Impaired Mitochondrial Morphology and Functionality in Lonp1wt/− Mice

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation and Genotyping of Lonp1wt/− Mice
2.2. Ethics Statement
2.3. Isolation of Organs and Tissue
2.4. Isolation and Culture of MEFs
2.5. Quantification of Mitochondrial DNA
2.6. Immunoblot
2.7. Hematoxylin and Eosin Staining
2.8. Confocal and Stimulated Emission Depletion (STED) Microscopy
2.9. Electron Microscopy
2.10. Morphological Analysis of Mitochondria
2.11. Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR)
2.12. Cytofluorimetric Analyses
2.13. Statistical Analysis
3. Results
3.1. Lonp1 Depletion Causes Early Embryonic Lethality
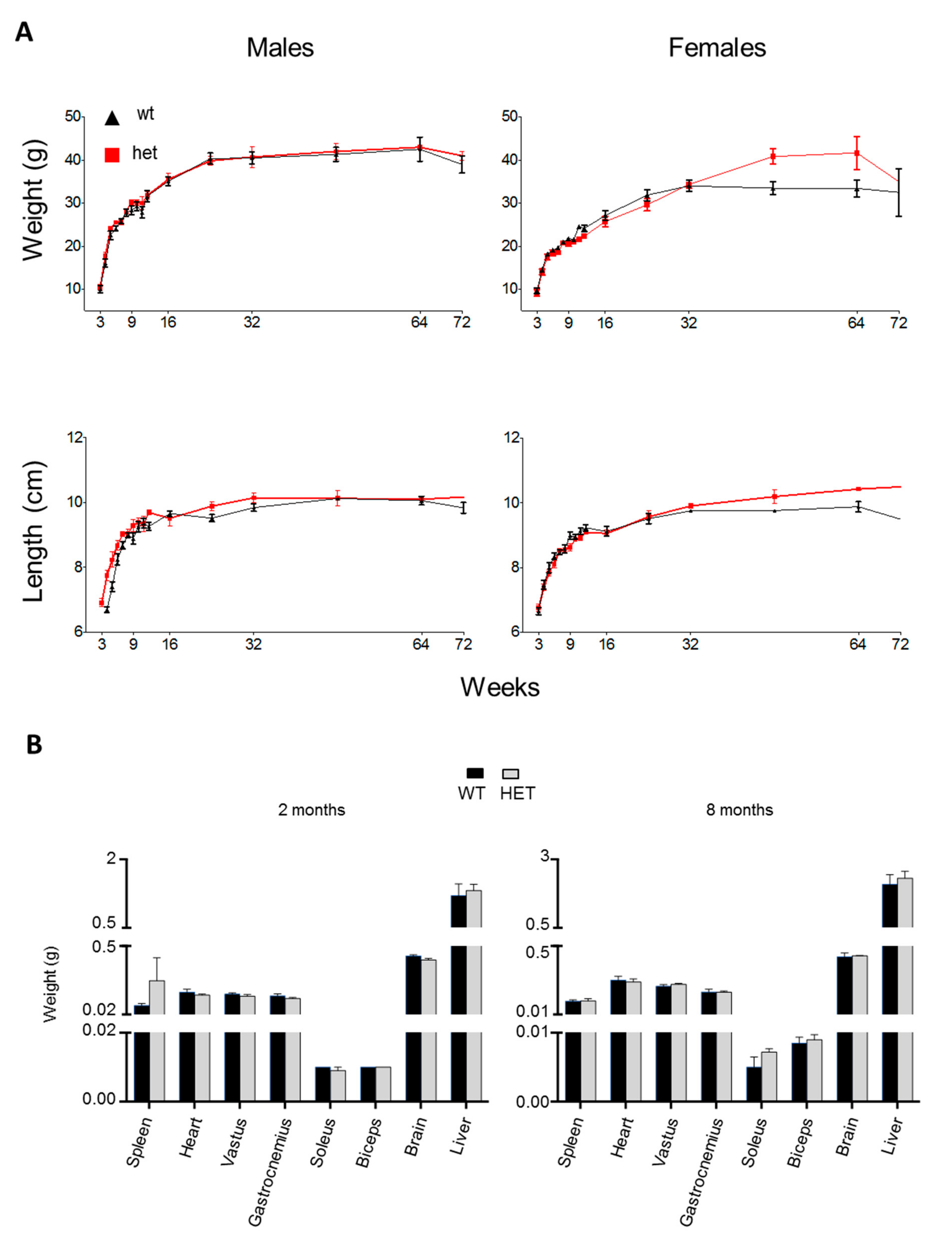
3.2. Phenotypic Characterization of the Lonp1wt/− Mouse
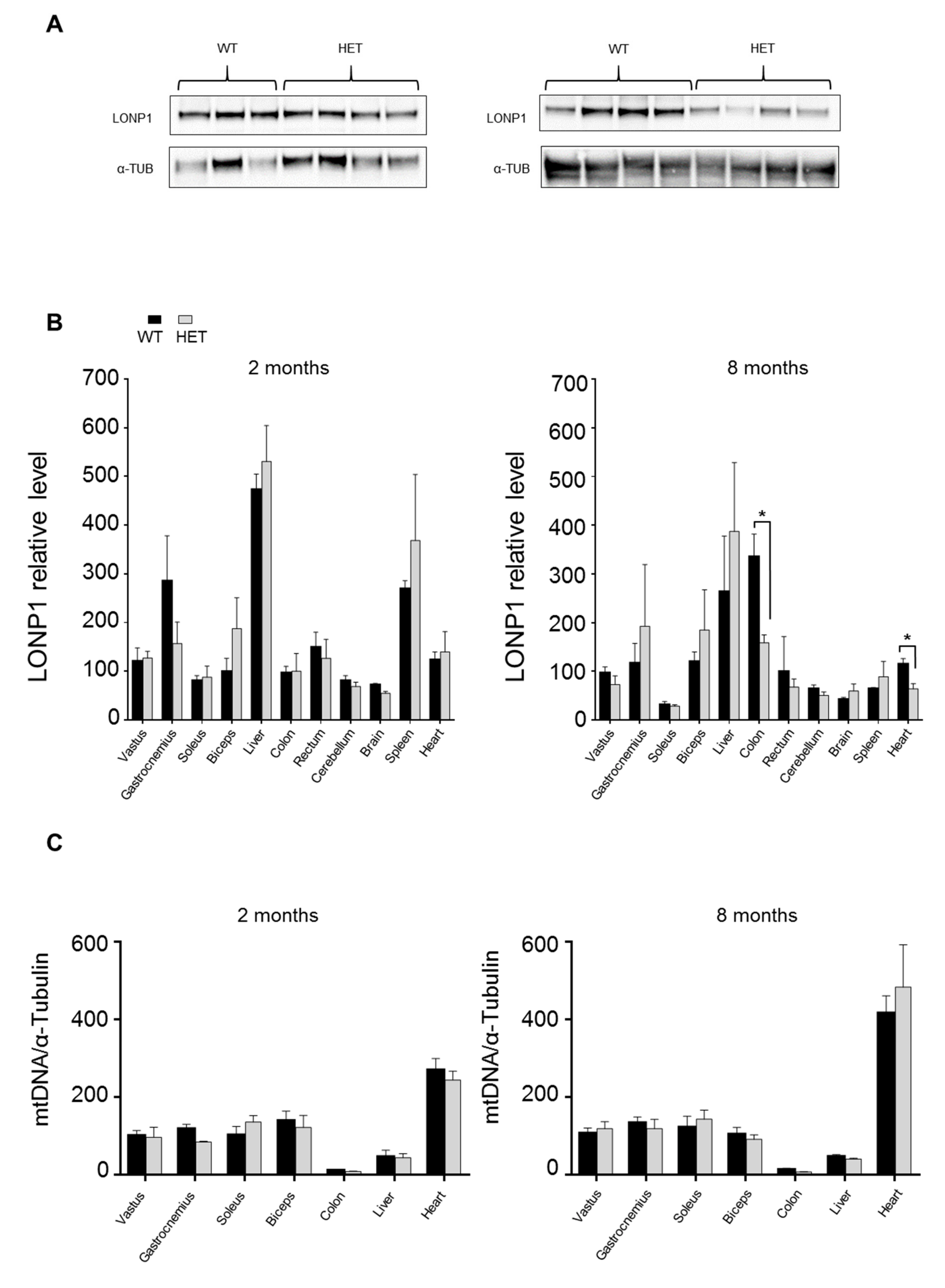
3.3. LONP1 and mtDNA Levels in Wild Type and Lonp1wt/− Mice
3.4. Histological Characterization of Lonp1wt/− Mouse
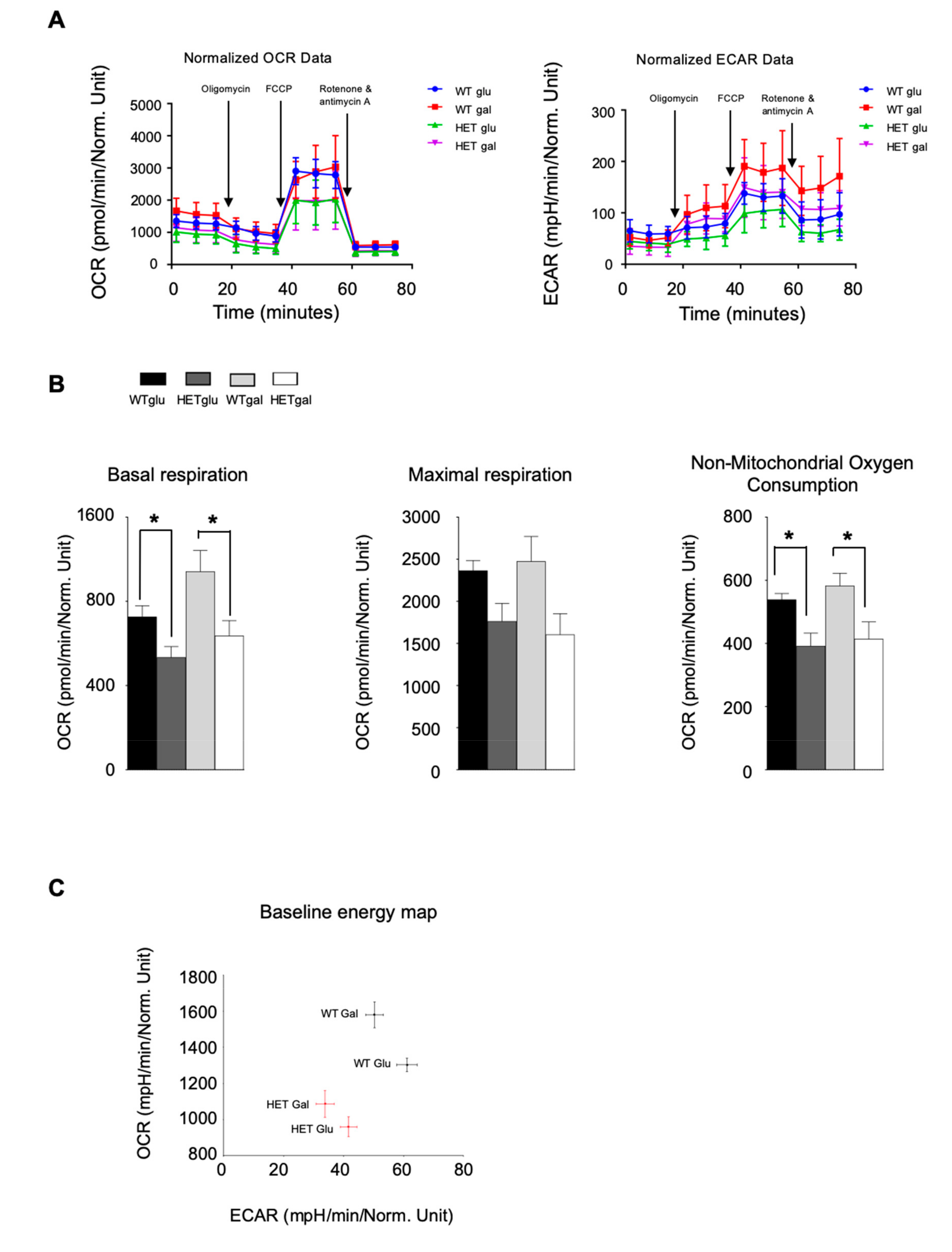
3.5. LONP1 and TFAM Levels Are Lower in Het Embryos, and Lead to mtDNA Reduction and Mitochondrial Impairment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Truscott, K.N.; Lowth, B.R.; Strack, P.R.; Dougan, D.A. Diverse functions of mitochondrial AAA+ proteins: Protein activation, disaggregation, and degradation. This paper is one of a selection of papers published in this special issue entitled 8th International Conference on AAA Proteins and has undergone the Journal’s usual peer review process. Biochem. Cell Biol. 2010, 88, 97–108. [Google Scholar] [CrossRef]
- Gottesman, S.; Wickner, S.; Maurizi, M.R. Protein quality control: triage by chaperones and proteases. Genes Dev. 1997, 11, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Rep, M.; Grivell, L.A. The role of protein degradation in mitochondrial function and biogenesis. Curr. Genet. 1996, 30, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, L.; Langer, T. ATP-dependent proteases controlling mitochondrial function in the yeast Saccharomyces cerevisiae. Cell. Mol. Life Sci. 1999, 56, 825–842. [Google Scholar] [CrossRef] [PubMed]
