, R.G. Protein-Coupled receptors mediate coronary flow- and agonist-Induced responses via lectin-Oligosaccharide interactions. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H699–H708. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.B.; Laughlin, M.H. Modulation of endothelial cell phenotype by physical activity: Impact on obesity-related endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1–H8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, M.; Xue, S.J.; Liu, D.H.; Tang, Y.B. Simvastatin ameliorates angiotensin II-Induced endothelial dysfunction through restoration of Rho-BH4-eNOS-NO pathway. Cardiovasc. Drugs Ther. 2012, 26, 31–40. [Google Scholar] [CrossRef]
- Chuaiphichai, S.; McNeill, E.; Douglas, G.; Crabtree, M.J.; Bendall, J.K.; Hale, A.B.; Alp, N.J.; Channon, K.M. Cell-Autonomous role of endothelial GTP cyclohydrolase 1 and tetrahydrobiopterin in blood pressure regulation. Hypertension 2014, 64, 530–540. [Google Scholar] [CrossRef]
- Porkert, M.; Sher, S.; Reddy, U.; Cheema, F.; Niessner, C.; Kolm, P.; Jones, D.P.; Hooper, C.; Taylor, W.R.; Harrison, D.; et al. Tetrahydrobiopterin: A novel antihypertensive therapy. J. Hum. Hypertens. 2008, 22, 401–407. [Google Scholar] [CrossRef]
- Maier, W.; Cosentino, F.; Lütolf, R.B.; Fleisch, M.; Seiler, C.; Hess, O.M.; Meier, B.; Lüscher, T.F. Tetrahydrobiopterin improves endothelial function in patients with coronary artery disease. J. Cardiovasc. Pharmacol. 2000, 35, 173–178. [Google Scholar] [CrossRef]
- Cosentino, F.; Hürlimann, D.; Delli Gatti, C.; Chenevard, R.; Blau, N.; Alp, N.J.; Channon, K.M.; Eto, M.; Lerch, P.; Enseleit, F.; et al. Chronic treatment with tetrahydrobiopterin reverses endothelial dysfunction and oxidative stress in hypercholesterolaemia. Heart 2008, 94, 487–492. [Google Scholar] [CrossRef]
- Heitzer, T.; Krohn, K.; Albers, S.; Meinertz, T. Tetrahydrobiopterin improves endothelium-Dependent vasodilation by increasing nitric oxide activity in patients with Type II diabetes mellitus. Diabetologia 2000, 43, 1435–1438. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Emrick, L.T.; Craigen, W.J.; Scaglia, F. Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol. Genet. Metab. 2012, 107, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Perticone, F.; Sciacqua, A.; Maio, R.; Perticone, M.; Maas, R.; Boger, R.H.R.H.; Tripepi, G.; Sesti, G.; Zoccali, C. Asymmetric dimethylarginine, L-Arginine, and endothelial dysfunction in essential hypertension. J. Am. Coll. Cardiol. 2005, 46, 518–523. [Google Scholar] [CrossRef]
- Lei, J.; Vodovotz, Y.; Tzeng, E.; Billiar, T.R. Nitric oxide, A protective molecule in the cardiovascular system. Nitric Oxide 2013, 35, 175–185. [Google Scholar] [CrossRef]
- Wilson, A.M.; Harada, R.; Nair, N.; Balasubramanian, N.; Cooke, J.P. L-Arginine supplementation in peripheral arterial disease: No benefit and possible harm. Circulation 2007, 116, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Scalera, F.; Closs, E.I.; Flick, E.; Martens-Lobenhoffer, J.; Boissel, J.P.; Lendeckel, U.; Heimburg, A.; Bode-Böger, S.M. Paradoxical effect of L-Arginine: Acceleration of endothelial cell senescence. Biochem. Biophys. Res. Commun. 2009, 386, 650–655. [Google Scholar] [CrossRef]
- Böger, R.H.; Zoccali, C. ADMA: A novel risk factor that explains excess cardiovascular event rate in patients with end-Stage renal disease. Atheroscler. Suppl. 2003, 4, 23–28. [Google Scholar] [CrossRef]
- Vallance, P.; Leiper, J. Cardiovascular biology of the asymmetric dimethylarginine: Dimethylarginine dimethylaminohydrolase pathway. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1023–1030. [Google Scholar] [CrossRef]
- Wilcox, C.S. Asymmetric dimethylarginine and reactive oxygen species: Unwelcome twin visitors to the cardiovascular and kidney disease tables. Hypertension 2012, 59, 375–381. [Google Scholar] [CrossRef]
- Dowsett, L.; Piper, S.; Slaviero, A.; Dufton, N.; Wang, Z.; Boruc, O.; Delahaye, M.; Colman, L.; Kalk, E.; Tomlinson, J.; et al. Endothelial dimethylarginine dimethylaminohydrolase 1 is an important regulator of angiogenesis but does not regulate vascular reactivity or hemodynamic homeostasis. Circulation 2015, 131, 2217–2225. [Google Scholar] [CrossRef]
- Zhang, Y.; Janssens, S.P.; Wingler, K.; Schmidt, H.H.H.W.; Moens, A.L. Modulating endothelial nitric oxide synthase: A new cardiovascular therapeutic strategy. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H634–H646. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- Solzbach, U.; Hornig, B.; Jeserich, M.; Just, H. Vitamin C improves endothelial dysfunction of epicardial coronary arteries in hypertensive patients. Circulation 1997, 96, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Erbs, S.; Gielen, S.; Linke, A.; Möbius-Winkler, S.; Adams, V.; Baither, Y.; Schuler, G.; Hambrecht, R. Improvement of peripheral endothelial dysfunction by acute vitamin C application: Different effects in patients with coronary artery disease, ischemic, and dilated cardiomyopathy. Am. Heart J. 2003, 146, 280–285. [Google Scholar] [CrossRef]
- Heitzer, T.; Just, H.; Münzel, T. Antioxidant Vitamin C Improves Endothelial Dysfunction in Chronic Smokers. Circulation 1996, 94, 6–9. [Google Scholar] [CrossRef]
- On, Y.K.; Kim, C.H.; Sohn, D.W.; Oh, B.H.; Lee, M.M.; Park, Y.B.; Choi, Y.S. Improvement of endothelial function by amlodipine and vitamin C in essential hypertension. Korean J. Intern. Med. 2002, 17, 131–137. [Google Scholar] [CrossRef]
- Ren, X.; Ren, L.; Wei, Q.; Shao, H.; Chen, L.; Liu, N. Advanced glycation end-Products decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Cardiovasc. Diabetol. 2017, 16, 52. [Google Scholar] [CrossRef]
- Stinghen, A.E.M.; Massy, Z.A.; Vlassara, H.; Striker, G.E.; Boullier, A. Uremic toxicity of advanced glycation end products in CKD. J. Am. Soc. Nephrol. 2016, 27, 354–370. [Google Scholar] [CrossRef]
- Ando, R.; Ueda, S.; Yamagishi, S.I.; Miyazaki, H.; Kaida, Y.; Kaifu, K.; Yokoro, M.; Nakayama, Y.; Obara, N.; Fukami, K.; et al. Involvement of advanced glycation end product-Induced asymmetric dimethylarginine generation in endothelial dysfunction. Diabetes Vasc. Dis. Res. 2013, 10, 436–441. [Google Scholar] [CrossRef]
- Kim, H.W.; Belin de Chantemèle, E.J.; Weintraub, N.L. Perivascular Adipocytes in Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2220–2227. [Google Scholar] [CrossRef]
