Immunoadsorption and Plasma Exchange in Seropositive and Seronegative Immune-Mediated Neuropathies

 , ,
, ,
Abstract
1. Introduction
2. Experimental Section
2.1. Paranodal Antibody (PNAb) Patient Cohort
2.2. IA Patient Cohort
2.2.1. CIDP
2.2.2. GBS
2.2.3. MS/CIS
2.3. IA Treatment
2.4. Sample Collection and Storage
2.5. Serological Analysis
3. Results
3.1. Nodal/Paranodal Antibody (PNAb) Diagnostic Cohort
3.1.1. Demographics, Clinical and Serological Characteristics
3.1.2. Physician-Reported Subjective Evaluation of Responses to Plasma Exchange or Immunoadsorption
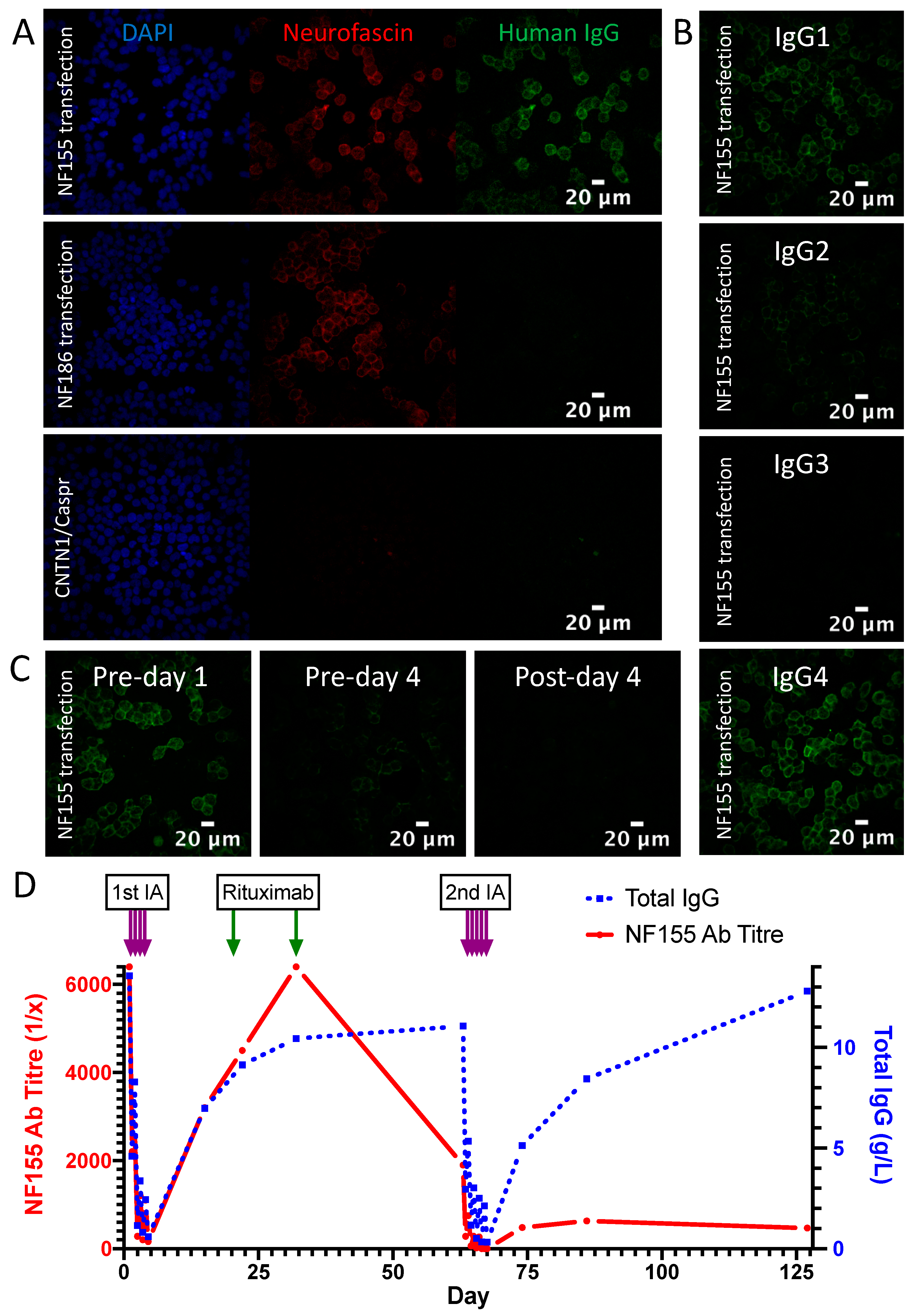
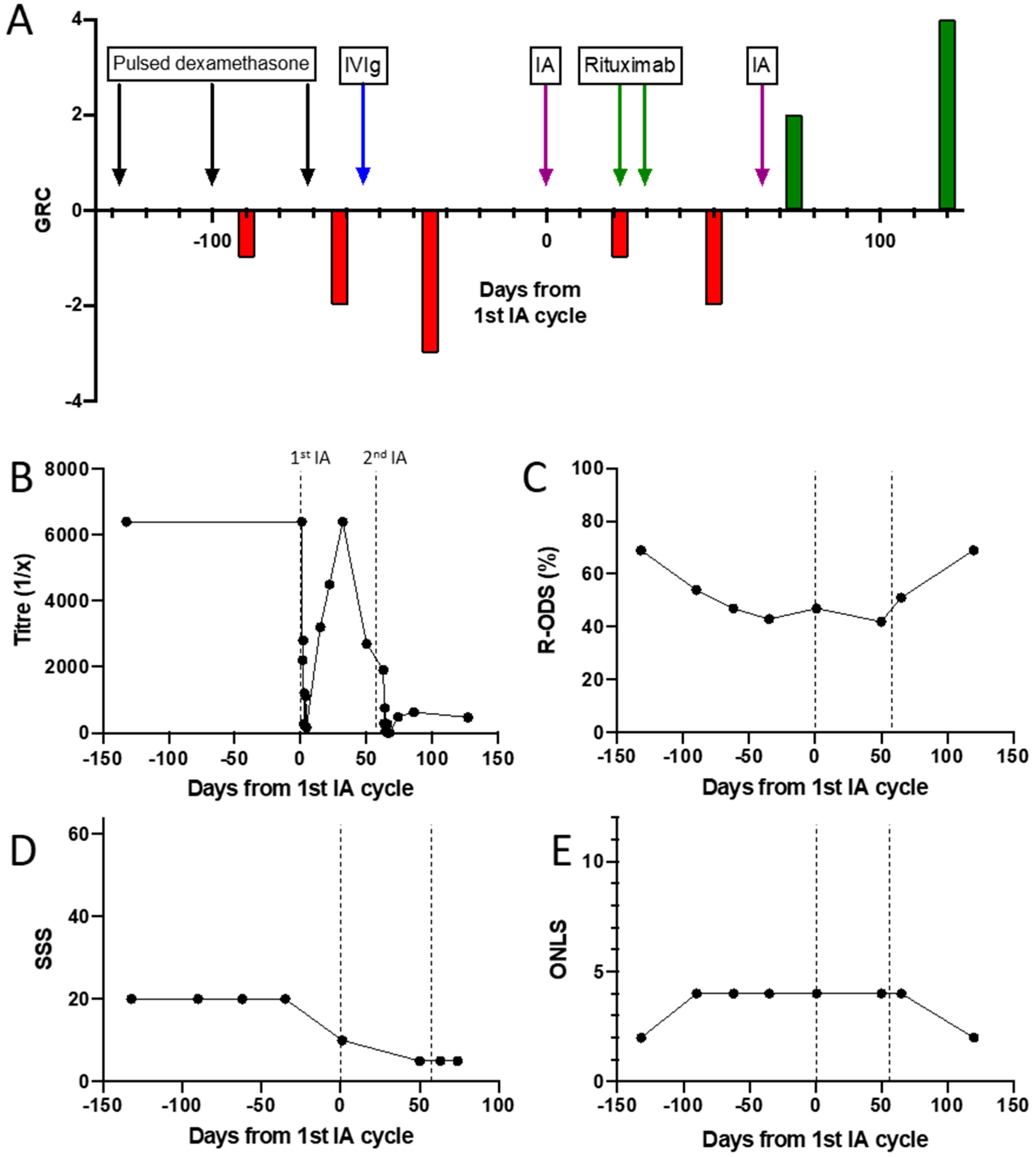
3.2. Detailed Profile of an NF155 Antibody Positive Patient Treated with Immunoadsorption
3.3. Demographics and Clinical Characteristics of the IA Treated Cohort
3.3.1. CIDP
3.3.2. GBS
3.3.3. MS/CIS
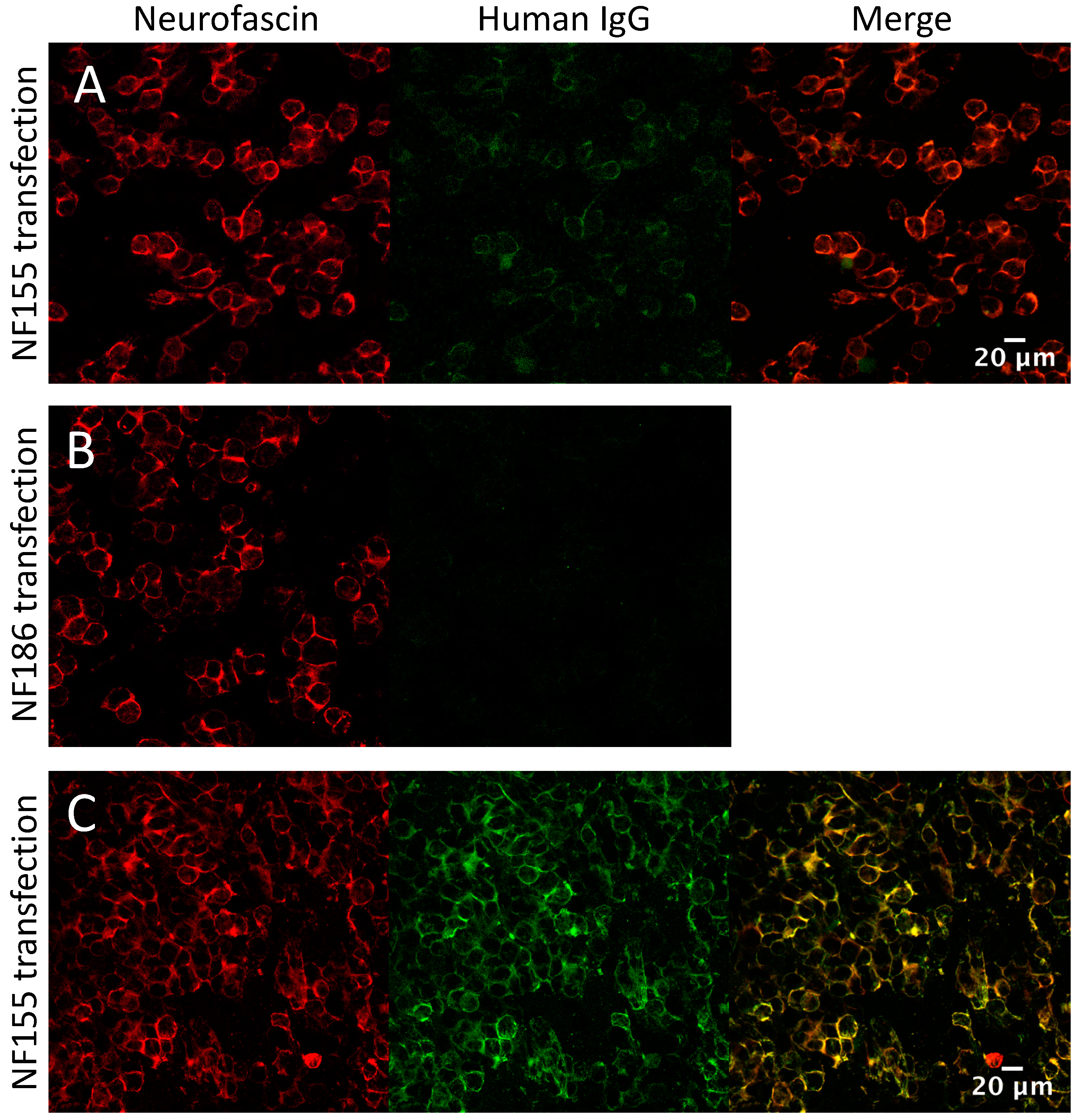
3.4. Glycolipid and Nodal/Paranodal Antibodies in the IA Cohort
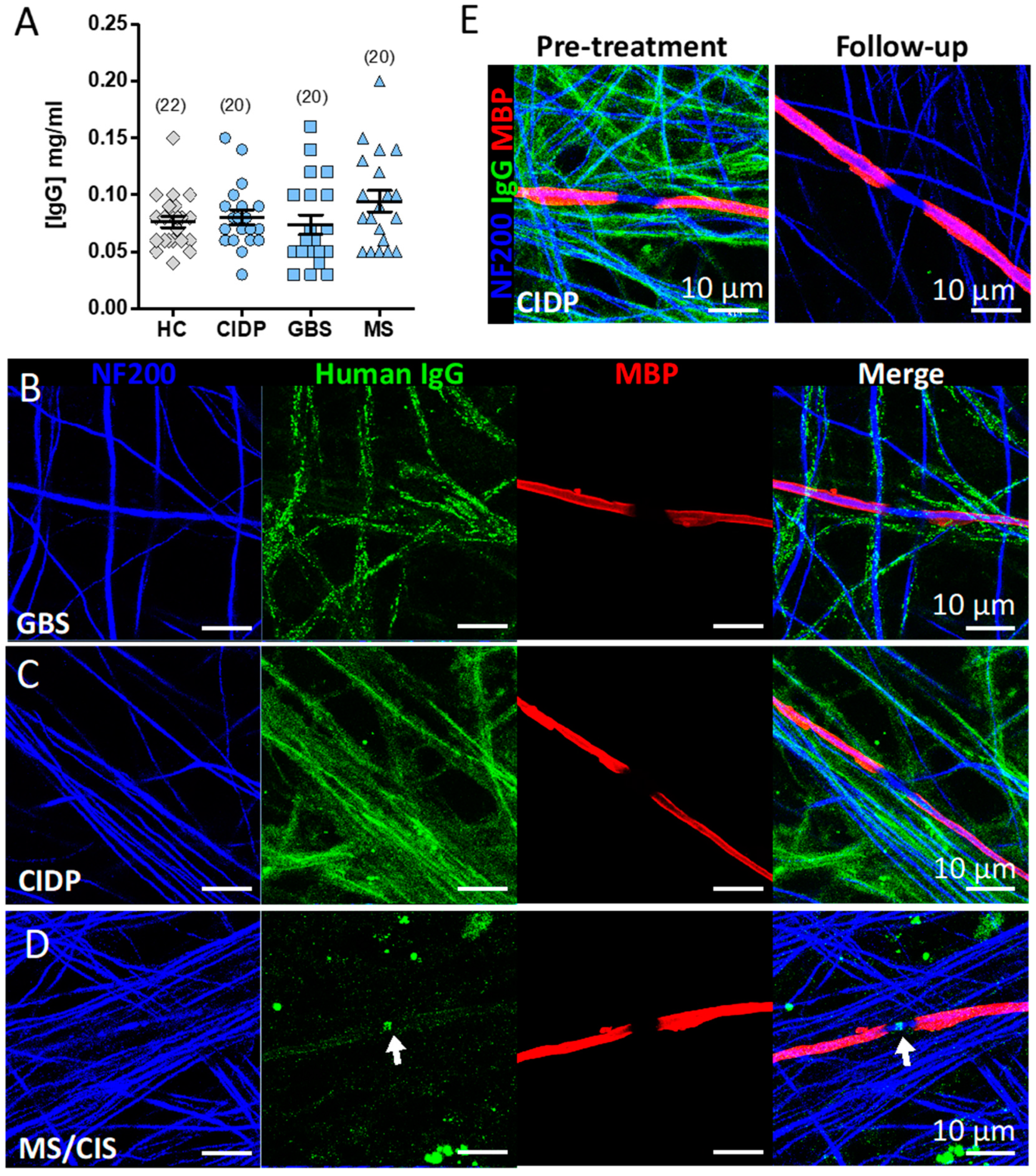
3.5. Screening the IA Eluates for Novel Antibodies Using Myelinating Co-Cultures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Detailed Experimental Methods
Appendix A.1. Nodal/Parnodal Cell-Based Assays
Appendix A.2. Nodal/Paranodal ELISA
Appendix A.3. Ganglioside and Sulfatide ELISA
Appendix A.4. Protein G IgG Purification
Appendix A.5. IgG ELISA
Appendix A.6. Myelinating Co-Cultures
Appendix A.7. Myelinated Co-Culture Immunreactivity Screening
Appendix B. Baseline Clinical Features of IA Cohorts and Clinical Vignettes of Patients with IgG Deposition in Co-Cultures

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Age | Sex | Disease Duration (mo) | Steroids Yes/No | IVIg Yes/No | Other Immunosuppression Used 1 | IA Cycles | CIDP-Score Baseline | CIDP Score at 2 Weeks | Progression before IA | Progression after IA |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 01 * | 59 | M | 83 | Y | Y | 1 | 296 | 298 | |||
| 02 * | 61 | M | 114 | Y | Y | AZA | 3 | 376 | 416 | 2.9 | 0 |
| 03 * | 58 | M | 158 | Y | N | AZA, CPM, MPM | 9 | 102 | 118 | 4.1 | 0.1 |
| 04 * | 65 | M | 114 | Y | N | 1 | 434 | 438 | |||
| 05 * | 67 | F | 112 | Y | Y | 1 | 435 | 435 | |||
| 06 * | 60 | M | 104 | Y | Y | 1 | 268 | 313 | |||
| 07 * | 80 | M | 66 | Y | N | AZA | 3 | 306 | 342 | 19.3 | 0.8 |
| 08 * | 62 | M | 73 | N | Y | 5 | 231 | 361 | 13.1 | 0.7 | |
| 09 * | 68 | M | 67 | Y | Y | 4 | 373 | 364 | 6.7 | 0 | |
| 10 * | 75 | M | 134 | Y | Y | 1 | 326 | 326 | |||
| 11 * | 66 | M | 166 | Y | Y | AZA, MTX | 1 | 308 | 316 | ||
| 12 * | 72 | M | 65 | N | Y | 3 | 314 | 330 | 8.7 | 2.0 | |
