Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development

 ,
,  and
and
Abstract
1. Introduction
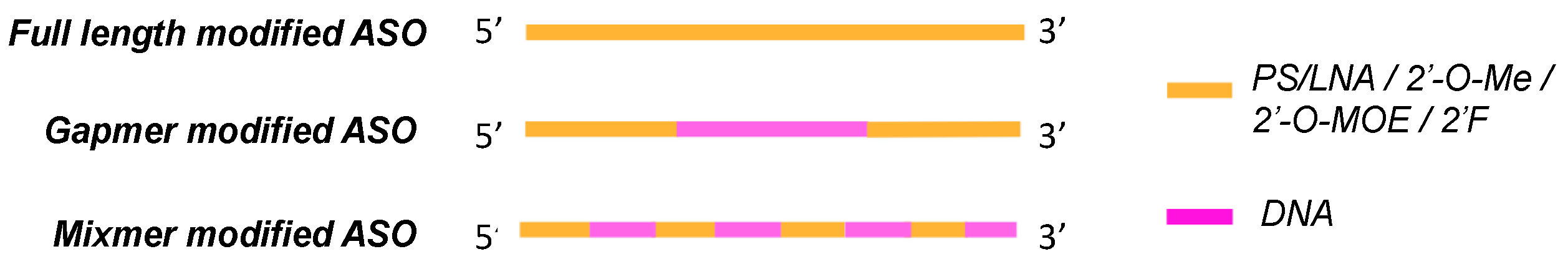
2. Oligonucleotide Modifications
2.1. Phosphorothioate (PS)
2.2. Phosphorodiamidate Morpholino Oligomer (PMO)
2.3. Peptide Nucleic Acids (PNA)
2.4. Locked Nucleic Acids (LNAs)
Ribose Modifications
2.5. Nucleobase Modification
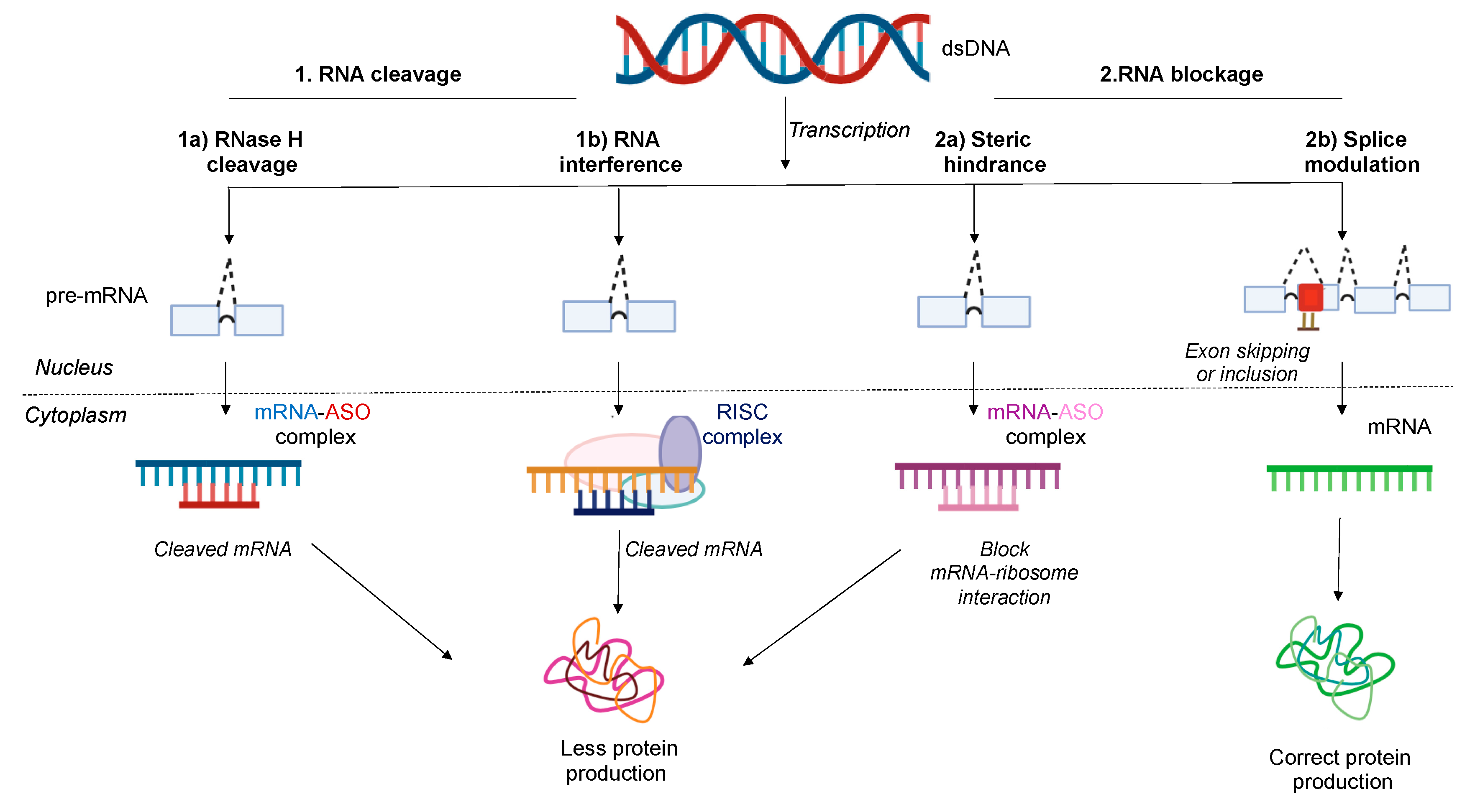
3. Antisense Mechanism of Action
3.1. RNase H1 Mediated Degradation
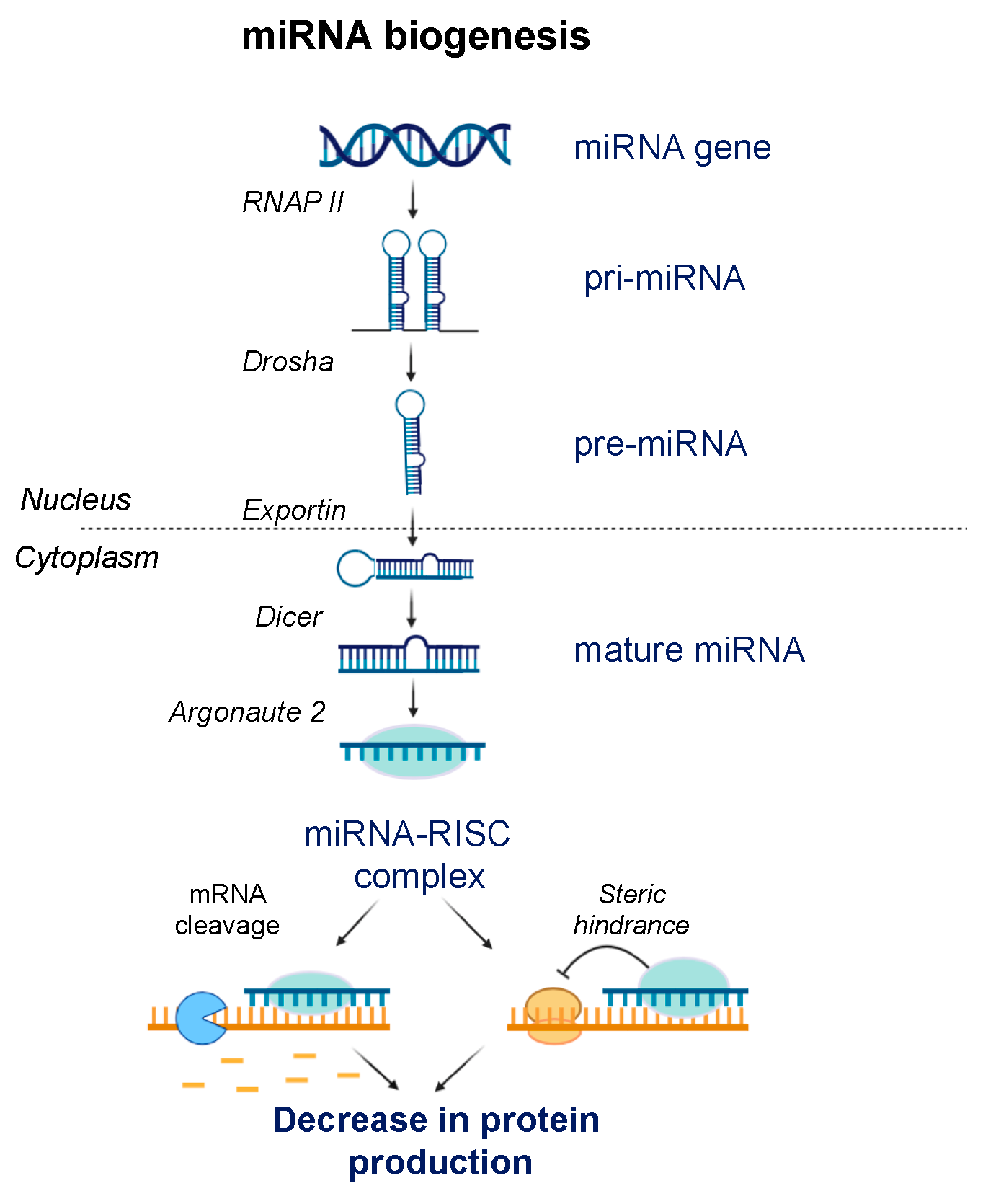
3.2. RNA Interference (RNAi)
3.3. Steric Blockage
3.3.1. Translation Arrest Due to Steric Hindrance
3.3.2. Splice Modulation or Splice Switching-Based Mechanism
4. Delivery of ASOs.
4.1. Enhanced Stabilization Chemistry-Based Delivery
4.2. Nanoformulation-Based Delivery
4.3. Lipid-Based Delivery Systems
5. FDA-Approved Formulations
5.1. Fomivirsen (VitraveneTM)
5.2. Mipomersen (KynamroTM)
5.3. Nusinersen (Spinraza®)
5.4. Patisiran (Onpattro®)
5.5. Inotersen (Tegsedi®)
5.6. Eteplirsen (Exondys 51®)
5.7. Golodirsen (Vyondys 53TM)
5.8. Givosiran (Givlaari®)
5.9. Milasen
6. Potential Drug Candidates in Clinical Trials
6.1. Tominersen
6.2. Tofersen
6.3. Volanesorsen (Waylivra®)
6.4. Alicaforsen
6.5. Vutrisiran
6.6. Fitusiran
6.7. Inclisiran
7. ASOs Targeting microRNA
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dá Mesquita, S.; Ferreira, A.C.; Sousa, J.C.; Correia-Neves, M.; Sousa, N.; Marques, F. Insights on the pathophysiology of Alzheimer’s disease: The crosstalk between amyloid pathology, neuroinflammation and the peripheral immune system. Neurosci. Biobehav. Rev. 2016, 68, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.L.; Burns, J.L.; Ramsey, B.W. Pathophysiology and Management of Pulmonary Infections in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 918–951. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting huntingtin expression in patients with Huntington’s disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease View project Sickle Cell Anemia View project. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Giegé, R. Toward a more complete view of tRNA biology. Nat. Struct. Mol. Biol. 2008, 15, 1007–1014. [Google Scholar] [CrossRef]
- Pink, R.C.; Wicks, K.; Caley, D.P.; Punch, E.K.; Jacobs, L.; Carter, D.R.F. Pseudogenes: Pseudo-functional or key regulators in health and diseasě. RNA 2011, 17, 792–798. [Google Scholar] [CrossRef]
- Girard, A.; Sachidanandam, R.; Hannon, G.J.; Carmell, M.A. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 2006, 442, 199–202. [Google Scholar] [CrossRef]
- Yao, R.W.; Wang, Y.; Chen, L.L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542–551. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Sharma, R.K.; Singh, S.K. Antisense oligonucleotides: Modifications and clinical trials. Medchemcomm 2014, 5, 1454–1471. [Google Scholar] [CrossRef]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of Antisense Drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Furdon, P.J.; Dominski, Z.; Kole, R. RNase H cleavage of RNA hybridized to oligonucleotides containing methylphosphonate, phosphorothioate and phosphodiester bonds. Nucleic Acids Res. 1989, 17, 9193–9204. [Google Scholar] [CrossRef]
- Eckstein, F. Phosphorothioate oligodeoxynucleotides: What is their origin and what is unique about them? Antisense Nucleic Acid Drug Dev. 2000, 10, 117–121. [Google Scholar] [CrossRef]
- Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387. [Google Scholar] [CrossRef]
- Crooke, S.T.; Bennett, C.F. Progress in antisense oligonucleotide therapeutics. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 107–129. [Google Scholar] [CrossRef]
- Agrawal, S.; Temsamani, J.; Tang, J.Y. Pharmacokinetics, biodistribution, and stability of oligodeoxynucleotide phosphorothioates in mice. Proc. Natl. Acad. Sci. USA 1991, 88, 7595–7599. [Google Scholar] [CrossRef]
- Agrawal, S.; Teamani, J.; Galbraith, W.; Tang, J. Pharmacokinetics of Antisense Oligonucleotides. Clin. Pharmacokinet. 1995, 28, 7–16. [Google Scholar] [CrossRef]
- Chiasson, B.J.; Armstrong, J.N.; Hooper, M.L.; Murphy, P.R.; Robertson, H.A. The application of antisense oligonucleotide technology to the brain: Some pitfalls. Cell. Mol. Neurobiol. 1994, 14, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Hudziak, R.M.; Barofsky, E.; Barofsky, D.F.; Weller, D.L.; Huang, S.; Weller, D.D. Resistance of Morpholino phosphorodiamidate oligomers to enzymatic degradation. Antisense Nucleic Acid Drug Dev. 1996, 6, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Devi, G.R.; Beer, T.M.; Corless, C.L.; Arora, V.; Weller, D.L.; Iversen, P.L. In vivo bioavailability and pharmacokinetics of a c-MYC antisense phosphorodiamidate morpholino oligomer, AVI-4126, in solid tumors. Clin. Cancer Res. 2005, 11, 3930–3938. [Google Scholar] [CrossRef]