- Shibata, R.; Ouchi, N.; Ohashi, K.; Murohara, T. The role of adipokines in cardiovascular disease. J. Cardiol. 2017, 70, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.; Schini, V.B.; Sundt, T.M.; Vanhoutte, P.M. Reduced production of cGMP underlies the loss of endothelium-Dependent relaxations in the canine basilar artery after subarachnoid hemorrhage. Circ. Res. 1992, 70, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, Z.; Leung, S.W.S.; Vanhoutte, P.M. Hypoxic Vasospasm Mediated by cIMP: When Soluble Guanylyl Cyclase Turns Bad. J. Cardiovasc. Pharmacol. 2015, 65, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Corretti, M.C.; Anderson, T.J.; Benjamin, E.J.; Ms, C.; Celermajer, D.; Charbonneau, F.; Creager, M.A.; Deanfield, J.; Drexler, H.; Gerhard-herman, M.; et al. Guidelines for the Ultrasound Assessment of Endothelial-Dependent Flow-Mediated Vasodilation of the Brachial Artery A Report of the International Brachial Artery Reactivity Task Force. J. Am. Coll. Cardiol. 2002, 39, 257–265. [Google Scholar] [CrossRef]
- Brocq, M.L.; Leslie, S.J.; Milliken, P.; Megson, I.L. Endothelial dysfunction: From molecular mechanisms to measurement, clinical implications, and therapeutic opportunities. Antioxid. Redox Signal. 2008, 10, 1631–1673. [Google Scholar] [CrossRef]
- Anderson, T.J.; Gerhard, M.D.; Meredith, I.T.; Charbonneau, F.; Delagrange, D.; Creager, M.A.; Selwyn, A.P.; Ganz, P. Systemic nature of endothelial dysfunction in atherosclerosis. Am. J. Cardiol. 1995, 75, 71B–74B. [Google Scholar] [CrossRef]
- Flammer, A.J.; Anderson, T.; Celermajer, D.S.; Creager, M.a.; Deanfield, J.; Ganz, P.; Hamburg, N.M.; Lüscher, T.F.; Shechter, M.; Taddei, S.; et al. The assessment of endothelial function: From research into clinical practice. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef]
- Nohria, A.; Kinlay, S.; Buck, J.S.; Redline, W.; Copeland-Halperin, R.; Kim, S.; Beckman, J.A. The effect of salsalate therapy on endothelial function in a broad range of subjects. J. Am. Heart Assoc. 2014, 3, e000609. [Google Scholar] [CrossRef]
- Kang, L.S.; Chen, B.; Reyes, R.A.; Leblanc, A.J.; Teng, B.; Mustafa, S.J.; Muller-Delp, J.M. Aging and estrogen alter endothelial reactivity to reactive oxygen species in coronary arterioles. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2105–H2115. [Google Scholar] [CrossRef]
- Kruger, A.; Stewart, J.; Sahityani, R.; O’Riordan, E.; Thompson, C.; Adler, S.; Garrick, R.; Vallance, P.; Goligorsky, M.S. Laser Doppler flowmetry detection of endothelial dysfunction in end-Stage renal disease patients: Correlation with cardiovascular risk. Kidney Int. 2006, 70, 157–164. [Google Scholar] [CrossRef]
- Stewart, J.; Kohen, A.; Brouder, D.; Rahim, F.; Adler, S.; Garrick, R.; Goligorsky, M.S. Noninvasive interrogation of microvasculature for signs of endothelial dysfunction in patients with chronic renal failure. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2687–H2696. [Google Scholar] [CrossRef] [PubMed]
- Strisciuglio, T.; De Luca, S.; Capuano, E.; Luciano, R.; Niglio, T.; Trimarco, B.; Galasso, G. Endothelial dysfunction: Its clinical value and methods of assessment. Curr. Atheroscler. Rep. 2014, 16, 417. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, I.B.; Webb, D.J. Venous occlusion plethysmography in cardiovascular research: Methodology and clinical applications. Br. J. Clin. Pharmacol. 2001, 52, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Blann, A.D. A reliable marker of vascular function: Does it exist? Trends Cardiovasc. Med. 2015, 25, 588–591. [Google Scholar] [CrossRef]
- Deng, F.; Wang, S.; Zhang, L. Endothelial microparticles act as novel diagnostic and therapeutic biomarkers of circulatory hypoxia-Related diseases: A literature review. J. Cell. Mol. Med. 2017, 21, 1698–1710. [Google Scholar] [CrossRef]
- Bulut, D.; Maier, K.; Bulut-Streich, N.; Börgel, J.; Hanefeld, C.; Mügge, A. Circulating Endothelial Microparticles Correlate Inversely With Endothelial Function in Patients With Ischemic Left Ventricular Dysfunction. J. Card. Fail. 2008, 14, 336–340. [Google Scholar] [CrossRef]
- Leite, A.R.; Borges-Canha, M.; Cardoso, R.; Neves, J.S.; Castro-Ferreira, R.; Leite-Moreira, A. Novel Biomarkers for Evaluation of Endothelial Dysfunction. Angiology 2020, 71, 397–410. [Google Scholar] [CrossRef]
- Kapur, N.K.; Heffernan, K.S.; Yunis, A.A.; Parpos, P.; Kiernan, M.S.; Sahasrabudhe, N.A.; Kimmelstiel, C.D.; Kass, D.A.; Karas, R.H.; Mendelsohn, M.E. Usefulness of soluble endoglin as a noninvasive measure of left ventricular filling pressure in heart failure. Am. J. Cardiol. 2010, 106, 1770–1776. [Google Scholar] [CrossRef]
- Blázquez-Medela, A.M.; García-Ortiz, L.; Gómez-Marcos, M.A.; Recio-Rodríguez, J.I.; Sánchez-Rodríguez, A.; López-Novoa, J.M.; Martínez-Salgado, C. Increased plasma soluble endoglin levels as an indicator of cardiovascular alterations in hypertensive and diabetic patients. BMC Med. 2010, 8, 86. [Google Scholar] [CrossRef]
- Canpolat, U.; Kocyigit, D.; Yildirim, A. Role of Endothelial Dysfunction and Endocan in Atherosclerosis: Point of Origin or End Point? Angiology 2020, 71, 477. [Google Scholar] [CrossRef]
- Zoccali, C. Endothelial dysfunction and the kidney: Emerging risk factors for renal insufficiency and cardiovascular outcomes in essential hypertension. J. Am. Soc. Nephrol. 2006, 17, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Fliser, D.; Wiecek, A.; Suleymanlar, G.; Ortiz, A.; Massy, Z.; Lindholm, B.; Martinez-Castelao, A.; Agarwal, R.; Jager, K.J.; Dekker, F.W.; et al. The dysfunctional endothelium in CKD and in cardiovascular disease: Mapping the origin(s) of cardiovascular problems in CKD and of kidney disease in cardiovascular conditions for a research agenda. Kidney Int. Suppl. 2011, 1, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Peyster, E.; Chen, J.; Feldman, H.I.; Go, A.S.; Gupta, J.; Mitra, N.; Pan, Q.; Porter, A.; Rahman, M.; Raj, D.; et al. Inflammation and Arterial Stiffness in Chronic Kidney Disease: Findings From the CRIC Study. Am. J. Hypertens. 2017, 30, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Dominic, E.A.; Fink, J.C.; Ojo, A.O.; Barrows, I.R.; Reilly, M.P.; Townsend, R.R.; Joffe, M.M.; Rosas, S.E.; Wolman, M.; et al. Association between Inflammation and Cardiac Geometry in Chronic Kidney Disease: Findings from the CRIC Study. PLoS ONE 2015, 10, e0124772. [Google Scholar] [CrossRef]
- Amdur, R.L.; Feldman, H.I.; Gupta, J.; Yang, W.; Kanetsky, P.; Shlipak, M.; Rahman, M.; Lash, J.P.; Townsend, R.R.; Ojo, A.; et al. Inflammation and Progression of CKD: The CRIC Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1–11. [Google Scholar] [CrossRef]