- Park, S.C.; Jia, B.; Yang, J.K.; Van, D.L.; Shao, Y.G.; Han, S.W.; Jeon, Y.J.; Chung, C.H.; Cheong, G.W. Oligomeric structure of the ATP-dependent protease La (Lon) of Escherichia coli. Mol. Cells 2006, 21, 129–134. [Google Scholar]
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. AAA+: A Class of Chaperone-Like ATPases Associated with the Assembly, Operation, and Disassembly of Protein Complexes. Genome Res. 1999, 9, 27–43. [Google Scholar] [CrossRef]
- Duman, R.E.; Löwe, J. Crystal Structures of Bacillus subtilis Lon Protease. J. Mol. Biol. 2010, 401, 653–670. [Google Scholar] [CrossRef]
- Gibellini, L.; Pinti, M.; Boraldi, F.; Giorgio, V.; Bernardi, P.; Bartolomeo, R.; Nasi, M.; De Biasi, S.; Missiroli, S.; Carnevale, G.; et al. Silencing of mitochondrial Lon protease deeply impairs mitochondrial proteome and function in colon cancer cells. FASEB J. 2014, 28, 5122–5135. [Google Scholar] [CrossRef]
- Lu, B.; Lee, J.; Nie, X.; Li, M.; Morozov, Y.I.; Venkatesh, S.; Bogenhagen, D.F.; Temiakov, D.; Suzuki, C.K. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol. Cell 2013, 49, 121–132. [Google Scholar] [CrossRef]
- Matsushima, Y.; Goto, Y.-i.; Kaguni, L.S. Mitochondrial Lon protease regulates mitochondrial DNA copy number and transcription by selective degradation of mitochondrial transcription factor A (TFAM). Proc. Natl. Acad. Sci. USA 2010, 107, 18410–18415. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Gibellini, L.; Nasi, M.; De Biasi, S.; Bortolotti, C.A.; Iannone, A.; Cossarizza, A. Emerging role of Lon protease as a master regulator of mitochondrial functions. Biochim. Biophys. Acta 2016, 1857, 1300–1306. [Google Scholar] [CrossRef] [PubMed]
- Bota, D.A.; Davies, K.J.A. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol. 2002, 4, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Kita, K.; Suzuki, T.; Ochi, T. Diphenylarsinic acid promotes degradation of glutaminase C by mitochondrial Lon protease. J. Biol. Chem. 2012, 287, 18163–18172. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Gibellini, L.; Liu, Y.; Xu, S.; Lu, B.; Cossarizza, A. Mitochondrial Lon protease at the crossroads of oxidative stress, ageing and cancer. Cell. Mol. Life Sci. 2015, 72, 4807–4824. [Google Scholar] [CrossRef] [PubMed]