| 13 * | 66 | F | 64 | Y | N | 1 | 314 | 282 | |||
| 14 * | 60 | M | 102 | Y | N | 2 | 297 | 382 | 3.0 | 0 | |
| 15 * | 66 | F | 86 | Y | Y | 1 | 393 | 405 | |||
| 16 * | 68 | M | 62 | Y | Y | MPM | 1 | 264 | 264 | ||
| 17 | 67 | M | 65 | Y | N | 3 | N/A | N/A | N/A | N/A | |
| 18 | 53 | M | 97 | Y | Y | 1 | N/A | N/A | N/A | N/A | |
| 19 | 67 | F | 94 | Y | Y | AZA | 2 | N/A | N/A | N/A | N/A |
| 20 | 67 | M | 201 | Y | Y | 1 | N/A | N/A | N/A | N/A |
| ID | Age | Sex | 1st/2nd/3rd-Line | PLEx Yes/No | IVIg Yes/No | PLEx / IA Cycles | Clinical Outcome |
|---|---|---|---|---|---|---|---|
| 01 | 76 | F | 2 | N | Y | 1 | 0 |
| 02 | 73 | M | 3 | Y | Y | 2 | (+) |
| 03 | 36 | F | 2 | N | Y | 1 | (+) |
| 04 | 76 | M | 2 | N | Y | 2 | ++ |
| 05 | 64 | M | 2 | N | Y | 1 | 0 |
| 06 | 31 | F | 1 | N | N | 1 | ++ |
| 07 | 52 | M | 1 | Y | N | 1 | + |
| 08 | 33 | F | 1 | N | N | 1 | 0 |
| 09 | 53 | F | 2 | N | Y | 1 | + |
| 10 | 38 | F | 2 | N | Y | 1 | ++ |
| 11 | 89 | M | 2 | N | Y | 1 | + |
| 12 | 75 | M | PLEx (1) | Y | N | 1 | + |
| 13 | 66 | F | PLEx (1) | Y | Y | 1 | 0 |
| 14 | 66 | F | PLEx (1) | Y | N | 1 | ++ |
| 15 | 42 | M | PLEx (1) | Y | N | 1 | + |
| 16 | 77 | F | PLEx (1) | Y | N | 1 | + |
| 17 | 67 | M | 2 | N | Y | 1 | ++ |
| 18 | 77 | M | PLEx (2) | Y | Y | 1 | (+) |
| 19 | 62 | M | PLEx (1) | Y | N | 1 | + |
| 20 | 66 | F | 2 | N | Y | 1 | + |
| ID | Age | Sex | Diagnosis | DMT | Symptoms | MP | IA Cycles | EDSS before IA | EDSS after IA |
|---|---|---|---|---|---|---|---|---|---|
| 01 | 44 | F | CIS | none | ON | 5×1g iv 5×2g iv | 1 | 2.0 | 0.0 |
| 02 | 21 | M | MS | none | Sensory deficits UE+LE | 5×1g iv | 1 | 4.0 | 3.0 |
| 03 | 48 | M | MS | none | Sensory deficits UE+LE | 5×1g iv | 1 | 4.0 | 3.0 |
| 04 | 18 | F | CIS | none | ON | 7×1g iv | 1 | 1.0 | 1.0 |
| 05 | 30 | F | MS | dimethyl fumarate | Sensory deficits UE | 2×5x1g iv 2×5x1g it | 1 | 1.0 | 1.0 |
| 06 | 46 | F | MS | none | Sensory deficits UE+LE, gait ataxia | 5×1g iv | 1 | 6.5 | 6.5 |
| 07 | 28 | F | MS | dimethyl fumarate | Sensomotoric deficits UE+LE | 5×1g iv 5×1g iv | 1 | 6.5 | 6.5 |
| 08 | 26 | F | MS | dimethyl fumarate | Sensory deficits UE | 4×1g iv | 1 | 3.0 | 3.0 |
| 09 | 19 | M | MS | none | Organic psycho syndrome | 5×1g iv | 2 | 5.5 | 3.0 |
| 10 | 20 | F | MS | none | ON | 5×1g iv | 1 | 2.0 | 1.0 |
| 11 | 47 | F | MS | fingolimod | Motor deficits UE+LE | 5×1g iv | 1 | 7.0 | 6.0 |
| 12 | 19 | F | MS | none | ON (bilateral), hemihypesthesia | 5×1g iv 5×2g iv | 1 | 2.5 | 2.5 |
| 13 | 49 | F | MS | fingolimod | ON, paraparesis | 5×1g iv | 1 | 4.5 | 4.5 |