- van der Vorm, L.N.; Visser, R.; Huskens, D.; Veninga, A.; Adams, D.L.; Remijn, J.A.; Hemker, H.C.; Rensma, P.L.; van Horssen, R.; de Laat, B. Circulating active von Willebrand factor levels are increased in chronic kidney disease and end-Stage renal disease. Clin. Kidney J. 2020, 13, 72–74. [Google Scholar] [CrossRef]
- Péquériaux, N.C.; Fijnheer, R.; Gemen, E.F.; Barendrecht, A.D.; Dekker, F.W.; Krediet, R.T.; Beutler, J.J.; Boeschoten, E.W.; Roest, M. Plasma concentration of von Willebrand factor predicts mortality in patients on chronic renal replacement therapy. Nephrol. Dial. Transplant. 2012, 27, 2452–2457. [Google Scholar] [CrossRef]
- Cuenca, M.V.; Van Bezu, J.; Beelen, R.H.J.; Vervloet, M.G.; Hordijk, P.L. Stabilization of cell-Cell junctions by active Vitamin D ameliorates uraemia-Induced loss of human endothelial barrier function. Nephrol. Dial. Transplant. 2019, 34, 252–264. [Google Scholar] [CrossRef]
- Himmelfarb, J.; Stenvinkel, P.; Ikizler, T.A.; Hakim, R.M. Perspectives in renal medicine: The elephant in uremia: Oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002, 62, 1524–1538. [Google Scholar] [CrossRef]
- London, G.M.; Pannier, B.; Agharazii, M.; Guerin, A.P.; Verbeke, F.H.M.; Marchais, S.J. Forearm reactive hyperemia and mortality in end-Stage renal disease. Kidney Int. 2004, 65, 700–704. [Google Scholar] [CrossRef]
- Zoccali, C.; Tripepi, G.; Cutrupi, S.; Pizzini, P.; Mallamaci, F. Low triiodothyronine: A new facet of inflammation in end-Stage renal disease. J. Am. Soc. Nephrol. 2005, 16, 2789–2795. [Google Scholar] [CrossRef] [PubMed]
- Meuwese, C.L.; Dekkers, O.M.; Stenvinkel, P.; Dekker, F.W.; Carrero, J.J. Nonthyroidal illness and the cardiorenal syndrome. Nat. Rev. Nephrol. 2013, 9, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.I.; Sonmez, A.; Karaman, M.; Ay, S.A.; Saglam, M.; Yaman, H.; Kilic, S.; Eyileten, T.; Caglar, K.; Oguz, Y.; et al. Low triiodothyronine alters flow-Mediated vasodilatation in advanced nondiabetic kidney disease. Am. J. Nephrol. 2011, 33, 25–32. [Google Scholar] [CrossRef]
- Miyata, T.; Wada, Y.; Cai, Z.; Iida, Y.; Horie, K.; Yasuda, Y.; Maeda, K.; Kurokawa, K.; Van Ypersele De Strihou, C. Implication of an increased oxidative stress in the formation of advanced glycation end products in patients with end-Stage renal failure. Kidney Int. 1997, 51, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Linden, E.; Cai, W.; He, J.C.; Xue, C.; Li, Z.; Winston, J.; Vlassara, H.; Uribarri, J. Endothelial dysfunction in patients with chronic kidney disease results from advanced glycation end products (AGE)-Mediated inhibition of endothelial nitric oxide synthase through RAGE activation. Clin. J. Am. Soc. Nephrol. 2008, 3, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Lutgers, H.L.; Graaff, R.; De Vries, R.; Smit, A.J.; Dullaart, R.P.F. Carotid artery intima media thickness associates with skin autofluoresence in non-Diabetic subjects without clinically manifest cardiovascular disease. Eur. J. Clin. Investig. 2010, 40, 812–817. [Google Scholar] [CrossRef]
- Basta, G.; Leonardis, D.; Mallamaci, F.; Cutrupi, S.; Pizzini, P.; Gaetano, L.; Tripepi, R.; Tripepi, G.; De Caterina, R.; Zoccali, C.; et al. Circulating soluble receptor of advanced glycation end product inversely correlates with atherosclerosis in patients with chronic kidney disease. Kidney Int. 2010, 77, 225–231. [Google Scholar] [CrossRef][Green Version]
- Leonardis, D.; Basta, G.; Mallamaci, F.; Cutrupi, S.; Pizzini, P.; Tripepi, R.; Tripepi, G.; De Caterina, R.; Zoccali, C. Circulating soluble receptor for advanced glycation end product (sRAGE) and left ventricular hypertrophy in patients with chronic kidney disease (CKD). Nutr. Metab. Cardiovasc. Dis. 2012, 22, 748–755. [Google Scholar] [CrossRef]
- Mallipattu, S.K.; Uribarri, J. Advanced glycation end product accumulation: A new enemy to target in chronic kidney disease? Curr. Opin. Nephrol. Hypertens. 2014, 23, 547–554. [Google Scholar] [CrossRef]
- Ueno, H.; Koyama, H.; Tanaka, S.; Fukumoto, S.; Shinohara, K.; Shoji, T.; Emoto, M.; Tahara, H.; Kakiya, R.; Tabata, T.; et al. Skin autofluorescence, a marker for advanced glycation end product accumulation, is associated with arterial stiffness in patients with end-Stage renal disease. Metabolism 2008, 57, 1452–1457. [Google Scholar] [CrossRef]
- Zoccali, C.; Mallamaci, F.; Asahia, K.; Benedetto, F.A.A.; Tripepi, G.; Tripepi, R.; Nicocia, G.; Buemi, M.; Miyata, T. Pentosidine, carotid atherosclerosis and alterations in left ventricular geometry in hemodialysis patients. J. Nephrol. 2001, 14, 293–298. [Google Scholar] [PubMed]
- Dalan, R.; Liew, H.; Tan, W.K.A.; Chew, D.E.K.; Leow, M.K.S. Vitamin D and the endothelium: Basic, translational and clinical research updates. IJC Metab. Endocr. 2014, 4, 4–17. [Google Scholar] [CrossRef]
- Zoccali, C.; Curatola, G.; Panuccio, V.; Tripepi, R.; Pizzini, P.; Versace, M.; Bolignano, D.; Cutrupi, S.; Politi, R.; Tripepi, G.; et al. Paricalcitol and endothelial function in chronic kidney disease trial. Hypertens 2014, 64, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Lundwall, K.; Jörneskog, G.; Jacobson, S.H.; Spaak, J. Paricalcitol, Microvascular and Endothelial Function in Non-Diabetic Chronic Kidney Disease: A Randomized Trial. Am. J. Nephrol. 2015, 42, 265–273. [Google Scholar] [CrossRef]
- Shuto, E.; Taketani, Y.; Tanaka, R.; Harada, N.; Isshiki, M.; Sato, M.; Nashiki, K.; Amo, K.; Yamamoto, H.; Higashi, Y.; et al. Dietary phosphorus acutely impairs endothelial function. J. Am. Soc. Nephrol. 2009, 20, 1504–1512. [Google Scholar] [CrossRef]
- Perticone, M.; Maio, R.; Sciacqua, A.; Cimellaro, A.; Andreucci, M.; Tripepi, G.; Zoccali, C.; Sesti, G.; Perticone, F. Serum phosphorus levels are associated with endothelial dysfunction in hypertensive patients. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Abbasian, N.; Burton, J.O.; Herbert, K.E.; Tregunna, B.E.; Brown, J.R.; Ghaderi-Najafabadi, M.; Brunskill, N.J.; Goodall, A.H.; Bevington, A. Hyperphosphatemia, phosphoprotein phosphatases, and microparticle release in vascular endothelial cells. J. Am. Soc. Nephrol. 2015, 26, 2152–2162. [Google Scholar] [CrossRef]
- Zoccali, C.; Torino, C.; Curatola, G.; Panuccio, V.; Tripepi, R.; Pizzini, P.; Versace, M.; Bolignano, D.; Cutrupi, S.; Ghiadoni, L.; et al. Serum phosphate modifies the vascular response to vitamin D receptor activation in chronic kidney disease (CKD) patients. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 581–589. [Google Scholar] [CrossRef]
- Block, G.a.; Wheeler, D.C.; Persky, M.S.; Kestenbaum, B.; Ketteler, M.; Spiegel, D.M.; Allison, M.a.; Asplin, J.; Smits, G.; Hoofnagle, A.N.; et al. Effects of phosphate binders in moderate CKD. J. Am. Soc. Nephrol. 2012, 23, 1407–1415. [Google Scholar] [CrossRef]
