Cohen Syndrome Patient iPSC-Derived Neurospheres and Forebrain-Like Glutamatergic Neurons Reveal Reduced Proliferation of Neural Progenitor Cells and Altered Expression of Synapse Genes

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Genetic Analysis of the CS Patient
2.2. Cell Culture and Generation of iPSCs
2.3. Rapid Neuronal Induction from iPSCs by Neurogenein2 (NGN2)
2.4. Immunocytochemistry
2.5. Reverse Transcription and Quantitative Real-Time (RT) PCR
2.6. Alkaline Phosphatase (AP) Staining and Karyotype Analysis
2.7. In Vivo Teratoma Formation
2.8. RNA Extraction and Sequencing
2.9. RNA-Seq Data Analysis
2.10. Data Availability
2.11. Electrophysiology
2.12. Western Blot Analysis
2.13. Generation of Neurospheres from CS iPSCs
3. Results
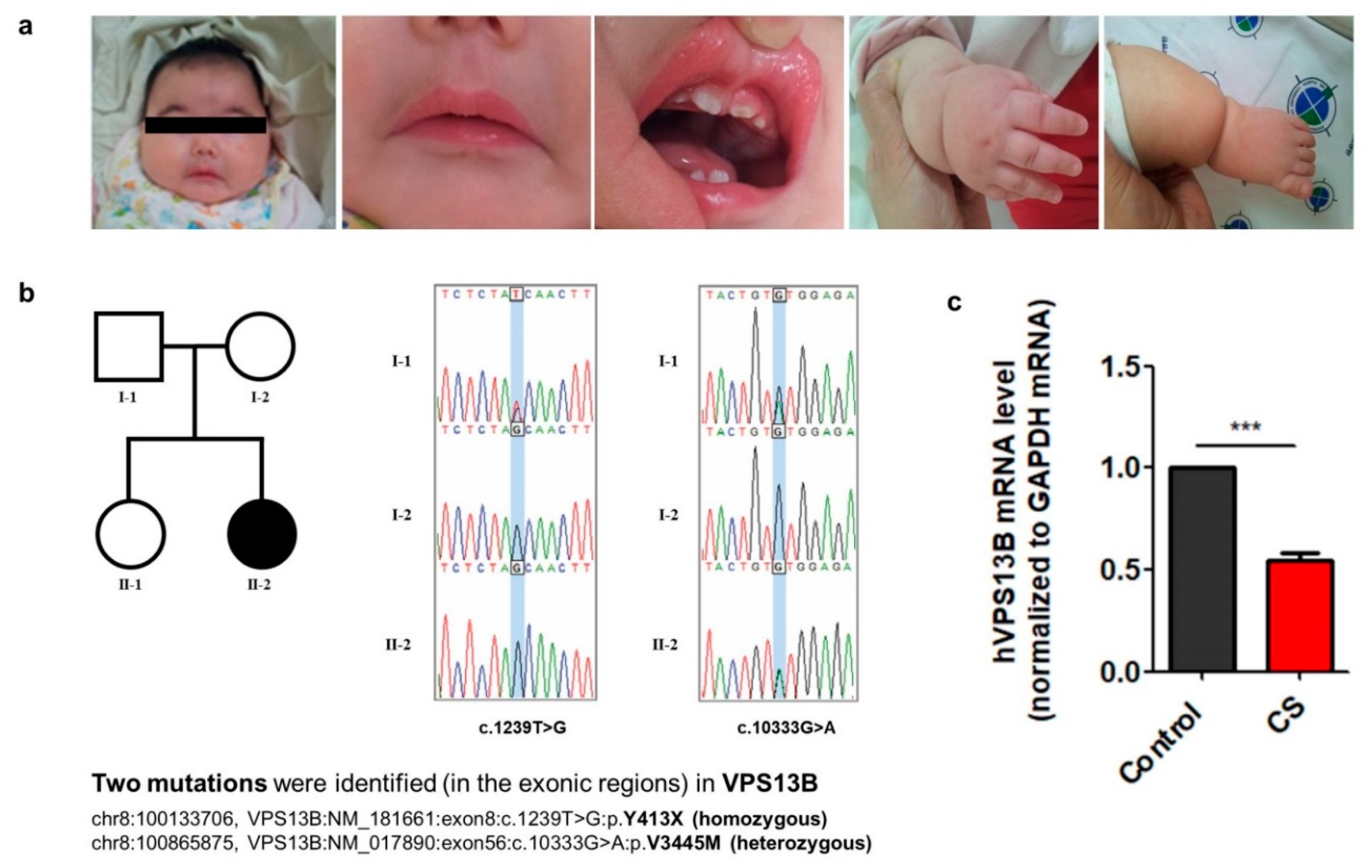