| 14 | 23 | F | CIS | none | ON | 5×1g iv 5×1g iv | 1 | 2.0 | 1.0 |
| 15 | 50 | F | MS | none | ON (bilateral), gait ataxia | 5×1g iv 5×2g iv | 1 | 5.0 | 4.0 |
| 16 | 15 | M | MS | none | Dysarthria, dysphagia | 12×1g iv | 1 | 4.0 | 3.0 |
| 17 | 17 | F | CIS | none | ON | 5×1g iv 5×1g iv | 1 | 1.0 | 1.0 |
| 18 | 46 | F | MS | interferon beta 1a | Sensory deficits UE+LE, gait ataxia | 5×1g iv | 1 | 3.5 | 3.0 |
| 19 | 57 | F | MS | none | Paraparesis, hemihypesthesia | 5×1g iv | 1 | 4.0 | 4.0 |
| 20 | 37 | M | MS | none | Paresis LE | 5×1g iv 5×2g iv | 1 | 6.0 | 5.5 |
Appendix B.1. CIDP (Patient 11)
Appendix B.2. GBS (Patient 07)
Appendix B.3. MS (Patient 13)
References
- Rinaldi, S.; Bennett, D.L. Pathogenic mechanisms in inflammatory and paraproteinaemic peripheral neuropathies. Curr. Opin. Neurol. 2014, 27, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Willison, H.J. The immunobiology of Guillain-Barre syndromes. J. Peripher. Nerv. Syst. 2005, 10, 94–112. [Google Scholar] [CrossRef] [PubMed]
- Fehmi, J.; Scherer, S.S.; Willison, H.J.; Rinaldi, S. Nodes, paranodes and neuropathies. J. Neurol. Neurosurg. Psychiatry 2017, 89, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Chevret, S.; Hughes, R.A.; Annane, D. Plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst. Rev. 2017, 2017, CD001798. [Google Scholar] [CrossRef]
- Mehndiratta, M.M.; Hughes, R.A.C.; Pritchard, J. Plasma exchange for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst. Rev. 2015, 2015, CD003906. [Google Scholar] [CrossRef]
- Eftimov, F.; Winer, J.B.; Vermeulen, M.; De Haan, R.; Van Schaik, I.N. Intravenous immunoglobulin for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst. Rev. 2013, CD001797. [Google Scholar] [CrossRef]
- Hughes, R.A.C.; Raphaël, J.C.; Swan, A.V.; A Doorn, P. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst. Rev. 2004, CD002063. [Google Scholar] [CrossRef]
- Lieker, I.; Slowinski, T.; Harms, L.; Hahn, K.; Klehmet, J. A prospective study comparing tryptophan immunoadsorption with therapeutic plasma exchange for the treatment of chronic inflammatory demyelinating polyneuropathy. J. Clin. Apher. 2017, 32, 486–493. [Google Scholar] [CrossRef]
- Zinman, L.; Sutton, D.; Ng, E.; Nwe, P.; Ngo, M.; Bril, V. A pilot study to compare the use of the Excorim staphylococcal protein immunoadsorption system and IVIG in chronic inflammatory demyelinating polyneuropathy. Transfus. Apher. Sci. 2005, 33, 317–324. [Google Scholar] [CrossRef]