- Vervloet, M. Renal and extrarenal effects of fibroblast growth factor 23. Nat. Rev. Nephrol. 2019, 15, 109–120. [Google Scholar] [CrossRef]
- Silswal, N.; Touchberry, C.D.; Daniel, D.R.; McCarthy, D.L.; Zhang, S.; Andresen, J.; Stubbs, J.R.; Wacker, M.J. FGF23 directly impairs endothelium-Dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E426–E436. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutiérrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.I.M.I.; Sonmez, A.; Saglam, M.; Yaman, H.; Kilic, S.; Demirkaya, E.; Eyileten, T.; Caglar, K.; Oguz, Y.; Vural, A.; et al. FGF-23 and vascular dysfunction in patients with stage 3 and 4 chronic kidney disease. Kidney Int. 2010, 78, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Coban, M.; Inci, A.; Yllmaz, U.; Asilturk, E. The association of fibroblast growth factor 23 with arterial stiffness and atherosclerosis in patients with autosomal dominant polycystic kidney disease. Kidney Blood Press. Res. 2018, 43, 1160–1173. [Google Scholar] [CrossRef]
- Gutiérrez, O.M.; Januzzi, J.L.; Isakova, T.; Laliberte, K.; Smith, K.; Collerone, G.; Sarwar, A.; Hoffmann, U.; Coglianese, E.; Christenson, R.; et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 2009, 119, 2545–2552. [Google Scholar] [CrossRef]
- Marthi, A.; Donovan, K.; Haynes, R.; Wheeler, D.C.; Baigent, C.; Rooney, C.M.; Landray, M.J.; Moe, S.M.; Yang, J.; Holland, L.; et al. Fibroblast Growth Factor-23 and Risks of Cardiovascular and Noncardiovascular Diseases: A Meta-Analysis. J. Am. Soc. Nephrol. 2018, 29, 2015–2027. [Google Scholar] [CrossRef]
- Olauson, H.; Vervloet, M.G.; Cozzolino, M.; Massy, Z.A.; Torres, P.U.; Larsson, T.E. New Insights Into the FGF23-Klotho Axis. Semin. Nephrol. 2014, 34, 586–597. [Google Scholar] [CrossRef]
- Nagai, R.; Saito, Y.; Ohyama, Y.; Aizawa, H.; Suga, T.; Nakamura, T.; Kurabayashi, M.; Kuro-O, M. Endothelial dysfunction in the klotho mouse and downregulation of klotho gene expression in various animal models of vascular and metabolic diseases. Cell. Mol. Life Sci. 2000, 57, 738–746. [Google Scholar] [CrossRef]
- Shimada, T.; Takeshita, Y.; Murohara, T.; Sasaki, K.I.; Egami, K.; Shintani, S.; Katsuda, Y.; Ikeda, H.; Nabeshima, Y.I.; Imaizumi, T. Angiogenesis and vasculogenesis are impaired in the precocious-Aging klotho mouse. Circulation 2004, 110, 1148–1155. [Google Scholar] [CrossRef]
- Six, I.; Okazaki, H.; Gross, P.; Cagnard, J.; Boudot, C.; Maizel, J.; Drueke, T.B.; Massy, Z. A direct, Acute effects of Klotho and FGF23 on vascular smooth muscle and endothelium. PLoS ONE 2014, 9, e93423. [Google Scholar] [CrossRef]
- Maekawa, Y.; Ishikawa, K.; Yasuda, O.; Oguro, R.; Hanasaki, H.; Kida, I.; Takemura, Y.; Ohishi, M.; Katsuya, T.; Rakugi, H. Klotho suppresses TNF-α-Induced expression of adhesion molecules in the endothelium and attenuates NF-κB activation. Endocrine 2009, 35, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Kusaba, T.; Okigaki, M.; Matui, A.; Murakami, M.; Ishikawa, K.; Kimuraa, T.; Sonomura, K.; Adachi, Y.; Shibuya, M.; Shirayama, T.; et al. Klotho is associated with VEGF receptor-2 and the transient receptor potential canonical-1 Ca2+ channel to maintain endothelial integrity. Proc. Natl. Acad. Sci. USA 2010, 107, 19308–19313. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Len, B.; Liu, W.; Wang, G. Suppression of apoptosis in human umbilical vein endothelial cells (HUVECs) by klotho protein is associated with reduced endoplasmic reticulum oxidative stress and activation of the PI3K/AKT pathway. Med. Sci. Monit. 2018, 24, 8489–8499. [Google Scholar] [CrossRef] [PubMed]
- Keles, N.; Caliskan, M.; Dogan, B.; Keles, N.N.; Kalcik, M.; Aksu, F.; Kostek, O.; Aung, S.M.; Isbilen, B.; Oguz, A. Low serum level of Klotho is an early predictor of atherosclerosis. Tohoku J. Exp. Med. 2015, 237, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Sugiyama, H.; Morinaga, H.; Inoue, T.; Takiue, K.; Ogawa, A.; Yamanari, T.; Kikumoto, Y.; Uchida, H.A.; Kitamura, S.; et al. A Decreased Level of Serum Soluble Klotho Is an Independent Biomarker Associated with Arterial Stiffness in Patients with Chronic Kidney Disease. PLoS ONE 2013, 8, e56695. [Google Scholar] [CrossRef]
- Kim, H.J.; Kang, E.; Oh, Y.K.; Kim, Y.H.; Han, S.H.; Yoo, T.H.; Chae, D.W.; Lee, J.; Ahn, C.; Oh, K.H. The association between soluble klotho and cardiovascular parameters in chronic kidney disease: Results from the KNOW-CKD study. BMC Nephrol. 2018, 19, 51. [Google Scholar] [CrossRef]
- Seiler, S.; Rogacev, K.S.; Roth, H.J.; Shafein, P.; Emrich, I.; Neuhaus, S.; Floege, J.; Fliser, D.; Heine, G.H. Associations of FGF-23 and sklotho with cardiovascular outcomes among patients with CKD stages 2–4. Clin. J. Am. Soc. Nephrol. 2014, 9, 1049–1058. [Google Scholar] [CrossRef]
- London, G.M.; Guérin, A.P.; Verbeke, F.H.; Pannier, B.; Boutouyrie, P.; Marchais, S.J.; Mëtivier, F. Mineral metabolism and arterial functions in end-Stage renal disease: Potential role of 25-Hydroxyvitamin D deficiency. J. Am. Soc. Nephrol. 2007, 18, 613–620. [Google Scholar] [CrossRef]
- Tentori, F.; Blayney, M.J.; Albert, J.M.; Gillespie, B.W.; Kerr, P.G.; Bommer, J.; Young, E.W.; Akizawa, T.; Akiba, T.; Pisoni, R.L.; et al. Mortality Risk for Dialysis Patients With Different Levels of Serum Calcium, Phosphorus, and PTH: The Dialysis Outcomes and Practice Patterns Study (DOPPS). Am. J. Kidney Dis. 2008, 52, 519–530. [Google Scholar] [CrossRef]
- Wolf, M.; Shah, A.; Gutierrez, O.; Ankers, E.; Monroy, M.; Tamez, H.; Steele, D.; Chang, Y.; Camargo, C.A.; Tonelli, M.; et al. Vitamin D levels and early mortality among incident hemodialysis patients. Kidney Int. 2007, 72, 1004–1013. [Google Scholar] [CrossRef]
- Gutiérrez, O.M.; Mannstadt, M.; Isakova, T.; Rauh-Hain, J.A.; Tamez, H.; Shah, A.; Smith, K.; Lee, H.; Thadhani, R.; Jüppner, H.; et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N. Engl. J. Med. 2008, 359, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Scialla, J.J.; Parekh, R.S.; Eustace, J.A.; Astor, B.C.; Plantinga, L.; Jaar, B.G.; Shafi, T.; Coresh, J.; Powe, N.R.; Melamed, M.L. Race, Mineral Homeostasis and Mortality in Patients with End-Stage Renal Disease on Dialysis. Am. J. Nephrol. 2015, 42, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, R.; Schuh, A.; Heine, G.H.; Brandenburg, V. FGF23 in cardiovascular disease: Innocent bystander or active mediator? Front. Endocrinol. (Lausanne) 2018, 9, 351. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.Y.; Xu, R.J.; Zhang, S.H.; Qiao, Q.; Shen, L.; Li, M.; Xu, D.Y.; Wang, Z.Y. Alteration of circulatory platelet microparticles and endothelial microparticles in patients with chronic kidney disease. Int. J. Clin. Exp. Med. 2015, 8, 16704–16708. [Google Scholar]