3.1. Clinical Features and Characterization of Mutations in the VPS13B Gene from a Cohen Syndrome Patient
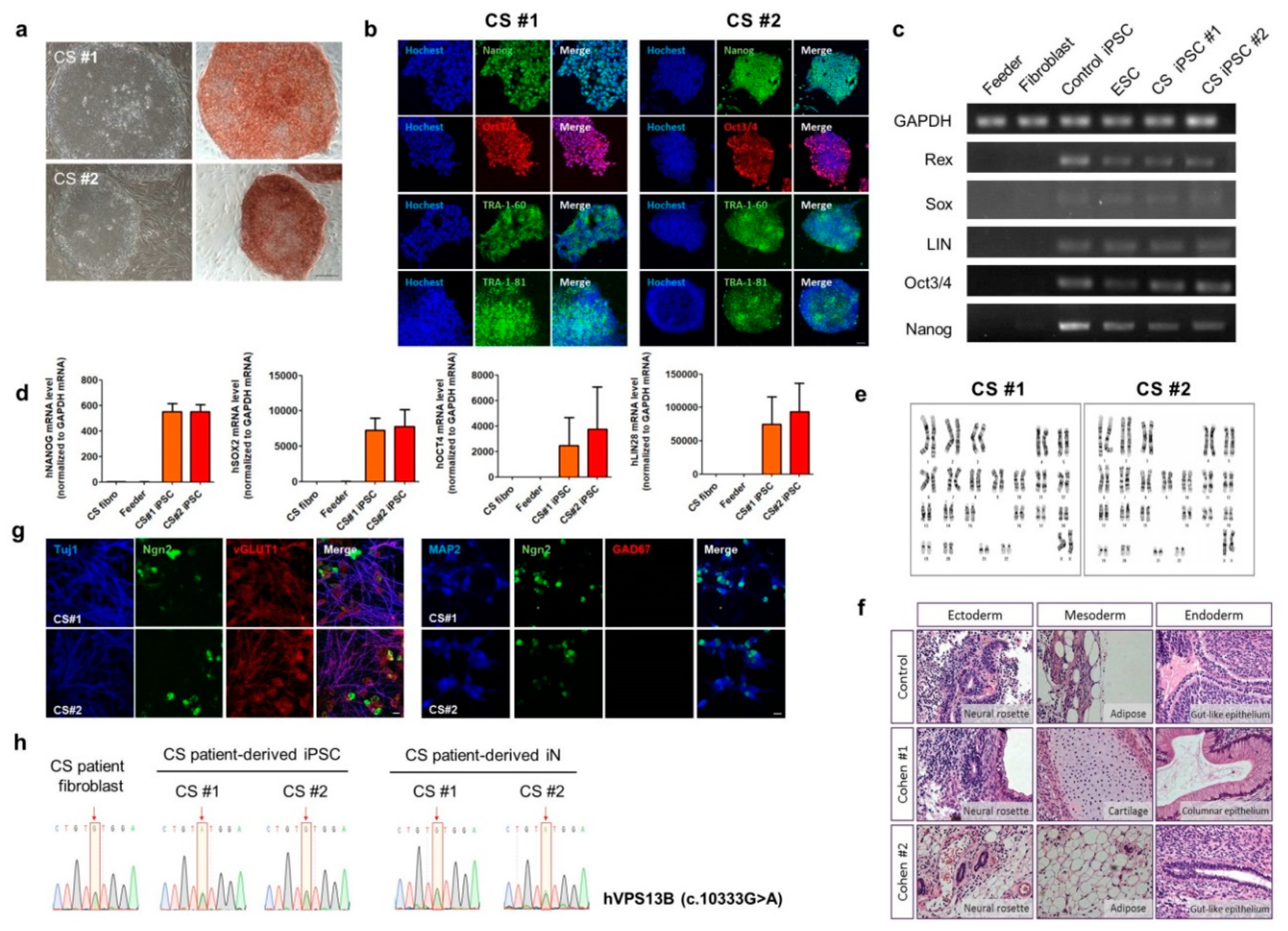
3.2. Generation of CS-Derived iPSC and Rapid Neuronal Induction of CS iPSCs by Neurogenin2 (NGN2)
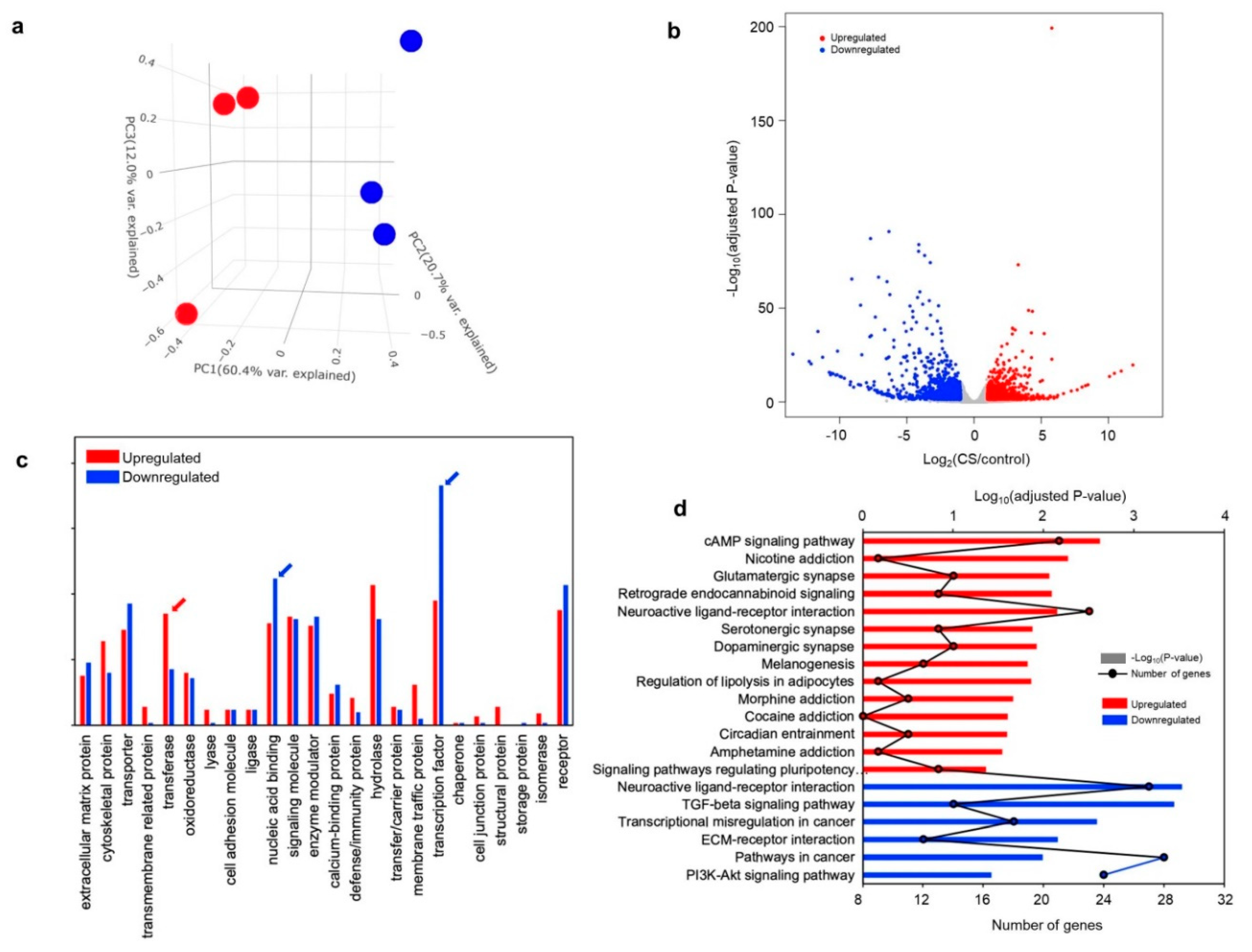
3.3. Transcriptomic Analysis in CS iPSC-Derived Glutamatergic Neurons
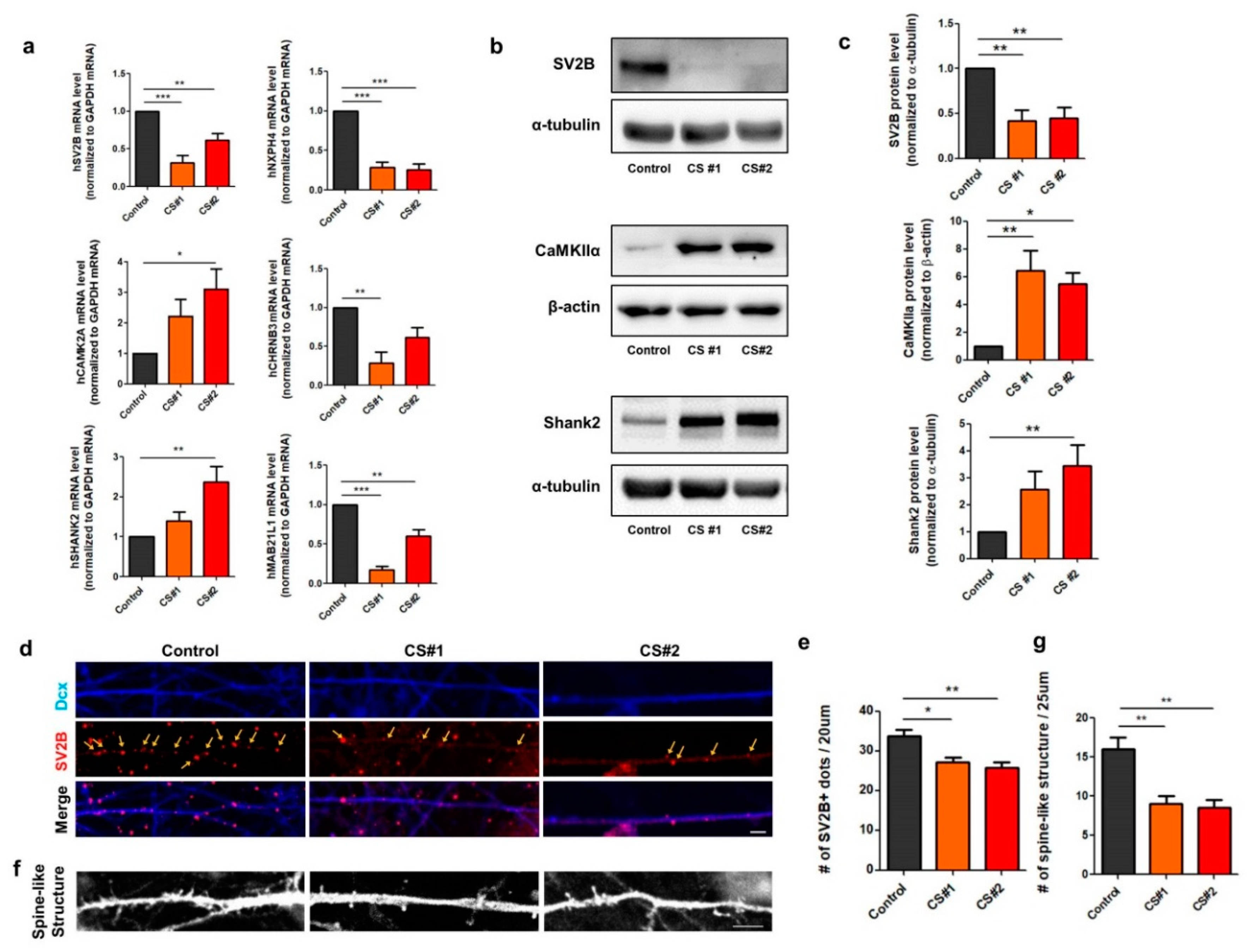