- Pellestor, F.; Paulasova, P. The peptide nucleic acids (PNAs), powerful tools for molecular genetics and cytogenetics. Eur. J. Hum. Genet. 2004, 12, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Lundin, P.; Johansson, H.; Guterstam, P.; Holm, T.; Hansen, M.; Langel, Ü.; Andaloussi, S.E.L. Distinct uptake routes of cell-penetrating peptide conjugates. Bioconjug. Chem. 2008, 19, 2535–2542. [Google Scholar] [CrossRef] [PubMed]
- Dragulescu-Andrasi, A.; Rapireddy, S.; Frezza, B.M.; Gayathri, C.; Gil, R.R.; Ly, D.H. A simple γ-backbone modification preorganizes peptide nucleic acid into a helical structure. J. Am. Chem. Soc. 2006, 128, 10258–10267. [Google Scholar] [CrossRef]
- Zhou, P.; Wang, M.; Du, L.; Fisher, G.W.; Waggoner, A.; Ly, D.H. Novel binding and efficient cellular uptake of guanidine-based peptide nucleic acids (GPNA). J. Am. Chem. Soc. 2003, 125, 6878–6879. [Google Scholar] [CrossRef]
- Swenson, C.S.; Heemstra, J.M. Peptide nucleic acids harness dual information codes in a single molecule. Chem. Commun. 2020, 56, 1926–1935. [Google Scholar] [CrossRef]
- Braasch, D.A.; Corey, D.R. Locked nucleic acid (LNA): Fine-tuning the recognition of DNA and RNA. Chem. Biol. 2001, 8, 1–7. [Google Scholar] [CrossRef]
- Marrosu, E.; Ala, P.; Muntoni, F.; Zhou, H. Gapmer Antisense Oligonucleotides Suppress the Mutant Allele of COL6A3 and Restore Functional Protein in Ullrich Muscular Dystrophy. Mol. Ther. Nucleic Acids 2017, 8, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, P.H.; Persson, R.; Funder, E.D.; Albæk, N.; Diemer, S.L.; Hansen, D.J.; Møller, M.R.; Papargyri, N.; Christiansen, H.; Hansen, B.R.; et al. Locked nucleic acid: Modality, diversity, and drug discovery. Drug Discov. Today 2018, 23, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Kurreck, J. Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res. 2002, 30, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Freier, S. The ups and downs of nucleic acid duplex stability: Structure-stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acids Res. 1997, 25, 4429–4443. [Google Scholar] [CrossRef] [PubMed]
- Hebb, M.O.; Robertson, H.A. End-capped antisense oligodeoxynucleotides effectively inhibit gene expression in vivo and offer a low-toxicity alternative to fully modified phosphorothioate oligodeoxynucleotides. Mol. Brain Res. 1997, 47, 223–228. [Google Scholar] [CrossRef]
- Shen, W.; De Hoyos, C.L.; Sun, H.; Vickers, T.A.; Liang, X.H.; Crooke, S.T. Acute hepatotoxicity of 2 fluoro-modified 5–10–5 gapmer phosphorothioate oligonucleotides in mice correlates with intracellular protein binding and the loss of DBHS proteins. Nucleic Acids Res. 2018, 46, 2204–2217. [Google Scholar] [CrossRef]
- Krieg, A.M. Antiinfective applications of toll-like receptor 9 agonists. Proc. Am. Thorac. Soc. 2007, 4, 289–294. [Google Scholar] [CrossRef]
- Ortega, J.A.; Blas, J.R.; Orozco, M.; Grandas, A.; Pedroso, E.; Robles, J. Binding affinities of oligonucleotides and PNAs containing phenoxazine and G-clamp cytosine analogues are unusually sequence-dependent. Org. Lett. 2007, 9, 4503–4506. [Google Scholar] [CrossRef]
- Chenna, V.; Rapireddy, S.; Sahu, B.; Ausin, C.; Pedroso, E.; Ly, D.H. A simple cytosine to G-clamp nucleobase substitution enables chiral γ-PNAs to invade mixed-sequence double-helical B-form DNA. ChemBioChem 2008, 9, 2388–2391. [Google Scholar] [CrossRef]
- Rapireddy, S.; Bahal, R.; Ly, D.H. Strand invasion of mixed-sequence, double-helical B-DNA by γ-peptide nucleic acids containing g-clamp nucleobases under physiological conditions. Biochemistry 2011, 50, 3913–3918. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [PubMed]
- De Mesmaeker, A.; HÄner, R.; Martin, P.; Moser, H.E. Antisense Oligonucleotides. Acc. Chem. Res. 1995, 28, 366–374. [Google Scholar] [CrossRef]
- Ward, A.J.; Norrbom, M.; Chun, S.; Bennett, C.F.; Rigo, F. Nonsense-mediated decay as a terminating mechanism for antisense oligonucleotides. Nucleic Acids Res. 2014, 42, 5871–5879. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.; Wood, M.J.A. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lima, W.F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S.T. Determination of the Role of the Human RNase H1 in the Pharmacology of DNA-like Antisense Drugs. J. Biol. Chem. 2004, 279, 17181–17189. [Google Scholar] [CrossRef]