- Amabile, N.; Guérin, A.P.; Leroyer, A.; Mallat, Z.; Nguyen, C.; Boddaert, J.; London, G.M.; Tedgui, A.; Boulanger, C.M. Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-Stage renal failure. J. Am. Soc. Nephrol. 2005, 16, 3381–3388. [Google Scholar] [CrossRef]
- Yilmaz, M.I.; Siriopol, D.; Saglam, M.; Kurt, Y.G.; Unal, H.U.; Eyileten, T.; Gok, M.; Cetinkaya, H.; Oguz, Y.; Sari, S.; et al. Plasma endocan levels associate with inflammation, vascular abnormalities, cardiovascular events, and survival in chronic kidney disease. Kidney Int. 2014, 86, 1213–1220. [Google Scholar] [CrossRef]
- Baylis, C. Arginine, arginine analogs and nitric oxide production in chronic kidney disease. Nat. Clin. Pract. Nephrol. 2006, 2, 209–220. [Google Scholar] [CrossRef]
- Meyer, C.; Heiss, C.; Drexhage, C.; Kehmeier, E.S.; Balzer, J.; Mühlfeld, A.; Merx, M.W.; Lauer, T.; Kühl, H.; Floege, J.; et al. Hemodialysis-Induced Release of Hemoglobin Limits Nitric Oxide Bioavailability and Impairs Vascular Function. J. Am. Coll. Cardiol. 2010, 55, 454–459. [Google Scholar] [CrossRef]
- Vanholder, R.; Baurmeister, U.; Brunet, P.; Cohen, G.; Glorieux, G.; Jankowski, J. A bench to bedside view of uremic toxins. J. Am. Soc. Nephrol. 2008, 19, 863–870. [Google Scholar] [CrossRef]
- Wang, F.; Xiong, R.; Feng, S.; Lu, X.; Li, H.; Wang, S. Association of Circulating Levels of ADMA with Carotid Intima-Media Thickness in Patients with CKD: A Systematic Review and Meta-Analysis. Kidney Blood Press. Res. 2018, 43, 25–33. [Google Scholar] [CrossRef]
- Ravani, P.; Tripepi, G.; Malberti, F.; Testa, S.; Mallamaci, F.; Zoccali, C. Asymmetrical dimethylarginine predicts progression to dialysis and death in patients with chronic kidney disease: A competing risks modeling approach. J. Am. Soc. Nephrol. 2005, 16, 2449–2455. [Google Scholar] [CrossRef] [PubMed]
- Emrich, I.E.; Zawada, A.M.; Martens-Lobenhoffer, J.; Fliser, D.; Wagenpfeil, S.; Heine, G.H.; Bode-Böger, S.M. Symmetric dimethylarginine (SDMA) outperforms asymmetric dimethylarginine (ADMA) and other methylarginines as predictor of renal and cardiovascular outcome in non-Dialysis chronic kidney disease. Clin. Res. Cardiol. 2018, 107, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Ronden, R.A.; Houben, A.J.H.M.; Teerlink, T.; Bakker, J.A.; Bierau, J.; Stehouwer, C.D.A.; de Leeuw, P.W.; Kroon, A.A. Reduced renal plasma clearance does not explain increased plasma asymmetric dimethylarginine in hypertensive subjects with mild to moderate renal insufficiency. Am. J. Physiol. - Ren. Physiol. 2012, 303, F149–F156. [Google Scholar] [CrossRef] [PubMed]
- Tutarel, O.; Denecke, A.; Bode-Böger, S.M.; Martens-Lobenhoffer, J.; Schieffer, B.; Westhoff-Bleck, M.; Kielstein, J.T. Symmetrical dimethylarginine outperforms CKD-EPI and MDRD-Derived eGFR for the assessment of renal function in patients with adult congenital heart disease. Kidney Blood Press. Res. 2011, 34, 41–45. [Google Scholar] [CrossRef] [PubMed]
- El-Khoury, J.M.; Bunch, D.R.; Hu, B.; Payto, D.; Reineks, E.Z.; Wang, S. Comparison of symmetric dimethylarginine with creatinine, cystatin C and their eGFR equations as markers of kidney function. Clin. Biochem. 2016, 49, 1140–1143. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, S.; Sonntag, S.R.; Lieb, W.; Maas, R. Asymmetric and Symmetric Dimethylarginine as Risk Markers for Total Mortality and Cardiovascular Outcomes: A Systematic Review and Meta-Analysis of Prospective Studies. PLoS ONE 2016, 11, e0165811. [Google Scholar] [CrossRef]
- Scalera, F.; Kielstein, J.T.; Martens-Lobenhoffer, J.; Postel, S.C.; Täger, M.; Bode-Böger, S.M. Erythropoietin increases asymmetric dimethylarginine in endothelial cells: Role of dimethylarginine dimethylaminohydrolase. J. Am. Soc. Nephrol. 2005, 16, 892–898. [Google Scholar] [CrossRef]
- Yokoro, M.; Nakayama, Y.; Yamagishi, S.I.; Ando, R.; Sugiyama, M.; Ito, S.; Yano, J.; Taguchi, K.; Kaida, Y.; Saigusa, D.; et al. Asymmetric dimethylarginine contributes to the impaired response to erythropoietin in CKD-Anemia. J. Am. Soc. Nephrol. 2017, 28, 2670–2680. [Google Scholar] [CrossRef]
- Obayashi, K.; Kurumatani, N.; Saeki, K. Gender differences in the relationships between chronic kidney disease, asymmetric dimethylarginine, and sleep quality: The HEIJO-KYO cohort. Nitric Oxide Biol. Chem. 2018, 79, 25–30. [Google Scholar] [CrossRef]
- Wetzel, M.D.; Gao, T.; Stanley, K.; Cooper, T.K.; Morris, S.M.; Awad, A.S. Enhancing kidney DDAH-1 expression by adenovirus delivery reduces ADMA and ameliorates diabetic nephropathy. Am. J. Physiol. Physiol. 2020, 318, F509–F517. [Google Scholar] [CrossRef]
- Tomlinson, J.A.P.; Caplin, B.; Boruc, O.; Bruce-Cobbold, C.; Cutillas, P.; Dormann, D.; Faull, P.; Grossman, R.C.; Khadayate, S.; Mas, V.R.; et al. Reduced renal methylarginine metabolism protects against progressive kidney damage. J. Am. Soc. Nephrol. 2015, 26, 3045–3059. [Google Scholar] [CrossRef] [PubMed]
- Fliser, D.; Kronenberg, F.; Kielstein, J.T.; Morath, C.; Bode-Böger, S.M.; Haller, H.; Ritz, E. Asymmetric dimethylarginine and progression of chronic kidney disease: The mild to moderate kidney disease study. J. Am. Soc. Nephrol. 2005, 16, 2456–2461. [Google Scholar] [CrossRef] [PubMed]
- Eiselt, J.; Rajdl, D.; Racek, J.; Vostrý, M.; Rulcová, K.; Wirth, J. Asymmetric dimethylarginine and progression of chronic kidney disease—A One-Year Follow-Up Study. Kidney Blood Press. Res. 2014, 39, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Tripepi, G.; Kollerits, B.; Leonardis, D.; Yilmaz, M.I.M.I.; Postorino, M.; Fliser, D.; Mallamaci, F.; Kronenberg, F.; Zoccali, C. Competitive Interaction Between Fibroblast Growth Factor 23 And Asymmetric Dimethylarginine in Patients With CKD. J. Am. Soc. Nephrol. 2014, 26, 1–10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shafi, T.; Hostetter, T.H.; Meyer, T.W.; Hwang, S.; Hai, X.; Melamed, M.L.; Banerjee, T.; Coresh, J.; Powe, N.R. Serum Asymmetric and Symmetric Dimethylarginine and Morbidity and Mortality in Hemodialysis Patients. Am. J. Kidney Dis. 2017, 70, 48–58. [Google Scholar] [CrossRef]
- Kalousová, M.; Kielstein, J.T.; Hodková, M.; Zima, T.; Dusilová-Sulková, S.; Martens-Lobenhoffer, J.; Bode-Boger, S.M. No benefit of hemodiafiltration over hemodialysis in lowering elevated levels of asymmetric dimethylarginine in ESRD patients. Blood Purif. 2006, 24, 439–444. [Google Scholar] [CrossRef]