3.4. Alteration of Genes Involved in Synapse Function in CS iPSC-Derived Glutamatergic Neurons
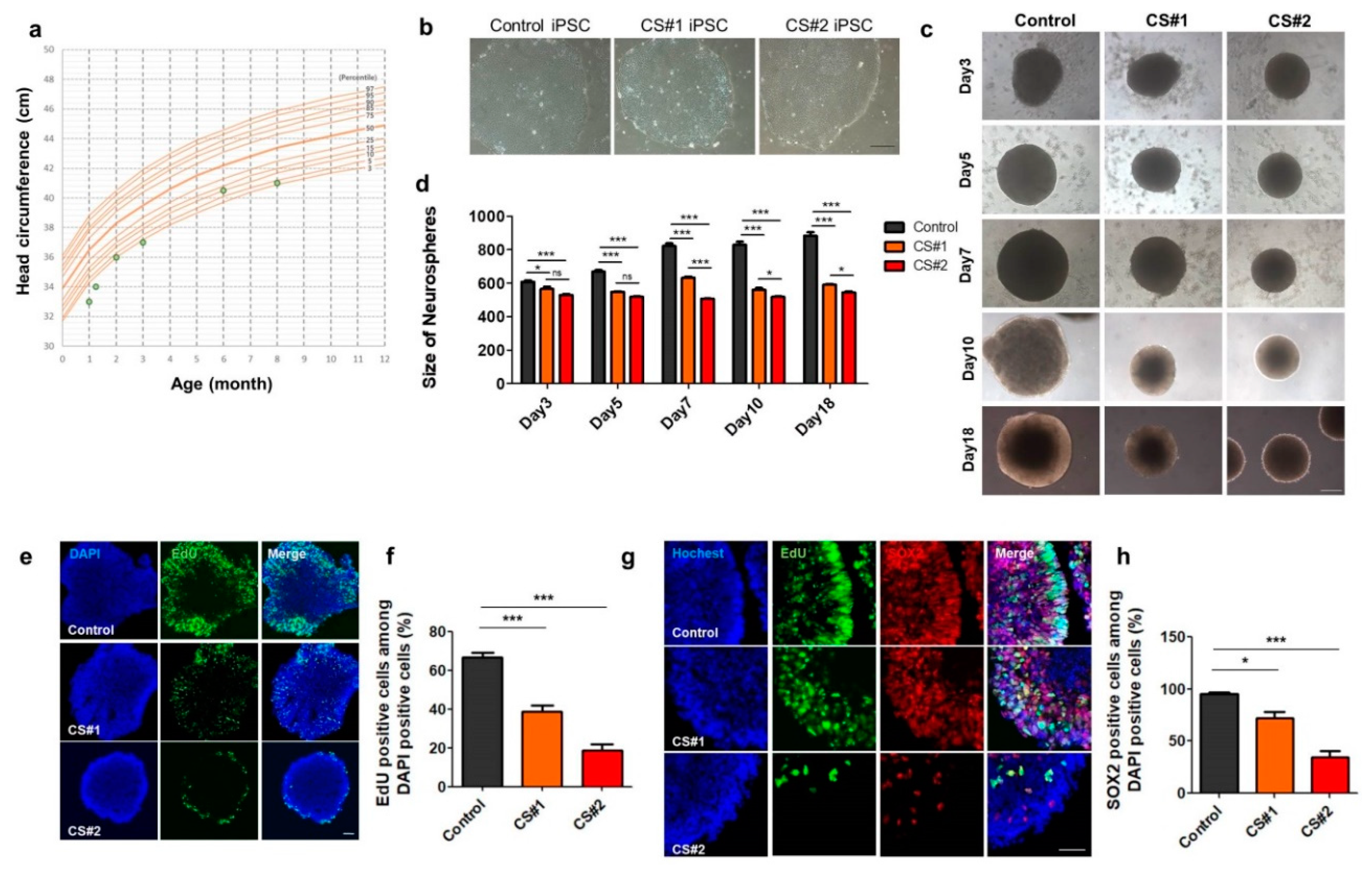
3.5. Small Size of Neurospheres Differentiated from CS iPSCs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, H.; Falk, M.J.; Wensel, C.; Traboulsi, E.I. Cohen Syndrome; GeneReviews [Interent]; University of Washington: Seattle, WA, USA, 1993–2020.