- Yin, W.; Rogge, M. Targeting RNA: A Transformative Therapeutic Strategy. Clin. Transl. Sci. 2019, 12, 98–112. [Google Scholar] [CrossRef]
- Crooke, S.T. Molecular mechanisms of action of antisense drugs. Biochim. Biophys. Acta Gene Struct. Exp. 1999, 1489, 31–44. [Google Scholar] [CrossRef]
- Zamore, P.D.; Tuschl, T.; Sharp, P.A.; Bartel, D.P. RNAi: Double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 2000, 101, 25–33. [Google Scholar] [CrossRef]
- Matranga, C.; Tomari, Y.; Shin, C.; Bartel, D.P.; Zamore, P.D. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 2005, 123, 607–620. [Google Scholar] [CrossRef]
- Rand, T.A.; Petersen, S.; Du, F.; Wang, X. Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell 2005, 123, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D.W.Y. RNAi therapeutics: A potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006, 2, 711–719. [Google Scholar] [CrossRef]
- Dias, N.; Dheur, S.; Nielsen, P.E.; Gryaznov, S.; Van Aerschot, A.; Herdewijn, P.; Hélène, C.; Saison-Behmoaras, T.E. Antisense PNA tridecamers targeted to the coding region of Ha-ras mRNA arrest polypeptide chain elongation. J. Mol. Biol. 1999, 294, 403–416. [Google Scholar] [CrossRef]
- Bennett, C.F.; Swayze, E.E. RNA Targeting Therapeutics: Molecular Mechanisms of Antisense Oligonucleotides as a Therapeutic Platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef] [PubMed]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in control of gene expression: An overview of nuclear functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef]
- Qi, L.L.; Rabinowitz, A.; Yun, C.C.; Yokota, T.; Yin, H.F.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restorers dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef]
- Svasti, S.; Suwanmanee, T.; Fucharoen, S.; Moulton, H.M.; Nelson, M.H.; Maeda, N.; Smithies, O.; Kole, R. RNA repair restores hemoglobin expression in IVS2-654 thalassemic mice. Proc. Natl. Acad. Sci. USA 2009, 106, 1205–1210. [Google Scholar] [CrossRef]
- Passini, M.A.; Bu, J.; Richards, A.M.; Kinnecom, C.; Sardi, S.P.; Stanek, L.M.; Hua, Y.; Rigo, F.; Matson, J.; Hung, G.; et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef]
- Siva, K.; Covello, G.; Denti, M.A. Exon-skipping antisense oligonucleotides to correct missplicing in neurogenetic diseases. Nucleic Acid Ther. 2014, 24, 69–86. [Google Scholar] [CrossRef]
- van der Wal, E.; Bergsma, A.J.; Pijnenburg, J.M.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. Antisense Oligonucleotides Promote Exon Inclusion and Correct the Common c.-32-13T>G GAA Splicing Variant in Pompe Disease. Mol. Ther. Nucleic Acids 2017, 7, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Pagani, F.; Baralle, F.E. Genomic variants in exons and introns: Identifying the splicing spoilers. Nat. Rev. Genet. 2004, 5, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P. Aberrant and alternative splicing in cancer. Cancer Res. 2004, 64, 7647–7654. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Duelli, D.M.; Hastings, M.L. Targeting RNA splicing for disease therapy. Wiley Interdiscip. Rev. RNA 2013, 4, 247–266. [Google Scholar] [CrossRef]
- Bauman, J.; Jearawiriyapaisarn, N.; Kole, R. Therapeutic potential of splice-switching oligonucleotides. Oligonucleotides 2009, 19, 1–13. [Google Scholar] [CrossRef]
- Dominski, Z.; Kole, R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1993, 18, 8673–8677. [Google Scholar] [CrossRef]
- Clark, A.J.; Davis, M.E. Increased brain uptake of targeted nanoparticles by adding an acid-cleavable linkage between transferrin and the nanoparticle core. Proc. Natl. Acad. Sci. USA 2015, 112, 12486–12491. [Google Scholar] [CrossRef]
- Lorenzer, C.; Dirin, M.; Winkler, A.M.; Baumann, V.; Winkler, J. Going beyond the liver: Progress and challenges of targeted delivery of siRNA therapeutics. J. Control. Release 2015, 203, 1–15. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Huang, Y. Preclinical and Clinical Advances of GalNAc-Decorated Nucleic Acid Therapeutics. Mol. Ther. Nucleic Acids 2017, 17, 116–132. [Google Scholar] [CrossRef]