- Beerenhout, C.H.; Luik, A.J.; Jeuken-Mertens, S.G.J.; Bekers, O.; Menheere, P.; Hover, L.; Klaassen, L.; van der Sande, F.M.; Cheriex, E.C.; Meert, N.; et al. Pre-Dilution On-Line haemofiltration vs. Low-Flux haemodialysis: A randomized prospective study. Nephrol. Dial. Transplant. 2005, 20, 1155–1163. [Google Scholar] [CrossRef][Green Version]
- Zoccali, C.; Enia, G.; Tripepi, G.; Panuccio, V.; Mallamaci, F. Clinical epidemiology of major nontraditional risk factors in peritoneal dialysis patients. Perit. Dial. Int. 2005, 25, 84–87. [Google Scholar] [CrossRef]
- Sahebkar, A.; Ponziani, M.C.; Goitre, I.; Bo, S. Does statin therapy reduce plasma VEGF levels in humans? A systematic review and meta-Analysis of randomized controlled trials. Metabolism 2015, 64, 1466–1476. [Google Scholar] [CrossRef]
- Sahebkar, A.; Kotani, K.; Serban, C.; Ursoniu, S.; Mikhailidis, D.P.; Jones, S.R.; Ray, K.K.; Blaha, M.J.; Rysz, J.; Toth, P.P.; et al. Statin therapy reduces plasma endothelin-1 concentrations: A meta-Analysis of 15 randomized controlled trials. Atherosclerosis 2015, 241, 433–442. [Google Scholar] [CrossRef]
- Serban, C.; Sahebkar, A.; Ursoniu, S.; Mikhailidis, D.P.; Rizzo, M.; Lip, G.Y.H.; Kees Hovingh, G.; Kastelein, J.J.P.; Kalinowski, L.; Rysz, J.; et al. A systematic review and meta-Analysis of the effect of statins on plasma asymmetric dimethylarginine concentrations. Sci. Rep. 2015, 5, 9902. [Google Scholar] [CrossRef] [PubMed]
- Katsiki, N.; Reiner, Ž.; Tedeschi Reiner, E.; Al-Rasadi, K.; Pirro, M.; Mikhailidis, D.P.; Sahebkar, A. Improvement of endothelial function by pitavastatin: A meta-Analysis. Expert Opin. Pharmacother. 2018, 19, 279–286. [Google Scholar] [CrossRef]
- Ando, M.; Sanaka, T.; Nihei, H. Eicosapentanoic acid reduces plasma levels of remnant lipoproteins and prevents in vivo peroxidation of LDL in dialysis patients. J. Am. Soc. Nephrol. 1999, 10, 2177–2184. [Google Scholar] [PubMed]
- Diepeveen, S.H.A.; Verhoeven, G.W.H.E.; Van Der Palen, J.; Dikkeschei, L.D.; Van Tits, L.J.; Kolsters, G.; Offerman, J.J.G.; Bilo, H.J.G.; Stalenhoef, A.F.H. Effects of atorvastatin and vitamin E on lipoproteins and oxidative stress in dialysis patients: A randomised-Controlled trial. J. Intern. Med. 2005, 257, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M.; Cappellari, G.G.; Barbetta, D.; Semolic, A.; Barazzoni, R. Omega 3 polyunsaturated fatty acids improve endothelial dysfunction in chronic renal failure: Role of eNOS activation and of oxidative stress. Nutrients 2017, 9, 895. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Lv, J.A.; Perkovic, V.; Yang, L.; Zhao, N.; Jardine, M.J.; Cass, A.; Zhang, H.; Wang, H. Effect of statin therapy on cardiovascular and renal outcomes in patients with chronic kidney disease: A systematic review and meta-Analysis. Eur. Heart J. 2013, 24, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Sabri, M.; Tavana, E.; Ahmadi, A.; Gheissari, A. Effect of vitamin C on endothelial function of children with chronic renal failure: An experimental study. Adv. Biomed. Res. 2015, 4, 260. [Google Scholar]
- Ghiadoni, L.; Cupisti, A.; Huang, Y.; Mattei, P.; Cardinal, H.; Favilla, S.; Rindi, P.; Barsotti, G.; Taddei, S.; Salvetti, A. Endothelial dysfunction and oxidative stress in chronic renal failure. J. Nephrol. 2004, 17, 512–519. [Google Scholar]
- Ramos, R.; Martínez-Castelao, A. Lipoperoxidation and hemodialysis. Metabolism 2008, 57, 1369–1374. [Google Scholar] [CrossRef]
- Nanayakkara, P.W.B.; Van Guldener, C.; Ter Wee, P.M.; Scheffer, P.G.; Van Ittersum, F.J.; Twisk, J.W.; Teerlink, T.; Van Dorp, W.; Stehouwer, C.D.A. Effect of a treatment strategy consisting of pravastatin, vitamin E, and homocysteine lowering on carotid intima-Media thickness, endothelial function, and renal function in patients with mild to moderate chronic kidney disease: Results from the Anti-Oxid. Arch. Intern. Med. 2007, 167, 1262–1270. [Google Scholar] [CrossRef]
- Yang, S.K.; Xiao, L.; Xu, B.; Xu, X.X.; Liu, F.Y.; Sun, L. Effects of vitamin E-coated dialyzer on oxidative stress and inflammation status in hemodialysis patients: A systematic review and meta-Analysis. Ren. Fail. 2014, 36, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yi, B.; Li, A.-M.; Zhang, H. Effects of vitamin E-Coated dialysis membranes on anemia, nutrition and dyslipidemia status in hemodialysis patients: A Meta-Analysis. Ren. Fail. 2015, 37, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Peters, H.; Border, W.A.; Noble, N.A. From rats to man: A perspective an dietary L-Arginine supplementation in human renal disease. Nephrol. Dial. Transplant. 1999, 14, 1640–1650. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hand, M.F.; Haynes, W.G.; Webb, D.J. Hemodialysis and L-Arginine, but not D-Arginine, correct renal failure-Associated endothelial dysfunction. Kidney Int. 1998, 53, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Bennett-Richards, K.J.; Kattenhorn, M.; Donald, A.E.; Oakley, G.R.; Varghese, Z.; Bruckdorfer, K.R.; Deanfield, J.E.; Rees, L. Oral L-Arginine does not improve endothelial dysfunction in children with chronic renal failure. Kidney Int. 2002, 62, 1372–1378. [Google Scholar] [CrossRef][Green Version]
- Cross, J.M.; Donald, A.E.; Kharbanda, R.; Deanfield, J.E.; Woolfson, R.G.; MacAllister, R.J. Acute administration of L-Arginine does not improve arterial endothelial function in chronic renal failure. Kidney Int. 2001, 60, 2318–2323. [Google Scholar] [CrossRef][Green Version]
- Wu-Wong, J.R.; Kawai, M.; Chen, Y.W.; Wessale, J.L.; Huang, C.J.; Wu, M.T.; Nakane, M. Two novel vitamin D receptor modulators with similar structures exhibit different hypercalcemic effects in 5/6 nephrectomized uremic rats. Am. J. Nephrol. 2013, 37, 310–319. [Google Scholar] [CrossRef]
- Wu-Wong, J.R.; Noonan, W.; Nakane, M.; Brooks, K.A.; Segreti, J.A.; Polakowski, J.S.; Cox, B. Vitamin D receptor activation mitigates the impact of uremia on endothelial function in the 5/6 nephrectomized rats. Int. J. Endocrinol. 2010, 2010, 1–10. [Google Scholar] [CrossRef][Green Version]
- Wu-Wong, J.R.; Li, X.; Chen, Y.W. Different vitamin D receptor agonists exhibit differential effects on endothelial function and aortic gene expression in 5/6 nephrectomized rats. J. Steroid Biochem. Mol. Biol. 2015, 148, 202–209. [Google Scholar] [CrossRef]
- Morishima Vitamin D Attenuates Endothelial Dysfunction in Uremic Rats and Maintains Human Endothelial Stability. J. Am. Heart Assoc. 2018, 7, e02709.