- Rodrigues, J.M.; Fernandes, H.D.; Caruthers, C.; Braddock, S.R.; Knutsen, A.P. Cohen Syndrome: Review of the Literature. Cureus 2018, 10, e3330. [Google Scholar] [CrossRef]
- Rzepnikowska, W.; Flis, K.; Munoz-Braceras, S.; Menezes, R.; Escalante, R.; Zoladek, T. Yeast and other lower eukaryotic organisms for studies of Vps13 proteins in health and disease. Traffic 2017, 18, 711–719. [Google Scholar] [CrossRef]
- Seifert, W.; Kuhnisch, J.; Maritzen, T.; Horn, D.; Haucke, V.; Hennies, H.C. Cohen syndrome-associated protein, COH1, is a novel, giant Golgi matrix protein required for Golgi integrity. J. Biol. Chem. 2011, 286, 37665–37675. [Google Scholar] [CrossRef]
- Seifert, W.; Kuhnisch, J.; Maritzen, T.; Lommatzsch, S.; Hennies, H.C.; Bachmann, S.; Horn, D.; Haucke, V. Cohen syndrome-associated protein COH1 physically and functionally interacts with the small GTPase RAB6 at the Golgi complex and directs neurite outgrowth. J. Biol. Chem. 2015, 290, 3349–3358. [Google Scholar] [CrossRef]
- Koike, S.; Jahn, R. SNAREs define targeting specificity of trafficking vesicles by combinatorial interaction with tethering factors. Nat. Commun. 2019, 10, 1608. [Google Scholar] [CrossRef]
- Balikova, I.; Lehesjoki, A.E.; de Ravel, T.J.; Thienpont, B.; Chandler, K.E.; Clayton-Smith, J.; Traskelin, A.L.; Fryns, J.P.; Vermeesch, J.R. Deletions in the VPS13B (COH1) gene as a cause of Cohen syndrome. Hum. Mutat. 2009, 30, E845–E854. [Google Scholar] [CrossRef]
- Seifert, W.; Holder-Espinasse, M.; Kuhnisch, J.; Kahrizi, K.; Tzschach, A.; Garshasbi, M.; Najmabadi, H.; Walter Kuss, A.; Kress, W.; Laureys, G.; et al. Expanded mutational spectrum in Cohen syndrome, tissue expression, and transcript variants of COH1. Hum. Mutat. 2009, 30, E404–E420. [Google Scholar] [CrossRef]
- Da Costa, R.; Bordessoules, M.; Guilleman, M.; Carmignac, V.; Lhussiez, V.; Courot, H.; Bataille, A.; Chlemaire, A.; Bruno, C.; Fauque, P.; et al. Vps13b is required for acrosome biogenesis through functions in Golgi dynamic and membrane trafficking. Cell Mol. Life Sci. 2020, 77, 511–529. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, R.U.; Oh, J.; Choi, J.E.; Kim, H.; Lee, K.; Hwang, S.K.; Lee, J.H.; Lee, J.A.; Kaang, B.K.; et al. Spatial Learning and Motor Deficits in Vacuolar Protein Sorting-associated Protein 13b (Vps13b) Mutant Mouse. Exp. Neurobiol. 2019, 28, 485–494. [Google Scholar] [CrossRef]
- Lim, C.S.; Yang, J.E.; Lee, Y.K.; Lee, K.; Lee, J.A.; Kaang, B.K. Understanding the molecular basis of autism in a dish using hiPSCs-derived neurons from ASD patients. Mol. Brain 2015, 8, 57. [Google Scholar] [CrossRef]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef]
- Lee, Y.K.; Jun, Y.W.; Choi, H.E.; Huh, Y.H.; Kaang, B.K.; Jang, D.J.; Lee, J.A. Development of LC3/GABARAP sensors containing a LIR and a hydrophobic domain to monitor autophagy. EMBO J. 2017, 36, 1100–1116. [Google Scholar] [CrossRef]
- Wang, H.; Gong, B.; Vadakkan, K.I.; Toyoda, H.; Kaang, B.K.; Zhuo, M. Genetic evidence for adenylyl cyclase 1 as a target for preventing neuronal excitotoxicity mediated by N-methyl-D-aspartate receptors. J. Biol. Chem. 2007, 282, 1507–1517. [Google Scholar] [CrossRef]
- Wen, Z.; Nguyen, H.N.; Guo, Z.; Lalli, M.A.; Wang, X.; Su, Y.; Kim, N.S.; Yoon, K.J.; Shin, J.; Zhang, C.; et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 2014, 515, 414–418. [Google Scholar] [CrossRef]
- Nelakanti, R.V.; Kooreman, N.G.; Wu, J.C. Teratoma formation: A tool for monitoring pluripotency in stem cell research. Curr. Protoc. Stem Cell Biol. 2015, 32, 4A.8.1–4A.8.17. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Torre, D.; Lachmann, A.; Ma’ayan, A. BioJupies: Automated Generation of Interactive Notebooks for RNA-Seq Data Analysis in the Cloud. Cell Syst. 2018, 7, 556–561. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Kang, S.J.; Kwak, C.; Lee, J.; Sim, S.E.; Shim, J.; Choi, T.; Collingridge, G.L.; Zhuo, M.; Kaang, B.K. Bidirectional modulation of hyperalgesia via the specific control of excitatory and inhibitory neuronal activity in the ACC. Mol. Brain 2015, 8, 81. [Google Scholar] [CrossRef]