- Hadden, R.D.M.; Bensa, S.; Lunn, M.; Hughes, R. Immunoadsorption inferior to plasma exchange in a patient with chronic inflammatory demyelinating polyradiculoneuropathy. J. Neurol. Neurosurg. Psychiatry 2002, 72, 644–646. [Google Scholar] [CrossRef]
- Ullrich, H.; Mansouri-Taleghani, B.; Lackner, K.J.; Schalke, B.; Bogdahn, U.; Schmitz, G. Chronic inflammatory demyelinating polyradiculoneuropathy: Superiority of protein A immunoadsorption over plasma exchange treatment. Transfus. Sci. 1998, 19, 33–38. [Google Scholar] [CrossRef]
- Galldiks, N.; Burghaus, L.; Dohmen, C.; Teschner, S.; Pollok, M.; Leebmann, J.; Frischmuth, N.; Höllinger, P.; Nazli, N.; Fassbender, C.; et al. Immunoadsorption in Patients with Chronic Inflammatory Demyelinating Polyradiculoneuropathy with Unsatisfactory Response to First-Line Treatment. Eur. Neurol. 2011, 66, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, T.; Suzuki, N. Can immunoadsorption plasmapheresis be used as the first choice therapy for neuroimmunological disorders? Ther. Apher. 1997, 1, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Pernat, A.M.; Svigelj, V.; Ponikvar, R.; Buturović-Ponikvar, J. Guillain-Barré Syndrome Treated by Membrane Plasma Exchange and/or Immunoadsorption. Ther. Apher. Dial. 2009, 13, 310–313. [Google Scholar] [CrossRef]
- Arakawa, H.; Yuhara, Y.; Todokoro, M.; Kato, M.; Mochizuki, H.; Tokuyama, K.; Kunimoto, F.; Morikawa, A. Immunoadsorption therapy for a child with Guillain-Barre syndrome subsequent to Mycoplasma infection: A case study. Brain Dev. 2005, 27, 431–433. [Google Scholar] [CrossRef]
- Okamiya, S.; Ogino, M.; Ogino, Y.; Irie, S.; Kanazawa, N.; Saito, T.; Sakai, F. Tryptophan-immobilized Column-based Immunoadsorption as the Choice Method for Plasmapheresis in Guillain-Barre Syndrome. Ther. Apher. Dial. 2004, 8, 248–253. [Google Scholar] [CrossRef]
- Haupt, W.; Rosenow, F.; Van Der Ven, C.; Birkmann, C. Immunoadsorption in Guillain-Barré syndrome and myasthenia gravis. Ther. Apher. 2000, 4, 195–197. [Google Scholar] [CrossRef]
- Uetakagaito, M.; Horikawa, H.; Yoshinaka, H.; Tagawa, Y.; Yuki, N. Two Patients with Acute Guillain-Barré Syndrome Treated with Different Apheresis Methods. Ther. Apher. 1997, 1, 340–342. [Google Scholar] [CrossRef]
- Hirai, K.; Kihara, M.; Nakajima, F.; Miyanomae, Y.; Yoshioka, H. Immunoadsorption Therapy in Guillain-Barré Syndrome. Pediatric Neurol. 1998, 19, 55–57. [Google Scholar] [CrossRef]
- Chida, K.; Takase, S.; Itoyama, Y. Development of facial palsy during immunoadsorption plasmapheresis in Miller Fisher syndrome: A clinical report of two cases. J. Neurol. Neurosurg. Psychiatry 1998, 64, 399–401. [Google Scholar] [CrossRef]