- Kumar, V.; Yadav, A.K.; Lal, A.; Kumar, V.; Singhal, M.; Billot, L.; Gupta, K.L.; Banerjee, D.; Jha, V. A randomized trial of Vitamin D supplementation on vascular function in CKD. J. Am. Soc. Nephrol. 2017, 28, 3100–3108. [Google Scholar] [CrossRef]
- Thadhani, R.; Appelbaum, E.; Pritchett, Y.; Chang, Y.; Wenger, J.; Tamez, H.; Bhan, I.; Agarwal, R.; Zoccali, C.; Wanner, C.; et al. Vitamin D therapy and cardiac structure and function in patients with chronic kidney disease: The PRIMO randomized controlled trial. JAMA 2012, 307, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Y.M.; Fang, F.; Chan, J.; Wen, Y.Y.; Qing, S.; Chan, I.H.S.; Lo, G.; Lai, K.N.; Lo, W.K.; Lam, C.W.K.; et al. Effect of paricalcitol on left ventricular mass and function in CKD-The OPERA trial. J. Am. Soc. Nephrol. 2014, 25, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Inaba, M.; Fukagawa, M.; Ando, R.; Emoto, M.; Fujii, H.; Fujimori, A.; Fukui, M.; Hase, H.; Hashimoto, T.; et al. Effect of Oral Alfacalcidol on Clinical Outcomes in Patients Without Secondary Hyperparathyroidism Receiving Maintenance Hemodialysis: The J-DAVID Randomized Clinical Trial. J. Am. Med. Assoc. 2018, 320, 2325–2334. [Google Scholar]
- Mazidi, M.; Karimi, E.; Rezaie, P.; Vatanparast, H. The impact of vitamin D supplement intake on vascular endothelial function; a systematic review and meta-Analysis of randomized controlled trials. Food Nutr. Res. 2017, 61, 1273574. [Google Scholar] [CrossRef]
- Beveridge, L.A.; Khan, F.; Struthers, A.D.; Armitage, J.; Barchetta, I.; Bressendorff, I.; Cavallo, M.G.; Clarke, R.; Dalan, R.; Dreyer, G.; et al. Effect of vitamin D supplementation on markers of vascular function: A systematic review and individual participant meta-Analysis. J. Am. Heart Assoc. 2018, 7, e008273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fang, F.; Tang, J.; Jia, L.; Feng, Y.; Xu, P.; Faramand, A. Association between vitamin D supplementation and mortality: Systematic review and meta-Analysis. BMJ 2019, 366, l4673. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Gillings, N.; Flores, B.; Takahashi, M.; Kuro-o, M.; Moe, O.W. Recombinant α-Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney Int. 2017, 91, 1104–1114. [Google Scholar] [CrossRef]
- Shi, M.; Flores, B.; Gillings, N.; Bian, A.; Cho, H.J.; Yan, S.; Liu, Y.; Levine, B.; Moe, O.W.; Hu, M.C. α-Klotho mitigates progression of aki to ckd through activation of autophagy. J. Am. Soc. Nephrol. 2016, 27, 2331–2345. [Google Scholar] [CrossRef]
- Buendía, P.; Carracedo, J.; Soriano, S.; Madueño, J.A.; Ortiz, A.; Martín-Malo, A.; Aljama, P.; Ramírez, R. Klotho Prevents NFκB Translocation and Protects Endothelial Cell from Senescence Induced by Uremia. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 70, 1198–1209. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, Y.; Zhang, Y.; Liu, C. Klotho ameliorates oxidized low density lipoprotein (Ox-LDL)-Induced oxidative stress via regulating LOX-1 and PI3K/Akt/eNOS pathways. Lipids Health Dis. 2017, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Haller, J.; Haffner, D.; Leifheit-nestler, M. Klotho modulates FGF23-Mediated NO synthesis and oxidative stress in human coronary artery endothelial cells. Pflügers Arch. Eur. J. Physiol. 2016, 468, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Nie, L.; Huang, Y.; Zhang, J.; Xiao, T.; Guan, X.; Zhao, J. Amelioration of uremic toxin indoxyl sulfate-Induced endothelial cell dysfunction by Klotho protein. Toxicol. Lett. 2012, 215, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.I.; Sonmez, A.; Saglam, M.; Yaman, H.; Kilic, S.; Eyileten, T.; Caglar, K.; Oguz, Y.; Vural, A.; Yenicesu, M.; et al. Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: A randomized clinical trial. Am. J. Kidney Dis. 2012, 59, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Uleri, S.; Magagna, A.; Salvetti, A. Lacidipine restores endothelium-Dependent vasodilation in essential hypertensive patients. Hypertension 1997, 30, 1606–1612. [Google Scholar] [CrossRef]
- Yavuz, D.; Koç, M.; Toprak, A.; Akpinar, I.; Velioğlu, A.; Deyneli, O.; Haklar, G.; Akalin, S. Effects of ACE inhibition and AT1-Receptor antagonism on endothelial function and insulin sensitivity in essential hypertensive patients. J. Renin. Angiotensin. Aldosterone. Syst. 2003, 4, 197–203. [Google Scholar] [CrossRef]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Mattei, P.; Salvetti, A. Effects of angiotensin converting enzyme inhibition on endothelium-Dependent vasodilatation in essential hypertensive patients. J. Hypertens. 1998, 16, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Flammer, A.J.; Hermann, F.; Wiesli, P.; Schwegler, B.; Chenevard, R.; Hürlimann, D.; Sudano, I.; Gay, S.; Neidhart, M.; Riesen, W.; et al. Effect of losartan, compared with atenolol, on endothelial function and oxidative stress in patients with type 2 diabetes and hypertension. J. Hypertens. 2007, 25, 785–791. [Google Scholar] [CrossRef]
- Prisant, L.M. Nebivolol: Pharmacologic profile of an ultraselective, vasodilatory β1-Blocker. J. Clin. Pharmacol. 2008, 48, 225–239. [Google Scholar] [CrossRef]
- Ding, Y.; Vaziri, N.D. Calcium channel blockade enhances nitric oxide synthase expression by cultured endothelial cells. Hypertension 1998, 32, 718–723. [Google Scholar] [CrossRef]
- Shetty, S.; Malik, A.H.; Feringa, H.; El Accaoui, R.; Girotra, S. Meta-Analysis Evaluating Calcium Channel Blockers and the Risk of Peripheral Arterial Disease in Patients With Hypertension. Am. J. Cardiol. 2020, 125, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Savarese, G.; Costanzo, P.; Cleland, J.G.F.; Vassallo, E.; Ruggiero, D.; Rosano, G.; Perrone-Filardi, P. A meta-Analysis reporting effects of angiotensin-Converting enzyme inhibitors and angiotensin receptor blockers in patients without heart failure. J. Am. Coll. Cardiol. 2013, 61, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.I.; Sonmez, A.; Saglam, M.; Yaman, H.; Cayci, T.; Kilic, S.; Eyileten, T.; Caglar, K.; Oguz, Y.; Vural, A.; et al. Reduced proteinuria using ramipril in diabetic CKD stage 1 decreases circulating cell death receptor activators concurrently with ADMA. A novel pathophysiological pathway? Nephrol. Dial. Transplant. 2010, 25, 3250–3256. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gamboa, J.L.; Pretorius, M.; Sprinkel, K.C.; Brown, N.J.; Ikizler, T.A. Angiotensin converting enzyme inhibition increases ADMA concentration in patients on maintenance hemodialysis—A Randomized Cross-Over study. BMC Nephrol. 2015, 16, 167. [Google Scholar] [CrossRef] [PubMed]