- Sim, S.E.; Lim, C.S.; Kim, J.I.; Seo, D.; Chun, H.; Yu, N.K.; Lee, J.; Kang, S.J.; Ko, H.G.; Choi, J.H.; et al. The Brain-Enriched MicroRNA miR-9-3p Regulates Synaptic Plasticity and Memory. J. Neurosci. 2016, 36, 8641–8652. [Google Scholar] [CrossRef]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Kang, Y.J.; Govindaiah, G.; Roselaar, N.; Cakir, B.; Kim, K.Y.; Lombroso, A.P.; Hwang, S.M.; et al. Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and Interneuron Migration. Cell Stem Cell 2017, 21, 383–398. [Google Scholar] [CrossRef]
- Volk, L.; Chiu, S.L.; Sharma, K.; Huganir, R.L. Glutamate synapses in human cognitive disorders. Annu. Rev. Neurosci. 2015, 38, 127–149. [Google Scholar] [CrossRef]
- van Bokhoven, H. Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 2011, 45, 81–104. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Horn, D.; Krebsova, A.; Kunze, J.; Reis, A. Homozygosity mapping in a family with microcephaly, mental retardation, and short stature to a Cohen syndrome region on 8q21.3–8q22.1: Redefining a clinical entity. Am. J. Med Genet. 2000, 92, 285–292. [Google Scholar] [CrossRef]
- Douzgou, S.; Petersen, M.B. Clinical variability of genetic isolates of Cohen syndrome. Clin. Genet. 2011, 79, 501–506. [Google Scholar] [CrossRef]
- Sala, C.; Vicidomini, C.; Bigi, I.; Mossa, A.; Verpelli, C. Shank synaptic scaffold proteins: Keys to understanding the pathogenesis of autism and other synaptic disorders. J. Neurochem. 2015, 135, 849–858. [Google Scholar] [CrossRef]
- Yoo, J.; Bakes, J.; Bradley, C.; Collingridge, G.L.; Kaang, B.K. Shank mutant mice as an animal model of autism. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130143. [Google Scholar] [CrossRef]
- Bear, M.F.; Cooke, S.F.; Giese, K.P.; Kaang, B.K.; Kennedy, M.B.; Kim, J.I.; Morris, R.G.M.; Park, P. In memoriam: John Lisman—Commentaries on CaMKII as a memory molecule. Mol. Brain 2018, 11, 76. [Google Scholar] [CrossRef]
- Kury, S.; van Woerden, G.M.; Besnard, T.; Proietti Onori, M.; Latypova, X.; Towne, M.C.; Cho, M.T.; Prescott, T.E.; Ploeg, M.A.; Sanders, S.; et al. De Novo Mutations in Protein Kinase Genes CAMK2A and CAMK2B Cause Intellectual Disability. Am. J. Hum. Genet. 2017, 101, 768–788. [Google Scholar] [CrossRef]
- Akita, T.; Aoto, K.; Kato, M.; Shiina, M.; Mutoh, H.; Nakashima, M.; Kuki, I.; Okazaki, S.; Magara, S.; Shiihara, T.; et al. De novo variants in CAMK2A and CAMK2B cause neurodevelopmental disorders. Ann. Clin. Transl. Neurol. 2018, 5, 280–296. [Google Scholar] [CrossRef]
- Wan, Q.F.; Zhou, Z.Y.; Thakur, P.; Vila, A.; Sherry, D.M.; Janz, R.; Heidelberger, R. SV2 acts via presynaptic calcium to regulate neurotransmitter release. Neuron 2010, 66, 884–895. [Google Scholar] [CrossRef]
- Ciruelas, K.; Marcotulli, D.; Bajjalieh, S.M. Synaptic vesicle protein 2: A multi-faceted regulator of secretion. Semin. Cell Dev. Biol. 2019, 95, 130–141. [Google Scholar] [CrossRef]
- Marsoner, F.; Koch, P.; Ladewig, J. Cortical organoids: Why all this hype? Curr. Opin. Genet. Dev. 2018, 52, 22–28. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef]
- Yoon, K.J.; Song, G.; Qian, X.; Pan, J.; Xu, D.; Rho, H.S.; Kim, N.S.; Habela, C.; Zheng, L.; Jacob, F.; et al. Zika-Virus-Encoded NS2A Disrupts Mammalian Cortical Neurogenesis by Degrading Adherens Junction Proteins. Cell Stem Cell 2017, 21, 349–358. [Google Scholar] [CrossRef]
- Ho, S.M.; Topol, A.; Brennand, K.J. From “directed differentiation” to “neuronal induction”: Modeling neuropsychiatric disease. Biomark. Insights 2015, 10, 31–41. [Google Scholar] [CrossRef]