- Ruiz, J.C.; Berciano, J.; Polo, J.M.; De Francisco, A.L.M.; Arias, M. Treatment of Guillain-Barré syndrome with protein-A immunoadsorption: Report of two cases. Ann. Neurol. 1992, 31, 574–575. [Google Scholar] [CrossRef] [PubMed]
- Takei, H.; Komaba, Y.; Araki, T.; Iino, Y.; Katayama, Y. Plasma Immunoadsorption Therapy for Guillain-Barré Syndrome: Critical Day for Initiation. J. Nippon. Med. Sch. 2002, 69, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Mahdi-Rogers, M.; Brassington, R.; A Gunn, A.; A Van Doorn, P.; Hughes, R.A. Immunomodulatory treatment other than corticosteroids, immunoglobulin and plasma exchange for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst. Rev. 2017, 2017, CD003280. [Google Scholar] [CrossRef] [PubMed]
- Rolfes, L.; Pfeuffer, S.; Ruck, T.; Melzer, N.; Pawlitzki, M.; Heming, M.; Brand, M.; Wiendl, H.; Meuth, S. Ruck Therapeutic Apheresis in Acute Relapsing Multiple Sclerosis: Current Evidence and Unmet Needs—A Systematic Review. J. Clin. Med. 2019, 8, 1623. [Google Scholar] [CrossRef] [PubMed]
- Lipphardt, M.; Wallbach, M.; Koziolek, M.J. Plasma Exchange or Immunoadsorption in Demyelinating Diseases: A Meta-Analysis. J. Clin. Med. 2020, 9, 1597. [Google Scholar] [CrossRef]
- Willison, H.J.; Yuki, N. Peripheral neuropathies and anti-glycolipid antibodies. Brain 2002, 125, 2591–2625. [Google Scholar] [CrossRef]
- Tagawa, Y.; Yuki, N.; Hirata, K. Ability to remove immunoglobulins and anti-ganglioside antibodies by plasma exchange, double-filtration plasmapheresis and immunoadsorption. J. Neurol. Sci. 1998, 157, 90–95. [Google Scholar] [CrossRef]
- Ng, J.K.M.; Malotka, J.; Kawakami, N.; Derfuss, T.; Khademi, M.; Olsson, T.; Linington, C.; Odaka, M.; Tackenberg, B.; Prüss, H.; et al. Neurofascin as a target for autoantibodies in peripheral neuropathies. Neurology 2012, 79, 2241–2248. [Google Scholar] [CrossRef]
- Querol, L.; Nogales-Gadea, G.; Rojas-García, R.; Martinez-Hernandez, E.; Diaz-Manera, J.; Suárez-Calvet, X.; Navas, M.; Araque, J.; Gallardo, E.; Illa, I. Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann. Neurol. 2012, 73, 370–380. [Google Scholar] [CrossRef]
- Querol, L.; Nogales-Gadea, G.; Rojas-Garcia, R.; Diaz-Manera, J.; Pardo, J.; Ortega-Moreno, A.; Sedano, M.J.; Gallardo, E.; Berciano, J.; Blesa, R.; et al. Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology 2014, 82, 879–886. [Google Scholar] [CrossRef]
- Doppler, K.; Appeltshauser, L.; Villmann, C.; Martin, C.; Peles, E.; Krämer, H.H.; Haarmann, A.; Buttmann, M.; Sommer, C. Auto-antibodies to contactin-associated protein 1 (Caspr) in two patients with painful inflammatory neuropathy. Brain 2016, 139, 2617–2630. [Google Scholar] [CrossRef]