- Moningka, N.C.; Tsarova, T.; Sasser, J.M.; Baylis, C. Protective actions of nebivolol on chronic nitric oxide synthase inhibition-Induced hypertension and chronic kidney disease in the rat: A comparison with angiotensin II receptor blockade. Nephrol. Dial. Transplant. 2012, 27, 913–920. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hewedy, W.A.; Mostafa, D.K. Nebivolol suppresses asymmetric dimethylarginine and attenuates cyclosporine-Induced nephrotoxicity and endothelial dysfunction in rats. Pharmacol. Reports 2016, 68, 1319–1325. [Google Scholar] [CrossRef]
- Wang, Y.; An, W.; Zhang, F.; Niu, M.; Liu, Y.; Shi, R. Nebivolol ameliorated kidney damage in Zucker diabetic fatty rats by regulation of oxidative stress/NO pathway: Comparison with captopril. Clin. Exp. Pharmacol. Physiol. 2018, 45, 1135–1148. [Google Scholar] [CrossRef]
- Wang, Y.; Niu, M.; Yin, S.; Zhang, F.; Shi, R. Nephroprotective effects of nebivolol in 2K1C rats through regulation of the kidney ROS-ADMA-NO pathway. Pharmacol. Rep. 2018, 70, 917–929. [Google Scholar] [CrossRef]
- Kolijn, D.; Pabel, S.; Tian, Y.; Lódi, M.; Herwig, M.; Carrizzo, A.; Zhazykbayeva, S.; Kovács, Á.; Fülöp, G.Á.; Falcão-Pires, I.; et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-Oxidative pathways and protein kinase Gα oxidation. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Ikonomidis, I.; Pavlidis, G.; Thymis, J.; Birba, D.; Kalogeris, A.; Kousathana, F.; Kountouri, A.; Balampanis, K.; Parissis, J.; Andreadou, I.; et al. Effects of Glucagon-Like Peptide-1 Receptor Agonists, Sodium-Glucose Cotransporter-2 Inhibitors, and Their Combination on Endothelial Glycocalyx, Arterial Function, and Myocardial Work Index in Patients With Type 2 Diabetes Mellitus After 12-Month Treatme. J. Am. Heart Assoc. 2020, 9, e015716. [Google Scholar] [CrossRef]
- Solini, A.; Rossi, C.; Duranti, E.; Taddei, S.; Natali, A.; Virdis, A. Saxagliptin prevents vascular remodeling and oxidative stress in db/db mice. Role of endothelial nitric oxide synthase uncoupling and cyclooxygenase. Vascul. Pharmacol. 2015, 76, 62–71. [Google Scholar] [CrossRef]
- Kocak, H.; Ceken, K.; Yavuz, A.; Yucel, S.; Gurkan, A.; Erdogan, O.; Ersoy, F.; Yakupoglu, G.; Demirbas, A.; Tuncer, M. Effect of renal transplantation on endothelial function in haemodialysis patients. Nephrol. Dial. Transplant. 2006, 21, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Oflaz, H.; Turkmen, A.; Turgut, F.; Pamukcu, B.; Umman, S.; Ucar, A.; Akyol, Y.; Uzun, S.; Kazancioglu, R.; Kurt, R.; et al. Changes in endothelial function before and after renal transplantation*. Transpl. Int. 2006, 19, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.I.I.; Sonmez, A.; Saglam, M.; Cayci, T.; Kilic, S.; Unal, H.U.U.; Karaman, M.; Cetinkaya, H.; Eyileten, T.; Gok, M.; et al. A Longitudinal Study of Inflammation, CKD-Mineral Bone Disorder, and Carotid Atherosclerosis after Renal Transplantation. Clin. J. Am. Soc. Nephrol. 2014, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nossaman, B.; Pankey, E.; Kadowitz, P. Stimulators and activators of soluble guanylate cyclase: Review and potential therapeutic indications. Crit. Care Res. Pract. 2012, 2012, 290805. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, P.W.; Pieske, B.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; Butler, J.; Lam, C.S.P.; Ponikowski, P.; Voors, A.A.; Jia, G.; et al. Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2020, 382, 1883–1893. [Google Scholar] [CrossRef]
- Shang, F.; Wang, S.C.; Hsu, C.Y.; Miao, Y.; Martin, M.; Yin, Y.; Wu, C.C.; Wang, Y.T.; Wu, G.; Chien, S.; et al. MicroRNA-92a mediates endothelial dysfunction in CKD. J. Am. Soc. Nephrol. 2017, 28, 3251–3261. [Google Scholar] [CrossRef]
- Chao, C.-T.; Liu, Y.-P.; Su, S.-F.; Yeh, H.-Y.; Chen, H.-Y.; Lee, P.-J.; Chen, W.-J.; Lee, Y.-M.; Huang, J.-W.; Chiang, C.-K.; et al. Circulating MicroRNA-125b Predicts the Presence and Progression of Uremic Vascular Calcification. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1402–1414. [Google Scholar] [CrossRef]
- Kétszeri, M.; Kirsch, A.; Frauscher, B.; Moschovaki-Filippidou, F.; Mooslechner, A.A.; Kirsch, A.H.; Schabhuettl, C.; Aringer, I.; Artinger, K.; Pregartner, G.; et al. MicroRNA-142-3p improves vascular relaxation in uremia. Atherosclerosis 2019, 280, 28–36. [Google Scholar] [CrossRef]
- Bello, A.K.; Alrukhaimi, M.; Ashuntantang, G.E.; Basnet, S.; Rotter, R.C.; Douthat, W.G.; Kazancioglu, R.; Kö Ttgen, A.; Nangaku, M.; Powe, N.R.; et al. Complications of chronic kidney disease: Current state, knowledge gaps, and strategy for action. Kidney Int. Suppl. 2017, 7, 122–129. [Google Scholar] [CrossRef]
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roumeliotis, S.; Mallamaci, F.; Zoccali, C. Endothelial Dysfunction in Chronic Kidney Disease, from Biology to Clinical Outcomes: A 2020 Update. J. Clin. Med. 2020, 9, 2359. https://doi.org/10.3390/jcm9082359
Roumeliotis S, Mallamaci F, Zoccali C. Endothelial Dysfunction in Chronic Kidney Disease, from Biology to Clinical Outcomes: A 2020 Update. Journal of Clinical Medicine. 2020; 9(8):2359. https://doi.org/10.3390/jcm9082359
Chicago/Turabian StyleRoumeliotis, Stefanos, Francesca Mallamaci, and Carmine Zoccali. 2020. "Endothelial Dysfunction in Chronic Kidney Disease, from Biology to Clinical Outcomes: A 2020 Update" Journal of Clinical Medicine 9, no. 8: 2359. https://doi.org/10.3390/jcm9082359
APA StyleRoumeliotis, S., Mallamaci, F., & Zoccali, C. (2020). Endothelial Dysfunction in Chronic Kidney Disease, from Biology to Clinical Outcomes: A 2020 Update. Journal of Clinical Medicine, 9(8), 2359. https://doi.org/10.3390/jcm9082359