- Shi, B.; Abrams, M.; Sepp-Lorenzino, L. Expression of Asialoglycoprotein Receptor 1 in Human Hepatocellular Carcinoma. J. Histochem. Cytochem. 2013, 61, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Xu, Y.; Solis, L.M.; Tao, W.; Wang, L.; Behrens, C.; Xu, X.; Zhao, L.; Liu, D.; Wu, J.; et al. Long-circulating siRNA nanoparticles for validating Prohibitin1-targeted non-small cell lung cancer treatment. Proc. Natl. Acad. Sci. USA 2015, 112, 7779–7784. [Google Scholar] [CrossRef] [PubMed]
- Schiffelers, R.M.; Ansari, A.; Xu, J.; Zhou, Q.; Tang, Q.; Storm, G.; Molema, G.; Lu, P.Y.; Scaria, P.V.; Woodle, M.C. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res. 2004, 32, e149. [Google Scholar] [CrossRef] [PubMed]
- Benjaminsen, R.V.; Mattebjerg, M.A.; Henriksen, J.R.; Moghimi, S.M.; Andresen, T.L. The possible “proton sponge “ effect of polyethylenimine (PEI) does not include change in lysosomal pH. Mol. Ther. 2013, 21, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Cai, C.; Xie, Y.; Wu, L.; Chen, X.; Liu, H.; Zhou, Y.; Zou, H.; Liu, D.; Zhao, Y.; Kong, X.; et al. PLGA-based dual targeted nanoparticles enhance miRNA transfection efficiency in hepatic carcinoma. Sci. Rep. 2017, 7, 46250. [Google Scholar] [CrossRef]
- Rehman, Z.U.; Hoekstra, D.; Zuhorn, I.S. Mechanism of polyplex- and lipoplex-mediated delivery of nucleic acids: Real-time visualization of transient membrane destabilization without endosomal lysis. ACS Nano 2013, 28, 3767–3777. [Google Scholar] [CrossRef]
- Mahmoodi Chalbatani, G.; Dana, H.; Gharagouzloo, E.; Grijalvo, S.; Eritja, R.; Logsdon, C.D.; Memari, F.; Miri, S.R.; Rad, M.R.; Marmari, V. Small interfering RNAs (siRNAs) in cancer therapy: A nano-based approach. Int. J. Nanomed. 2019, 2019, 3111–3128. [Google Scholar] [CrossRef]
- Kumar, V.; Qin, J.; Jiang, Y.; Duncan, R.G.; Brigham, B.; Fishman, S.; Nair, J.K.; Akinc, A.; Barros, S.A.; Kasperkovitz, P.V. Shielding of lipid nanoparticles for siRNA delivery: Impact on physicochemical properties, cytokine induction, and efficacy. Mol. Ther. Nucleic Acids 2014, 3, e210. [Google Scholar] [CrossRef]
- Azad, R.F.; Driver, V.B.; Tanaka, K.; Crooke, R.M.; Anderson, K.P. Antiviral activity of a phosphorothioate oligonucleotide complementary to RNA of the human cytomegalovirus major immediate-early region. Antimicrob. Agents Chemother. 1993, 37, 1945–1954. [Google Scholar] [CrossRef]
- Geary, R.S.; Henry, S.P.; Grillone, L.R. Fomivirsen: Clinical pharmacology and potential drug interactions. Clin. Pharmacokinet. 2002, 41, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Hutcherson, S.L.; Lanz, R. A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with aids. Am. J. Ophthalmol. 2002, 133, 467–474. [Google Scholar] [CrossRef]
- Goldberg, A.C.; Hopkins, P.N.; Toth, P.P.; Ballantyne, C.M.; Rader, D.J.; Robinson, J.G.; Daniels, S.R.; Gidding, S.S.; De Ferranti, S.D.; Ito, M.K.; et al. Familial hypercholesterolemia: Screening, diagnosis and management of pediatric and adult patients: Clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J. Clin. Lipidol. 2011, 5, S1–S8. [Google Scholar] [CrossRef]
- Raal, F.J.; Santos, R.D.; Blom, D.J.; Marais, A.D.; Charng, M.J.; Cromwell, W.C.; Lachmann, R.H.; Gaudet, D.; Tan, J.L.; Chasan-Taber, S.; et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: A randomised, double-blind, placebo-controlled trial. Lancet 2010, 375, 998–1006. [Google Scholar] [CrossRef]
- Hair, P.; Cameron, F.; McKeage, K. Mipomersen sodium: First global approval. Drugs 2013, 73, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal muscular atrophy: A timely review. Arch. Neurol. 2011, 68, 979–984. [Google Scholar] [CrossRef]
- Hua, Y.; Liu, Y.H.; Sahashi, K.; Rigo, F.; Frank Bennett, C.; Krainer, A.R. Motor neuron cell-nonautonomous rescue of spinal muscular atrophy phenotypes in mild and severe transgenic mouse models. Genes Dev. 2015, 29, 288–297. [Google Scholar] [CrossRef]
- Rigo, F.; Chun, S.J.; Norris, D.A.; Hung, G.; Lee, S.; Matson, J.; Fey, R.A.; Gaus, H.; Hua, Y.; Grundy, J.S.; et al. Pharmacology of a central nervous system delivered 2′-O-methoxyethyl- modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J. Pharmacol. Exp. Ther. 2014, 350, 46–55. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Wurster, C.D.; Winter, B.; Wollinsky, K.; Ludolph, A.C.; Uzelac, Z.; Witzel, S.; Schocke, M.; Schneider, R.; Kocak, T. Intrathecal administration of nusinersen in adolescent and adult SMA type 2 and 3 patients. J. Neurol. 2019, 266, 183–194. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Effect of Nusinersen on Adults with Spinal Muscular Atrophy. Available online: https://clinicaltrials.gov/ct2/show/NCT03878030 (accessed on 19 March 2020).