- Delmont, E.; Manso, C.; Querol, L.; Cortese, A.; Berardinelli, A.; Lozza, A.; Belghazi, M.; Malissart, P.; Labauge, P.; Taieb, G.; et al. Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain 2017, 140, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- Dorst, J.; Ludolph, A.C.; Senel, M.; Tumani, H. Short-term and long-term effects of immunoadsorption in refractory chronic inflammatory demyelinating polyneuropathy: A prospective study in 17 patients. J. Neurol. 2018, 265, 2906–2915. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M.; Suzuki, H.; Oka, N.; Ogata, H.; Yanagimoto, S.; Sadakane, S.; Fukumoto, Y.; Yamana, M.; Yuhara, Y.; Yoshikawa, K.; et al. ELectron microscopic abnormality and therapeutic efficacy in chronic inflammatory demyelinating polyneuropathy with anti-neurofascin155 immunoglobulin G4 antibody. Muscle Nerve 2017, 57, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Süfke, S.; Lehnert, H.; Gebauer, F.; Uhlenbusch-Körwer, I. Safety Aspects of Immunoadsorption in IgG Removal Using a Single-Use, Multiple-pass Protein A Immunoadsorber (LIGASORB): Clinical Investigation in Healthy Volunteers. Ther. Apher. Dial. 2017, 21, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Chida, K.; Watanabe, S.; Okita, N.; Takase, S.; Tagawa, Y.; Yuki, N. Immunoadsorption therapy for Fisher’s syndrome: Analysis of the recovery process of external ophthalmoplegia and the removal ability of anti-GQ1b antibodies. Rinsho Shinkeigaku 1996, 36, 551–556. [Google Scholar]
- Belak, M.; Borberg, H.; Jimenez, C.; Oette, K. Technical and clinical experience with Protein A Immunoadsorption columns. Transfus. Sci. 1994, 15, 419–422. [Google Scholar] [CrossRef]
- Merkies, I.S.; Schmitz, P.I.M.; A Van Der Meché, F.G.; Samijn, J.; A Van Doorn, P. Clinimetric evaluation of a new overall disability scale in immune mediated polyneuropathies. J. Neurol. Neurosurg. Psychiatry 2002, 72, 596–601. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2017, 17, 162–173. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Petzold, A.; Bennett, J.L.; Berven, F.S.; Brundin, L.; Comabella, M.; Franciotta, D.; Frederiksen, J.L.; Fleming, J.O.; Furlan, R.; et al. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology 2009, 73, 1914–1922. [Google Scholar] [CrossRef]
- Pns, J.T.F.O.T.E.A.T.; Efns, J.T.F.O.T.; Pns, T. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society-First Revision. J. Peripher. Nerv. Syst. 2010, 15, 1–9. [Google Scholar] [CrossRef]