- Charette, M.F.; Henderson, G.W.; Markovitz, A. ATP hydrolysis-dependent protease activity of the lon (capR) protein of Escherichia coli K-12. Proc. Natl. Acad. Sci. USA 1981, 78, 4728–4732. [Google Scholar] [CrossRef]
- Chung, C.H.; Goldberg, A.L. The product of the lon (capR) gene in Escherichia coli is the ATP-dependent protease, protease La. Proc. Natl. Acad. Sci. USA 1981, 78, 4931–4935. [Google Scholar] [CrossRef]
- Pinti, M.; Gibellini, L.; De Biasi, S.; Nasi, M.; Roat, E.; O’Connor, J.E.; Cossarizza, A. Functional characterization of the promoter of the human Lon protease gene. Mitochondrion 2010. [Google Scholar] [CrossRef]
- Ngo, J.K.; Davies, K.J. Mitochondrial Lon protease is a human stress protein. Free Radic. Biol. Med. 2009, 46, 1042–1048. [Google Scholar] [CrossRef]
- Ngo, J.K.; Pomatto, L.C.; Davies, K.J. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 2013, 1, 258–264. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef]
- Zurita Rendon, O.; Shoubridge, E.A. LONP1 Is Required for Maturation of a Subset of Mitochondrial Proteins, and Its Loss Elicits an Integrated Stress Response. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Bezawork-Geleta, A.; Saiyed, T.; Dougan, D.A.; Truscott, K.N. Mitochondrial matrix proteostasis is linked to hereditary paraganglioma: LON-mediated turnover of the human flavinylation factor SDH5 is regulated by its interaction with SDHA. FASEB J. 2014, 28, 1794–1804. [Google Scholar] [CrossRef] [PubMed]
- Felk, S.; Ohrt, S.; Kussmaul, L.; Storch, A.; Gillardon, F. Activation of the mitochondrial protein quality control system and actin cytoskeletal alterations in cells harbouring the MELAS mitochondrial DNA mutation. J. Neurol. Sci. 2010, 295, 46–52. [Google Scholar] [CrossRef]