- Sekijima, Y. Transthyretin (ATTR) amyloidosis: Clinical spectrum, molecular pathogenesis and disease-modifying treatments. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.J. Cell and Molecular Biology of Transthyretin and Thyroid Hormones. Int. Rev. Cytol. 2007, 258, 137–193. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Koike, H.; Slama, M.; Coelho, T. Hereditary transthyretin amyloidosis: A model of medical progress for a fatal disease. Nat. Rev. Neurol. 2019, 15, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Cullis, P.R.; Van Der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Patisiran in Patients with Hereditary Transthyretin-Mediated Amyloidosis (hATTR Amyloidosis) Disease Progression Post-Liver Transplant. Available online: https://clinicaltrials.gov/ct2/show/NCT03862807 (accessed on 19 March 2020).
- ClinicalTrials.gov. The Study of an Investigational Drug, Patisiran (ALN-TTR02), for the Treatment of Transthyretin (TTR)-Mediated Amyloidosis in Patients Who Have Already been Treated with ALN-TTR02 (Patisiran). Available online: https://clinicaltrials.gov/ct2/show/NCT02510261 (accessed on 19 March 2020).
- ClinicalTrials.gov. HELIOS-A: A Study of Vutrisiran (ALN-TTRSC02) in Patients with Hereditary Transthyretin Amyloidosis (hATTR Amyloidosis). Available online: https://clinicaltrials.gov/ct2/show/NCT03759379 (accessed on 19 March 2020).
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen treatment for patients with Hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef]
- McClorey, G.; Moulton, H.M.; Iversen, P.L.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006, 13, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Study of Eteplirsen in Young Patients with DMD Amenable to Exon 51 Kkipping. Available online: https://clinicaltrials.gov/ct2/show/NCT03218995 (accessed on 19 March 2020).
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020, 94, e2270–e2282. [Google Scholar] [CrossRef] [PubMed]
- Bissell, D.M.; Anderson, K.E.; Bonkovsky, H.L. Porphyria. N. Engl. J. Med. 2017, 377, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Givosiran: First Approval. Drugs 2020, 80, 335–339. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Study to Evaluate Long-Term Safety and Clinical Activity of Givosiran (ALN-AS1) in Patient with Acute Intermittent Porphyria (AIP). Available online: https://clinicaltrials.gov/ct2/show/NCT02949830 (accessed on 19 March 2020).
- Kim, J.; Hu, C.; El Achkar, C.M.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Finkbeiner, S. Huntington’s disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a007476. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Biogen. Biogen to Present New Interim Data from its Phase 1/2 Clinical Stidy of Tofersen (BIIB067) for the Potential Treatment of a Subtype of Familial Amyotrophic Lateral Sclerosis (ALS). Available online: https://investors.biogen.com/news-releases/news-release-details/biogen-present-new-interim-data-its-phase-12-clinical-study (accessed on 11 May 2020).
- Witztum, J.L.; Gaudet, D.; Freedman, S.D.; Alexander, V.J.; Digenio, A.; Williams, K.R.; Yang, Q.; Hughes, S.G.; Geary, R.S.; Arca, M.; et al. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome. N. Engl. J. Med. 2019, 381, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Van Deventer, S.J.H.; Wedel, M.K.; Baker, B.F.; Xia, S.; Chuang, E.; Miner, P.B. A Phase II dose ranging, double-blind, placebo-controlled study of alicaforsen enema in subjects with acute exacerbation of mild to moderate left-sided ulcerative colitis. Aliment. Pharmacol. Ther. 2006, 23, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Shilling, R.; Karsten, V.; Silliman, N.; Chen, J.; Li, W.; Vest, J. Study design and rationale of HELIOS-B: A Phase 3 study to evaluate the clinical efficacy and safety of Vutrisiran in patients with ATTR amyloidosis with cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 3579. [Google Scholar] [CrossRef]
- Mannucci, P.M.; Tuddenham, E.G.D. The hemophilias—From royal genes to gene therapy. N. Engl. J. Med. 2001, 344, 1773–1779. [Google Scholar] [CrossRef] [PubMed]
- Machin, N.; Ragni, M.V. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J. Blood Med. 2018, 9, 135–140. [Google Scholar] [CrossRef]
- Bruikman, C.S.; Hovingh, G.K.; Kastelein, J.J.P. Molecular basis of familial hypercholesterolemia. Curr. Opin. Cardiol. 2017, 32, 262–266. [Google Scholar] [CrossRef]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the treatment of heterozygous familial hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. ARRx in Combination with Enzalutamide in Metastatic Castration Resistant Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03300505 (accessed on 19 March 2020).
- ClinicalTrials.gov. A Study of RG-012 in Subjects With Alport Syndrome. Available online: https://clinicaltrials.gov/ct2/show/NCT03373786 (accessed on 19 March 2020).
- ClinicalTrials.gov. Exploratory Study to Evaluate QR-010 in Subjects with Cystic Fibrosis ΔF508 CFTR Mutation. Available online: https://clinicaltrials.gov/ct2/show/NCT02564354 (accessed on 19 March 2020).
- ClinicalTrials.gov. Phase I dose Escalation Study to Investigate the Safety of ISTH0036 in Subjects sith Glaucoma Undergoing Trabeculectomy. Available online: https://clinicaltrials.gov/ct2/show/NCT02406833 (accessed on 19 March 2020).
- ClinicalTrials.gov. Study of ARO-APOC3 in Healthy Volunteers, Hypertriglyceridemic Patients and Patients with Familial Chylomicronemia Syndrome (FCS). Available online: https://clinicaltrials.gov/ct2/show/NCT03783377 (accessed on 19 March 2020).
- ClinicalTrials.gov. Study of ARO-ANG3 in Healthy Volunteers and in Dyslipidemic Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT03747224 (accessed on 19 March 2020).