- Querol, L.; Rojas-García, R.; Diaz-Manera, J.; Barcena, J.; Pardo, J.; Ortega-Moreno, A.; Sedano, M.J.; Seró-Ballesteros, L.; Carvajal, A.; Ortiz-Castellon, N.; et al. Rituximab in treatment-resistant CIDP with antibodies against paranodal proteins. Neurol. Neuroimmunol. Neuroinflammation 2015, 2, e149. [Google Scholar] [CrossRef] [PubMed]
- Demichelis, C.; Franciotta, D.; Cortese, A.; Callegari, I.; Serrati, C.; Mancardi, G.L.; Schenone, A.; Leonardi, A.; Benedetti, L. Remarkable Rituximab Response on Tremor Related to Acute-Onset Chronic Inflammatory Demyelinating Polyradiculoneuropathy in an Antineurofascin155 Immunoglobulin G4-Seropositive Patient. Mov. Disord. Clin. Pr. 2018, 5, 559–560. [Google Scholar] [CrossRef] [PubMed]
- Cortese, A.; Lombardi, R.; Briani, C.; Callegari, I.; Benedetti, L.; Manganelli, F.; Luigetti, M.; Ferrari, S.; Clerici, A.M.; Marfia, G.A.; et al. Antibodies to neurofascin, contactin-1, and contactin-associated protein 1 in CIDP: Clinical relevance of IgG isotype. Neurol. Neuroimmunol. Neuroinflammation 2019, 7, e639. [Google Scholar] [CrossRef]
- Stich, O.; Perera, S.; Berger, B.; Jarius, S.; Wildemann, B.; Baumgartner, A.; Rauer, S. Prevalence of neurofascin-155 antibodies in patients with multiple sclerosis. J. Neurol. Sci. 2016, 364, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Häusser-Kinzel, S.; Weber, M.S. The Role of B Cells and Antibodies in Multiple Sclerosis, Neuromyelitis Optica, and Related Disorders. Front. Immunol. 2019, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Prineas, J.W.; Parratt, J.D. Multiple sclerosis: Serum anti-CNS autoantibodies. Mult. Scler. J. 2017, 24, 610–622. [Google Scholar] [CrossRef]
- Willison, H.J.; Veitch, J.; Swan, A.V.; Baumann, N.; Comi, G.; Gregson, N.A.; Iiia, I.; Jacobs, B.C.; Zielasek, J.; Hughes, R.A.C. Inter-laboratory validation of an ELISA for the determination of serum anti-ganglioside antibodies. Eur. J. Neurol. 1999, 6, 71–77. [Google Scholar] [CrossRef]
- Clark, A.; Kaller, M.; Galino, J.; Willison, H.J.; Rinaldi, S.; Bennett, D.L. Co-cultures with stem cell-derived human sensory neurons reveal regulators of peripheral myelination. Brain 2017, 140, 898–913. [Google Scholar] [CrossRef]





| PNAb Positive (n = 21) | PNAb Negative (n = 33) | Significance (PNAb+ v PNAb-neg) | |||
|---|---|---|---|---|---|
| Age: median, (range) | 58 (35–79) | 62 (5–90) | ns | p = 0.94 | Mann-Whitney |
| Male sex: n, (%) | 16 (76.2%) | 23 (69.7%) | ns | p = 0.76 | Fisher’s exact |
| Initial clinical diagnosis: | |||||
| ● GBS: n (%) | 6 (28.6%) | 10 (30.3%) | ns | p > 0.99 | Fisher’s exact (GBS or not) |