- Mertens, J.; Reid, D.; Lau, S.; Kim, Y.; Gage, F.H. Aging in a Dish: iPSC-Derived and Directly Induced Neurons for Studying Brain Aging and Age-Related Neurodegenerative Diseases. Annu. Rev. Genet. 2018, 52, 271–293. [Google Scholar] [CrossRef]
- Trujillo, C.A.; Muotri, A.R. Brain Organoids and the Study of Neurodevelopment. Trends Mol. Med. 2018, 24, 982–990. [Google Scholar] [CrossRef]
- Homem, C.C.; Repic, M.; Knoblich, J.A. Proliferation control in neural stem and progenitor cells. Nat. Rev. Neurosci. 2015, 16, 647–659. [Google Scholar] [CrossRef]
- Li, C.; Ito, H.; Fujita, K.; Shiwaku, H.; Qi, Y.; Tagawa, K.; Tamura, T.; Okazawa, H. Sox2 transcriptionally regulates PQBP1, an intellectual disability-microcephaly causative gene, in neural stem progenitor cells. PLoS ONE 2013, 8, e68627. [Google Scholar] [CrossRef]
- Ito, H.; Shiwaku, H.; Yoshida, C.; Homma, H.; Luo, H.; Chen, X.; Fujita, K.; Musante, L.; Fischer, U.; Frints, S.G.; et al. In utero gene therapy rescues microcephaly caused by Pqbp1-hypofunction in neural stem progenitor cells. Mol. Psychiatry 2015, 20, 459–471. [Google Scholar] [CrossRef]
- Brickner, J.H.; Fuller, R.S. SOI1 encodes a novel, conserved protein that promotes TGN-endosomal cycling of Kex2p and other membrane proteins by modulating the function of two TGN localization signals. J. Cell Biol. 1997, 139, 23–36. [Google Scholar] [CrossRef]
- Dalton, L.E.; Bean, B.D.M.; Davey, M.; Conibear, E. Quantitative high-content imaging identifies novel regulators of Neo1 trafficking at endosomes. Mol. Biol. Cell 2017, 28, 1539–1550. [Google Scholar] [CrossRef]
- De, M.; Oleskie, A.N.; Ayyash, M.; Dutta, S.; Mancour, L.; Abazeed, M.E.; Brace, E.J.; Skiniotis, G.; Fuller, R.S. The Vps13p-Cdc31p complex is directly required for TGN late endosome transport and TGN homotypic fusion. J. Cell Biol. 2017, 216, 425–439. [Google Scholar] [CrossRef]
- Foller, M.; Hermann, A.; Gu, S.; Alesutan, I.; Qadri, S.M.; Borst, O.; Schmidt, E.M.; Schiele, F.; vom Hagen, J.M.; Saft, C.; et al. Chorein-sensitive polymerization of cortical actin and suicidal cell death in chorea-acanthocytosis. FASEB J. 2012, 26, 1526–1534. [Google Scholar] [CrossRef]
- Munoz-Braceras, S.; Calvo, R.; Escalante, R. TipC and the chorea-acanthocytosis protein VPS13A regulate autophagy in Dictyostelium and human HeLa cells. Autophagy 2015, 11, 918–927. [Google Scholar] [CrossRef]
- Duplomb, L.; Duvet, S.; Picot, D.; Jego, G.; El Chehadeh-Djebbar, S.; Marle, N.; Gigot, N.; Aral, B.; Carmignac, V.; Thevenon, J.; et al. Cohen syndrome is associated with major glycosylation defects. Hum. Mol. Genet. 2014, 23, 2391–2399. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KEGG Pathway | Number of Genes | Adjusted p-Value | Genes |
|---|---|---|---|
| Upregulated | |||
| cAMP signaling pathway | 21 | 0.00236638 | FXYD2, PTGER3, ADCY8, ADCY5, PDE3A, GRIN3A, TNNI3, VIPR2, VAV1, BDNF, GRIN2B, PPP1R1B, GRIA1, HTR6, PIK3R5, PRKACB, ARAP3, HTR1D, CAMK2A, AKT3, HTR1E |
| Nicotine addiction | 9 | 0.00529695 | SLC17A8, GABRG2, GABRE, GRIN2B, GABRA4, GRIA1, GABRB2, GRIN3A, GABRQ |
| Glutamatergic synapse | 14 | 0.00864085 | ADCY8, ADCY5, GRIK4, GRIN3A, SHANK2, SLC17A8, GRIN2B, GRM8, GRIA1, SLC1A7, PRKACB, PLA2G4C, PLCB2, PLA2G4D |
| Retrograde endocannabinoid signaling | 13 | 0.00803098 | SLC17A8, GABRG2, GABRE, KCNJ9, GABRA4, GRIA1, ADCY8, GABRB2, ADCY5, FAAH, PRKACB, PLCB2, GABRQ |
| Neuroactive ligand-receptor interaction | 23 | 0.00703938 | GABRG2, GABRE, PTGER1, PTGER3, THRB, GABRA4, DRD3, GABRB2, CYSLTR2, GRIK4, TSHB, GRIN3A, VIPR2, CRHR2, GRIN2B, GRM8, GRIA1, HTR6, MAS1, CALCRL, HTR1D, GABRQ, HTR1E |
| Serotonergic synapse | 13 | 0.01312029 | KCND2, KCNJ9, GABRB2, ADCY5, HTR6, PTGS1, MAOB, PRKACB, HTR1D, PLA2G4C, PLCB2, HTR1E, PLA2G4D |