- ClinicalTrials.gov. Safety Study of a Single IVT Injection of QPI-1007 in Chronic Optic Nerve Atrophy and Recent Onset NAION Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT01064505 (accessed on 19 March 2020).
- ClinicalTrials.gov. A Study of ALN-AAT02 in Healthy Participants and Participants with ZZ Type alpha-1 Antitrypsin Deficiency Liver Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03767829 (accessed on 19 March 2020).
- ClinicalTrials.gov. A Study of MEDI1191 in Sequential and Concurrent Combination with Durvalumab in Subjects with Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT03946800 (accessed on 19 March 2020).
- ClinicalTrials.gov. A Safety, Tolerability, PK, and PD Study of Once Weekly ISIS-FGFR4RX SC in Obese Patients (FGFR4-CS2). Available online: https://clinicaltrials.gov/ct2/show/NCT02476019 (accessed on 19 March 2020).
- ClinicalTrials.gov. Safety, Tolerability, and Pharmacodynamics of IONIS-DGAT2Rx in Adult Patients with Type 2 Diabetes. Available online: https://clinicaltrials.gov/ct2/show/NCT03334214 (accessed on 19 March 2020).
- ClinicalTrials.gov. An Extension Study of IONIS-PKK-LRx in Participants with Hereditary Angioedema. Available online: https://clinicaltrials.gov/ct2/show/NCT04307381 (accessed on 19 March 2020).
- ClinicalTrials.gov. Safety, Tolerability and Efficacy of ISIS-GCGRRx in Patients with Type 2 Diabetes. Available online: https://clinicaltrials.gov/ct2/show/NCT02583919 (accessed on 19 March 2020).
- ClinicalTrials.gov. Evaluation of Safety and Feasibility of OGX-011 in Combination with 2nd-line Chemotherapy in Patients with HRPC. Available online: https://clinicaltrials.gov/ct2/show/NCT00327340 (accessed on 19 March 2020).
- ClinicalTrials.gov. A Phase 2 Study Comparing Chemotherapy in Combination with OGX-427 or Placebo in Patients with Bladder Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT01454089 (accessed on 19 March 2020).
- ClinicalTrials.gov. Phase 2 Study of ISIS 681257 (AKCEA-APO(a)-LRx) in Patients with Hyperlipoproteinemia(a) and Cardiovascular Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03070782 (accessed on 19 March 2020).
- ClinicalTrials.gov. Study of AKCEA-ANGPTL3-LRX (ISIS 703802) in Patients with Familial Partial Lipodystrophy (FPL). Available online: https://clinicaltrials.gov/ct2/show/NCT03514420 (accessed on 19 March 2020).
- ClinicalTrials.gov. Study of ISIS 678354 (AKCEA-APOCIII-LRx) in Patients with Hypertriglyceridemia and Established Ardiovascular Disease (CVD). Available online: https://clinicaltrials.gov/ct2/show/NCT03385239 (accessed on 19 March 2020).
- ClinicalTrials.gov. Danvatirsen and Durvalumab in Treating Patients with Advanced and Refractory Pancreatic, Non-Small Cell Lung Cancer, and Mismatch Repair Deficient Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02983578 (accessed on 19 March 2020).
- ClinicalTrials.gov. SOLAR: Efficacy and Safety of Cobomarsen (MRG-106) vs. Active Comparator in Subjects with Mycosis Fungoides (SOLAR). Available online: https://clinicaltrials.gov/ct2/show/NCT03713320 (accessed on 19 March 2020).
- ClinicalTrials.gov. Efficacy, Safety, and Tolerability of Remlarsen (MRG-201) Following Intradermal Injection in Subjects with a History of Keloids. Available online: https://clinicaltrials.gov/ct2/show/NCT03601052 (accessed on 19 March 2020).
- ClinicalTrials.gov. A Study of Cemdisiran in Adults with Immunoglobulin A Nephropathy (IgAN). Available online: https://clinicaltrials.gov/ct2/show/NCT03841448 (accessed on 19 March 2020).
- ClinicalTrials.gov. Assessment of Changes in a Novel Histological Activity Scale in Response to ARO-AAT. Available online: https://clinicaltrials.gov/ct2/show/NCT03946449 (accessed on 19 March 2020).
- ClinicalTrials.gov. PF-04523655 dose Escalation Study, and Evaluation of PF-04523655 with/without Ranibizumab in Diabetic Macular Edema (DME) (MATISSE). Available online: https://clinicaltrials.gov/ct2/show/NCT01445899 (accessed on 19 March 2020).
- ClinicalTrials.gov. AZD8601 Study in CABG Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT03370887 (accessed on 19 March 2020).
- ClinicalTrials.gov. Study of DS-5141b in Patients with Duchenne Muscular Dystrophy. Available online: https://clinicaltrials.gov/ct2/show/NCT02667483 (accessed on 19 March 2020).
- ClinicalTrials.gov. Safety, Efficacy, PK, and PD Characteristics of Orally Inhaled SB010 in Male Patients with Mild Asthma (Multiple Dose). Available online: https://clinicaltrials.gov/ct2/show/NCT01743768 (accessed on 19 March 2020).
- ClinicalTrials.gov. Miravirsen Study in Null Responder to Pegylated Interferon Alpha Plus Ribavirin Subjects with Chronic Hepatitis C. Available online: https://clinicaltrials.gov/ct2/show/NCT01727934 (accessed on 19 March 2020).
- ClinicalTrials.gov. Clinical Trial of BP1001 (Liposomal Grb2 Antisense Oligonucleotide) in Combination with Dasatinib in Patients with Ph + CML Who Have Failed TKI, Ph+ AML, Ph+ MDS. Available online: https://clinicaltrials.gov/ct2/show/NCT02923986 (accessed on 19 March 2020).
- ClinicalTrials.gov. An Open-Label Extension Study to Evaluate Long-Term Safety and Tolerability of RO7234292 (RG6042) in Huntington’s Disease Patients Who Participated in Prior Roche and Genentech Sponsored Studies. Available online: https://clinicaltrials.gov/ct2/show/NCT03842969 (accessed on 19 March 2020).
- ClinicalTrials.gov. An Efficacy, Safety, Tolerability, Pharmacokinetics and Pharmacodynamics Study of BIIB067 in Adults with Inherited Mmyotrophic Lateral Sclerosis (ALS) (VALOR (Part C)). Available online: https://clinicaltrials.gov/ct2/show/NCT02623699 (accessed on 19 March 2020).
- ClinicalTrials.gov. Open-Label Extension Assessing Long Term Safety and Efficacy of IONIS-TTR Rx in Familial Amyloid Polyneuropathy (FAP). Available online: https://clinicaltrials.gov/ct2/show/NCT02175004 (accessed on 19 March 2020).
- ClinicalTrials.gov. The Approach Open Label Study: A Study of Volanesorsen (Formerly IONIS-APOCIIIRx) in Patients with Familial Chylomicronemia Syndrome. Available online: https://clinicaltrials.gov/ct2/show/NCT02658175 (accessed on 19 March 2020).