| ● CIDP: n (%) | 14 (66.7%) | 18 (54.5%) | ns | p = 0.41 | Fisher’s exact (CIDP or not) |
| ● Other: n (%) | 1 (4.7%) | 5 (15.1%) | ns | p = 0.39 | Fisher’s exact (Other or not) |
| Peak severity/nadir mRs (median, range) | 5 (2–6) | 4 (2–5) | ns | p = 0.10 | Mann-Whitney |
| Other serology: n/n (%) | |||||
| Any ganglioside Ab | 1/16 (6.3%) | 3/18 (16.7%) | ns | p = 0.60 | Fisher’s exact |
| ● GM1 | 1/16 (6.3%) | 2/18 (11.1%) | ns | p > 0.99 | Fisher’s exact |
| ● GQ1b | 0/16 | 1/18 (5.6%) | ns | p > 0.99 | Fisher’s exact |
| MAG | 0/4 | 1/8 (12.5%) | ns | p > 0.99 | Fisher’s exact |
| Paraprotein | 2/17 (11.8%) | 6/26 (23.1%) | ns | p = 0.45 | Fisher’s exact |
| ● IgM | 0/17 | 5/26 (19.2%) | ns | p = 0.14 | Fisher’s exact |
| ● IgG | 2/17 (11.8%) | 1/26 (3.8%) | ns | p = 0.55 | Fisher’s exact |
| Treatment % treated (% of those judged to have good response) | Difference in proportion treated/proportion with good response | ||||
| IVIg | 90.5 (5.3%) | 87.9% (3.4%) | ns/ns | p > 0.99/p > 0.99 | Fisher’s exact |
| Steroids | 85.7% (0) | 75.8% (8%) | ns/ns | p = 0.50/p = 0.50 | Fisher’s exact |
| PLEx | 95.2% (0) | 100% (24.2%) | ns/* | p = 0.39/*p = 0.01 | Fisher’s exact |
| IA | 19% (0) | 0 (0) | ***/ns | ***p < 0.001/ p > 0.99 | Fisher’s exact |
| Rituximab | 66.7% (64.3%) | 18.2% (16.7%) | ***/ns | ***p < 0.002/p = 0.14 | Fisher’s exact |
| Other immuno-suppression | 33.3% (28.6%) | 24.2% (12.5%) | ns/ns | p = 0.54/p = 0.47 | Fisher’s exact |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davies, A.J.; Fehmi, J.; Senel, M.; Tumani, H.; Dorst, J.; Rinaldi, S. Immunoadsorption and Plasma Exchange in Seropositive and Seronegative Immune-Mediated Neuropathies. J. Clin. Med. 2020, 9, 2025. https://doi.org/10.3390/jcm9072025
Davies AJ, Fehmi J, Senel M, Tumani H, Dorst J, Rinaldi S. Immunoadsorption and Plasma Exchange in Seropositive and Seronegative Immune-Mediated Neuropathies. Journal of Clinical Medicine. 2020; 9(7):2025. https://doi.org/10.3390/jcm9072025
Chicago/Turabian StyleDavies, Alexander J., Janev Fehmi, Makbule Senel, Hayrettin Tumani, Johannes Dorst, and Simon Rinaldi. 2020. "Immunoadsorption and Plasma Exchange in Seropositive and Seronegative Immune-Mediated Neuropathies" Journal of Clinical Medicine 9, no. 7: 2025. https://doi.org/10.3390/jcm9072025
APA StyleDavies, A. J., Fehmi, J., Senel, M., Tumani, H., Dorst, J., & Rinaldi, S. (2020). Immunoadsorption and Plasma Exchange in Seropositive and Seronegative Immune-Mediated Neuropathies. Journal of Clinical Medicine, 9(7), 2025. https://doi.org/10.3390/jcm9072025