- Guillon, B.; Bulteau, A.-L.; Wattenhofer-Donzé, M.; Schmucker, S.; Friguet, B.; Puccio, H.; Drapier, J.-C.; Bouton, C. Frataxin deficiency causes upregulation of mitochondrial Lon and ClpP proteases and severe loss of mitochondrial Fe–S proteins. FEBS J. 2009, 276, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Corydon, T.J.; Palmfeldt, J.; Dürr, A.; Fontaine, B.; Nielsen, M.N.; Christensen, J.H.; Gregersen, N.; Bross, P. Decreased expression of the mitochondrial matrix proteases Lon and ClpP in cells from a patient with hereditary spastic paraplegia (SPG13). Neuroscience 2008, 153, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Gibellini, L.; Guaraldi, G.; Orlando, G.; Gant, T.W.; Morselli, E.; Nasi, M.; Salomoni, P.; Mussini, C.; Cossarizza, A. Upregulation of nuclear-encoded mitochondrial Lon protease in Haart-treated Hiv-positive patients with lipodystrophy: Implications for the pathogenesis of the disease. Aids 2010, 24, 841–850. [Google Scholar] [CrossRef]
- Wu, S.-B.; Ma, Y.-S.; Wu, Y.-T.; Chen, Y.-C.; Wei, Y.-H. Mitochondrial DNA Mutation-Elicited Oxidative Stress, Oxidative Damage, and Altered Gene Expression in Cultured Cells of Patients with MERRF Syndrome. Mol. Neurobiol. 2010, 41, 256–266. [Google Scholar] [CrossRef]
- Bernstein, S.H.; Venkatesh, S.; Li, M.; Lee, J.; Lu, B.; Hilchey, S.P.; Morse, K.M.; Metcalfe, H.M.; Skalska, J.; Andreeff, M.; et al. The mitochondrial ATP-dependent Lon protease: A novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood 2012, 119, 3321–3329. [Google Scholar] [CrossRef]
- Cheng, C.W.; Kuo, C.Y.; Fan, C.C.; Fang, W.C.; Jiang, S.S.; Lo, Y.K.; Wang, T.Y.; Kao, M.C.; Lee, A.Y.L. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis. 2013, 4, e681. [Google Scholar] [CrossRef]
- Nie, X.; Li, M.; Lu, B.; Zhang, Y.; Lan, L.; Chen, L.; Lu, J. Down-Regulating Overexpressed Human Lon in Cervical Cancer Suppresses Cell Proliferation and Bioenergetics. PLoS ONE 2013, 8, e81084. [Google Scholar] [CrossRef]
- Liu, Y.; Lan, L.; Huang, K.; Wang, R.; Xu, C.; Shi, Y.; Wu, X.; Wu, Z.; Zhang, J.; Chen, L.; et al. Inhibition of Lon blocks cell proliferation, enhances chemosensitivity by promoting apoptosis and decreases cellular bioenergetics of bladder cancer: Potential roles of Lon as a prognostic marker and therapeutic target in baldder cancer. Oncotarget 2014, 5, 11209–11224. [Google Scholar] [CrossRef]