- ClinicalTrials.gov. CARDIO-TTRansform: A Study to Evaluate the Efficacy and Safety of AKCEA-TTR-LRx in Participants with Transthyretin-Mediated Amyloid Cardiomyopathy (ATTR CM). Available online: https://clinicaltrials.gov/ct2/show/NCT04136171 (accessed on 19 March 2020).
- ClinicalTrials.gov. Efficacy of Alicaforsen in Pouchitis Patients Who Have Failed to Respond to at Least One Course of Antibiotics. Available online: https://clinicaltrials.gov/ct2/show/NCT02525523 (accessed on 19 March 2020).
- ClinicalTrials.gov. HELIOS-B: A Study to Evaluate Vutrisiran in Patients with Transthyretin Amyloidosis with Cardiomyopathy. Available online: https://clinicaltrials.gov/ct2/show/NCT04153149 (accessed on 19 March 2020).
- ClinicalTrials.gov. A Study of Fitusiran in Severe Hemophilia A and B Patients Previously Receiving Factor or Bypassing Agent Prophylaxis (ATLAS-PPX). Available online: https://clinicaltrials.gov/ct2/show/NCT03549871 (accessed on 19 March 2020).
- ClinicalTrials.gov. QPI-1002 Phase 3 for Prevention of Major Adverse Kidney Events (MAKE) in Subjects at High Risk for AKI Following Cardiac Surgery. Available online: https://clinicaltrials.gov/ct2/show/NCT03510897 (accessed on 19 March 2020).
- ClinicalTrials.gov. Trial to Assess the Effect of Long Term Dosing of Inclisiran in Subjects with High CV Risk and Elevated LDL-C (ORION-8). Available online: https://clinicaltrials.gov/ct2/show/NCT03814187 (accessed on 19 March 2020).
- Ottosen, S.; Parsley, T.B.; Yang, L.; Zeh, K.; Van Doorn, L.J.; Van Der Veer, E.; Raney, A.K.; Hodges, M.R.; Patick, A.K. In Vitro antiviral activity and preclinical and clinical resistance profile of miravirsen, a novel anti-hepatitis C virus therapeutic targeting the human factor miR-122. Antimicrob. Agents Chemother. 2015, 59, 599–608. [Google Scholar] [CrossRef]
- Gomez, I.G.; MacKenna, D.A.; Johnson, B.G.; Kaimal, V.; Roach, A.M.; Ren, S.; Nakagawa, N.; Xin, C.; Newitt, R.; Pandya, S.; et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Investig. 2015, 125, 141–156. [Google Scholar] [CrossRef]
- Shah, M.Y.; Ferrajoli, A.; Sood, A.K.; Lopez-Berestein, G.; Calin, G.A. microRNA Therapeutics in Cancer—An Emerging Concept. EBioMedicine 2016, 12, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Eis, P.S.; Tam, W.; Sun, L.; Chadburn, A.; Li, Z.; Gomez, M.F.; Lund, E.; Dahlberg, J.E. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc. Natl. Acad. Sci. USA 2005, 102, 3627–3632. [Google Scholar] [CrossRef]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef] [PubMed]
- Seto, A.G.; Beatty, X.; Lynch, J.M.; Hermreck, M.; Tetzlaff, M.; Duvic, M.; Jackson, A.L. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br. J. Haematol. 2018, 183, 428–444. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Watts, J.; Corey, D. Gene silencing by siRNAs and antisense oligonucleotides in the laboratory and the clinic. J. Pathol. 2012, 226, 365–379. [Google Scholar] [CrossRef]
- Spurgers, K.B.; Sharkey, C.M.; Warfield, K.L.; Bavari, S. Oligonucleotide antiviral therapeutics: Antisense and RNA interference for highly pathogenic RNA viruses. Antivir. Res. 2008, 78, 26–36. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef]
- Rossi, J.J.; Rossi, D. Oligonucleotides and the COVID-19 Pandemic: A Perspective. Nucleic Acid Ther. 2020, 30, 129–132. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Mechanism | Properties |
|---|---|---|---|
| Phosphate modification | |||
| Phosphorothioate (PS) |  | RNase H1 cleavage | Enzymatic stability |
| Sugar phosphatemodification | |||
| Phosphorodiamidate morpholino (PMO) |  | Steric hindrance/splice modulation | Improved aqueous solubility, higher binding affinity |
| Peptide nucleic acid (PNA) |  | Steric hindrance/splice modulation | Enzymatic stability, higher binding affinity, no immune activation |
| Sugar modification | |||
| Locked nucleic acid (LNA) |  | Steric hindrance/RNase H1 cleavage | Higher binding affinity, enzymatic stability |
| 2′-O-methyl (2′-O-Me) |  | Steric hindrance/splice modulation | Higher binding affinity, enzymatic stability, reduced immune stimulation |
| 2′-O-methoxyethyl (2′-O-MOE) |  | Steric hindrance/splice modulation | Higher binding affinity, enzymatic stability, reduced immune stimulation |
| 2′fluoro (2′ F) |  | Steric hindrance/splice modulation | Higher binding affinity |
| NucleoBase modification | |||
| 5′methylcytosine |  | RNase H1 cleavage | Higher binding affinity, no immune stimulation |
| G-clamp |  | Steric hindrance | Higher binding affinity |
| Drug | Chemistry | Route | Target | Indication | Approval | Designation | Company |
|---|---|---|---|---|---|---|---|
| Fomivirsen (VitraveneTM) | PS | IVT | CMV mRNA | CMV infection | FDA (1998) | - | Ionis |
| Mipomersen (KynamroTM) | 2′-O-MOE, PS, 5-methyl cytosine | SC | apo-B-100 mRNA | HoFH | FDA (2013) | Orphan | Genzyme |
| Nusinersen (Spinraza®) | 2′-O-MOE, PS, 5-methyl cytosine | ITH | SMN2 pre-mRNA | SMA | FDA (2016), EMA (2017) | Orphan | Biogen |
| Patisiran (Onpattro®) | siRNA | IV | TTR mRNA | hATTR | FDA (2018), EMA (2018) | Orphan | Alnylam |
| Inotersen (Tegsedi®) | 2′-O-MOE, PS | SC | TTR mRNA | hATTR | FDA (2018), EMA (2018) | Orphan | Ionis |
| Eteplirsen (Exondys 51®) | PMO | IV | exon 51 | DMD | FDA (2016), EMA (2018) | Orphan | Sarepta |
| Golodirsen (Vyondys 53TM) | PMO | IV | DMD pre-mRNA | DMD | FDA (2019) | Orphan | Sarepta |
| Givosiran (Givlaari®) | siRNA | SC | ALS1 mRNA | AHP | FDA (2019), EMA (2020) | Orphan | Alnylam |