| Dopaminergic synapse | 14 | 0.01189212 | CALY, DRD3, PPP2R5A, ADCY5, TH, MAOB, GRIN2B, KCNJ9, GRIA1, PPP1R1B, PRKACB, CAMK2A, PLCB2, AKT3 |
| Melanogenesis | 12 | 0.01494844 | WNT5A, WNT4, ADCY8, ADCY5, FZD1, PRKACB, WNT7A, CAMK2A, PLCB2, TCF7L2, WNT8A, WNT2B |
| Regulation of lipolysis in adipocytes | 9 | 0.01360123 | IRS4, PTGER3, ADCY8, ADCY5, PTGS1, TSHB, PIK3R5, PRKACB, AKT3 |
| Morphine addiction | 11 | 0.02151968 | GABRG2, GABRE, PDE7B, KCNJ9, GABRA4, ADCY8, GABRB2, ADCY5, PDE3A, PRKACB, GABRQ |
| Cocaine addiction | 8 | 0.02474648 | BDNF, GRIN2B, PPP1R1B, ADCY5, MAOB, TH, PRKACB, GRIN3A |
| Circadian entrainment | 11 | 0.02502941 | RPS6KA5, NOS1, GRIN2B, KCNJ9, GRIA1, ADCY8, ADCY5, GUCY1A2, PRKACB, CAMK2A, PLCB2 |
| Amphetamine addiction | 9 | 0.02830907 | GRIN2B, GRIA1, PPP1R1B, ADCY5, MAOB, TH, PRKACB, GRIN3A, CAMK2A |
| Signaling pathways regulating pluripotency of stem cells | 13 | 0.04332556 | WNT5A, FZD1, POU5F1B, WNT2B, PCGF5, WNT4, POU5F1, PIK3R5, JAK3, FGF2, WNT7A, WNT8A, AKT3 |
| Downregulated | |||
| Neuroactive ligand-receptor interaction | 27 | 2.93E-04 | CGA, OPRK1, GLRA2, BDKRB2, S1PR3, APLNR, CHRNA9, GALR1, CHRNA5, CHRNA6, CHRNA1, CHRNA3, GABRG3, GABRA1, PTH2R, GRM4, GABRR1, CHRM4, CHRM3, PRLR, GRM6, MC4R, CHRNB4, ADRA1A, GPR50, CHRNB3, CHRNG |
| Transforming growth factor (TGF)-beta signaling pathway | 14 | 3.57E-04 | SMAD9, SMAD7, TGFBR2, TGFB3, TGFB1, ID2, INHBE, ID3, BAMBI, MYC, CHRD, BMP5, PITX2, ACVR1 |
| Transcriptional misregulation in cancer | 18 | 0.00256055 | LMO2, TGFBR2, RXRG, SIX4, ZBTB16, MEIS1, FEV, JUP, CDKN1A, ID2, NTRK1, PAX8, SIX1, ETV1, NGFR, TLX3, MYC, TLX1 |
| Extracellular Matrix (ECM)-receptor interaction | 12 | 0.00695211 | ITGA9, COL6A6, COL27A1, TNC, ITGA8, COL3A1, ITGB6, SV2B, COL2A1, LAMB1, COL5A2, CHAD |
| Pathways in cancer | 28 | 0.01023292 | FGF18, ADCY2, PGF, TGFB3, KITLG, FGF10, FGF13, ZBTB16, BDKRB2, MMP2, TGFB1, TCF7L1, FLT3LG, CXCR4, PAX8, TGFA, LAMB1, MYC, EGFR, FZD8, EPAS1, TGFBR2, RXRG, MECOM, JUP, CDKN1A, FZD10, NTRK1 |
| PI3K-Akt signaling pathway | 24 | 0.03734476 | EGFR, FGF18, PGF, TNC, COL3A1, KITLG, FGF10, COL2A1, FGF13, COL5A2, CHAD, ITGA9, CDKN1A, COL6A6, PRLR, ITGA8, COL27A1, IL4R, ITGB6, PDGFC, NGFR, LAMB1, MYC, IL3RA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-K.; Hwang, S.-K.; Lee, S.-K.; Yang, J.-e.; Kwak, J.-H.; Seo, H.; Ahn, H.; Lee, Y.-S.; Kim, J.; Lim, C.-S.; et al. Cohen Syndrome Patient iPSC-Derived Neurospheres and Forebrain-Like Glutamatergic Neurons Reveal Reduced Proliferation of Neural Progenitor Cells and Altered Expression of Synapse Genes. J. Clin. Med. 2020, 9, 1886. https://doi.org/10.3390/jcm9061886
Lee Y-K, Hwang S-K, Lee S-K, Yang J-e, Kwak J-H, Seo H, Ahn H, Lee Y-S, Kim J, Lim C-S, et al. Cohen Syndrome Patient iPSC-Derived Neurospheres and Forebrain-Like Glutamatergic Neurons Reveal Reduced Proliferation of Neural Progenitor Cells and Altered Expression of Synapse Genes. Journal of Clinical Medicine. 2020; 9(6):1886. https://doi.org/10.3390/jcm9061886
Chicago/Turabian StyleLee, You-Kyung, Su-Kyeong Hwang, Soo-Kyung Lee, Jung-eun Yang, Ji-Hye Kwak, Hyunhyo Seo, Hyunjun Ahn, Yong-Seok Lee, Janghwan Kim, Chae-Seok Lim, and et al. 2020. "Cohen Syndrome Patient iPSC-Derived Neurospheres and Forebrain-Like Glutamatergic Neurons Reveal Reduced Proliferation of Neural Progenitor Cells and Altered Expression of Synapse Genes" Journal of Clinical Medicine 9, no. 6: 1886. https://doi.org/10.3390/jcm9061886
APA StyleLee, Y.-K., Hwang, S.-K., Lee, S.-K., Yang, J.-e., Kwak, J.-H., Seo, H., Ahn, H., Lee, Y.-S., Kim, J., Lim, C.-S., Kaang, B.-K., Lee, J.-H., Lee, J.-A., & Lee, K. (2020). Cohen Syndrome Patient iPSC-Derived Neurospheres and Forebrain-Like Glutamatergic Neurons Reveal Reduced Proliferation of Neural Progenitor Cells and Altered Expression of Synapse Genes. Journal of Clinical Medicine, 9(6), 1886. https://doi.org/10.3390/jcm9061886