- Quirós, P.M.; Español, Y.; Acín-Pérez, R.; Rodríguez, F.; Bárcena, C.; Watanabe, K.; Calvo, E.; Loureiro, M.; Fernández-García, M.S.; Fueyo, A.; et al. ATP-Dependent Lon Protease Controls Tumor Bioenergetics by Reprogramming Mitochondrial Activity. Cell Rep. 2014, 8, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; Losi, L.; De Biasi, S.; Nasi, M.; Lo Tartaro, D.; Pecorini, S.; Patergnani, S.; Pinton, P.; De Gaetano, A.; Carnevale, G.; et al. LonP1 Differently Modulates Mitochondrial Function and Bioenergetics of Primary Versus Metastatic Colon Cancer Cells. Front. Oncol. 2018, 8, 254. [Google Scholar] [CrossRef] [PubMed]
- Shebib, S.M.; Reed, M.H.; Shuckett, E.P.; Cross, H.G.; Perry, J.B.; Chudley, A.E. Newly recognized syndrome of cerebral, ocular, dental, auricular, skeletal anomalies: CODAS syndrome—A case report. Am. J. Med. Genet. 1991, 40, 88–93. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, J.C.; Vargas, F.R.; Barbosa-Neto, J.G.; Llerena, J.C. CODAS syndrome: A new distinct MCA/MR syndrome with radiological changes of spondyloepiphyseal dysplasia. Another case report. Am. J. Med. Genet. 1995, 55, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Dikoglu, E.; Alfaiz, A.; Gorna, M.; Bertola, D.; Chae, J.H.; Cho, T.-J.; Derbent, M.; Alanay, Y.; Guran, T.; Kim, O.-H.; et al. Mutations in LONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am. J. Med. Genet. 2015, 167, 1501–1509. [Google Scholar] [CrossRef]
- Innes, A.M.; Chudley, A.E.; Reed, M.H.; Shuckett, E.P.; Hildes-Ripstein, G.E.; Greenberg, C.R. Third case of cerebral, ocular, dental, auricular, skeletal anomalies (CODAS) syndrome, further delineating a new malformation syndrome: First report of an affected male and review of literature. Am. J. Med. Genet. 2001, 102, 44–47. [Google Scholar] [CrossRef]
- Marlin, S.; Ducou Le Pointe, H.; Le Merrer, M.; Portnoi, M.F.; Chantot, S.; Jonard, L.; Mantel-Guiochon, A.; Siffroi, J.P.; Garabedian, E.N.; Denoyelle, F. Fourth case of cerebral, ocular, dental, auricular, skeletal syndrome (CODAS), description of new features and molecular analysis. Am. J. Med. Genet. Part A 2010, 152A, 1510–1514. [Google Scholar] [CrossRef]