| Milasen | 2′-O-MOE, PS, 5-methyl cytosine | ITH | intron 6 spice acceptor cryptic site | CLN7 | FDA * (2018) | Orphan | Boston Children’s Hospital |
| Drug Candidate NCT ID | Chemistry/Delivery | Target | MOA | Route | Company | Indication | Ref. |
|---|---|---|---|---|---|---|---|
| Phase I status | |||||||
| ARRx 03300505 | cEt gapmer | Androgen receptor mRNA | RNase H1 | IV | Rogel Cancer Center | Prostate cancer | [126] |
| RG-012 03373786 | - | miR-21 | antimiR | SC | Genzyme | Alport syndrome | [127] |
| QR-010 02564354 | - | CFTR mRNA | Splice modulation | IN | ProQR | Cystic fibrosis | [128] |
| ISTH0036 02406833 | LNA | TGF beta 2 | RNase H1 | IVT | Isarna | Primary open angle glaucoma | [129] |
| ARO-APOC3 03783377 | siRNA-GalNAc | ApoC-III mRNA | RNAi | SC | Arrow head | HTG, FCS | [130] |
| ARO-ANG3 03747224 | siRNA-GalNAc | Angiopoietin-like protein 3 mRNA | RNAi | SC | Arrow head | Dyslipidemias, FH, HTG | [131] |
| QPI-1007 01064505 | siRNA | Caspase 2 mRNA | RNAi | IVT | Quark | Anterior ischemic optic neuropathy, glaucoma | [132] |
| ALN-AAT02 03767829 | siRNA-GalNAc | Alpha-1 antitrypsin mRNA | RNAi | SC | Alnylam | Alpha-1 antitrypsin deficiency liver disease | [133] |
| MEDI1191 03946800 | mRNA LNP | IL-12 | coding mRNA | IT | Med Immune | Solid tumors | [134] |
| Phase II status | |||||||
| ISIS-FGFR4RX 02476019 | 2′-O-MOE-PS | FGFR4 mRNA | RNase H1 | SC | Ionis | Obesity | [135] |
| IONIS DGAT2Rx 03334214 | 2′-O-MOE-PS | DGAT 2 mRNA | RNase H1 | SC | Ionis | Hepatic steatosis | [136] |
| IONIS-PKK Rx 04307381 | 2′-O-MOE-PS | Pre kallikrein mRNA | RNase H1 | SC | Ionis | Hereditary angioedema | [137] |
| ISIS-GCGRRx 02583919 | 2′-O-MOE GalNAc | Glucagon receptor mRNA | RNase H1 | SC | Ionis | Type 2 diabetes | [138] |
| Custirsen 00327340 | siRNA | Clusterin mRNA | RNase H1 | IV | Achieve Life Sciences | Prostate cancer | [139] |
| OGX-427 01454089 | LNA | Hsp27 mRNA | RNase H1 | IV | Achieve Life Sciences | Metastatic bladder cancer, urinary tract neoplasms | [140] |
| ISIS681257 03070782 | 2′-O-MOE-PS | Lp(a) mRNA | RNase H1 | SC | Akcea | Elevated lipoprotein (a), cardiovascular disease | [141] |
| AKCEA-ANGPTL3-LRx 03514420 | cEt gapmer | ANGPTL3 mRNA | RNase H1 | SC | Akcea | Familial partial lipodystrophy | [142] |
| ISIS678354 03385239 | GalNAc-ASO | ApoC-III mRNA | mRNA inhibitor | SC | Akcea | HTG, cardiovascular diseases | [143] |
| Danvatirsen 02983578 | 2′-O-MOE-PS | STAT3 mRNA | RNase H1 | IV | M.D. Anderson Cancer Center | Refractory pancreatic, NSCLC, colorectal cancer | [144] |
| Cobomarsen (MRG106) 03713320 | LNA | miR-155 | antimiR | IT | miRagen | Cutaneous T-cell lymphoma | [145] |
| Remlarsen 03601052 | 2′-O-MOE | miR-29 | miRNA mimic | ID | miRagen | Keloid | [146] |
| Cemdisiran 03841448 | siRNA-GalNAc | C5 mRNA | RNAi | SC | Alnylam | IgA nephropathy glomerulo nephritis | [147] |
| ARO-AAT 03946449 | siRNA-GalNAc | Alpha-1 antitrypsin mRNA | RNAi | SC | Arrow head | Alpha 1-antitrypsin deficiency | [148] |
| PF-655 01445899 | siRNA | RTP801 | RNAi | IVT | Quark | Diabetic macular edema | [149] |
| AZD8601 03370887 | mRNA LNP | VEGF-A mRNA | coding mRNA | EI | Astra Zeneca | Heart failure | [150] |
| DS-5141b 02667483 | ENA | Dystrophin mRNA exon 45 | Splice modulation | SC | Daiichi Sankyo | DMD | [151] |
| SB010 01743768 | - | GATA-3 | DNAzyme | I | Sterna Bio. | Asthma | [152] |
| Miravirsen 01727934 | LNA | miR-122 | antimiR | SC | Santaris | Hepatitis C | [153] |
| BP1001 02923986 | LNA | Grb2 | - | IV | Bio-Path Holdings | Leukemia | [154] |
| Phase III status | |||||||
| Tominersen 03842969 | 2′-O-MOE-PS | HTT mRNA | RNase H1 | ITH | Ionis | Huntington’s disease | [155] |
| Tofersen 02623699 | 2′-O-MOE-PS | SOD1 mRNA | RNase H1 | ITH | Ionis | Amyotrophic lateral sclerosis | [156] |
| IONIS-TTR RX 02175004 | 2′-O-MOE-PS | TTR mRNA | RNase H1 | SC | Ionis | Familial amyloid poly neuropathy | [157] |
| Volanesorsen 02658175 | 2′-O-MOE-PS | ApoC-III mRNA | RNase H1 | SC | Ionis | FCS hyperlipo proteinemia type 1 | [158] |
| AKCEA-TTR-LRx 04136171 | siRNA GalNAc | TTR mRNA | RNase H1 | SC | Ionis | ATTR cardio myopathy | [159] |
| Alicaforsen 02525523 | PS | ICAM-1 mRNA | RNase H1 | E | Atlantic | Pouchitis | [160] |
| Vutrisiran 04153149 | siRNA-GalNAc | TTR mRNA | RNAi | SC | Alnylam | ATTR with cardio myopathy | [161] |
| Fitusiran 03549871 | siRNA-GalNAc | Anti-thrombin mRNA | RNAi | SC | Genzyme | Hemophilia | [162] |
| QPI-1002 03510897 | siRNA | p53 mRNA | RNAi | IV | Quark | Cardiac surgery | [163] |
| Inclisiran 03814187 | siRNA-GalNAc | PCSK9 mRNA | RNAi | SC | The Medicines Company | Heterozygous FH | [164] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. https://doi.org/10.3390/jcm9062004
Dhuri K, Bechtold C, Quijano E, Pham H, Gupta A, Vikram A, Bahal R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. Journal of Clinical Medicine. 2020; 9(6):2004. https://doi.org/10.3390/jcm9062004
Chicago/Turabian StyleDhuri, Karishma, Clara Bechtold, Elias Quijano, Ha Pham, Anisha Gupta, Ajit Vikram, and Raman Bahal. 2020. "Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development" Journal of Clinical Medicine 9, no. 6: 2004. https://doi.org/10.3390/jcm9062004
APA StyleDhuri, K., Bechtold, C., Quijano, E., Pham, H., Gupta, A., Vikram, A., & Bahal, R. (2020). Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. Journal of Clinical Medicine, 9(6), 2004. https://doi.org/10.3390/jcm9062004