- Strauss, K.A.; Jinks, R.N.; Puffenberger, E.G.; Venkatesh, S.; Singh, K.; Cheng, I.; Mikita, N.; Thilagavathi, J.; Lee, J.; Sarafianos, S.; et al. CODAS Syndrome Is Associated with Mutations of LONP1, Encoding Mitochondrial AAA+ Lon Protease. Am. J. Hum. Genet. 2015, 96, 121–135. [Google Scholar] [CrossRef]
- Peter, B.; Waddington, C.L.; Oláhová, M.; Sommerville, E.W.; Hopton, S.; Pyle, A.; Champion, M.; Ohlson, M.; Siibak, T.; Chrzanowska-Lightowlers, Z.M.A.; et al. Defective mitochondrial protease LonP1 can cause classical mitochondrial disease. Hum. Mol. Genet. 2018, 27, 1743–1753. [Google Scholar] [CrossRef]
- Nimmo, G.A.M.; Venkatesh, S.; Pandey, A.K.; Marshall, C.R.; Hazrati, L.-N.; Blaser, S.; Ahmed, S.; Cameron, J.; Singh, K.; Ray, P.N.; et al. Bi-allelic mutations of LONP1 encoding the mitochondrial LonP1 protease cause pyruvate dehydrogenase deficiency and profound neurodegeneration with progressive cerebellar atrophy. Hum. Mol. Genet. 2018, 28, 290–306. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Li, M.; Saito, T.; Tong, M.; Rashed, E.; Mareedu, S.; Zhai, P.; Barcena, C.; Lopez-Otin, C.; Yehia, G.; et al. Mitochondrial LonP1 protects cardiomyocytes from ischemia/reperfusion injury in vivo. J. Mol. Cell. Cardiol. 2019, 128, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Key, J.; Kohli, A.; Bárcena, C.; López-Otín, C.; Heidler, J.; Wittig, I.; Auburger, G. Global Proteome of LonP1+/− Mouse Embryonal Fibroblasts Reveals Impact on Respiratory Chain, but No Interdependence between Eral1 and Mitoribosomes. Int. J. Mol. Sci. 2019, 20, 4523. [Google Scholar] [CrossRef] [PubMed]
- Boraldi, F.; Lofaro, F.D.; Accorsi, A.; Ross, E.; Malagoli, D. Toward the Molecular Deciphering of Pomacea canaliculata Immunity: First Proteomic Analysis of Circulating Hemocytes. Proteomics 2019, 19, e1800314. [Google Scholar] [CrossRef]
- Inui, T.; Anzai, M.; Takezawa, Y.; Endo, W.; Kakisaka, Y.; Kikuchi, A.; Onuma, A.; Kure, S.; Nishino, I.; Ohba, C.; et al. A novel mutation in the proteolytic domain of LONP1 causes atypical CODAS syndrome. J. Hum. Genet. 2017, 62, 653–655. [Google Scholar] [CrossRef]
- Delaval, E.; Perichon, M.; Friguet, B. Age-related impairment of mitochondrial matrix aconitase and ATP-stimulated protease in rat liver and heart. Eur. J. Biochem. 2004, 271, 4559–4564. [Google Scholar] [CrossRef]
- Ngo, J.K.; Davies, K.J. Importance of the lon protease in mitochondrial maintenance and the significance of declining lon in aging. Ann. N. Y. Acad. Sci. 2007, 1119, 78–87. [Google Scholar] [CrossRef]
- Gibellini, L.; Pinti, M.; Bartolomeo, R.; De Biasi, S.; Cormio, A.; Musicco, C.; Carnevale, G.; Pecorini, S.; Nasi, M.; De Pol, A.; et al. Inhibition of Lon protease by triterpenoids alters mitochondria and is associated to cell death in human cancer cells. Oncotarget 2015, 6, 25466–25483. [Google Scholar] [CrossRef]
- Bayot, A.; Gareil, M.; Chavatte, L.; Hamon, M.P.; L’Hermitte-Stead, C.; Beaumatin, F.; Priault, M.; Rustin, P.; Lombes, A.; Friguet, B.; et al. Effect of Lon protease knockdown on mitochondrial function in HeLa cells. Biochimie 2013. [Google Scholar] [CrossRef]
- Dott, W.; Mistry, P.; Wright, J.; Cain, K.; Herbert, K.E. Modulation of mitochondrial bioenergetics in a skeletal muscle cell line model of mitochondrial toxicity. Redox Biol. 2014, 2, 224–233. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Male | Female | ||||
|---|---|---|---|---|---|
| Type of Mating | Total | Born | Expected | Born | Expected |
| wt/− ♂ x wt/− ♀ | 110.0 | 58.0 | 55.0 | 52.0 | 55.0 |
| wt/− ♂ x wt/wt ♀ | 67.0 | 36.0 | 33.5 | 31.0 | 33.5 |
| wt/wt ♂ x wt/− ♀ | 52.0 | 27.0 | 26.0 | 25.0 | 26.0 |
| Total | 229.0 | 121.0 | 114.5 | 108.0 | 114.5 |
| Total | Male | Female | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Wild Type | Heterozygous | Wild Type | Heterozygous | Wild Type | Heterozygous | ||||||||
| Type of mating | Total | Born | Expected | Born | Expected | Born | Expected | Born | Expected | Born | Expected | Born | Expected |
| wt/− ♂ x wt/− ♀ | 110.0 | 45.0 | 36.7 | 65.0 | 73.3 | 24.0 | 19.3 | 34.0 | 38.7 | 21.0 | 17.3 | 31.0 | 34.7 |
| wt/− ♂ x wt/wt ♀ | 67.0 | 33.0 | 33.5 | 34.0 | 33.5 | 21.0 | 18.0 | 15.0 | 18.0 | 12.0 | 15.5 | 19.0 | 15.5 |
| wt/wt ♂ x wt/− ♀ | 52.0 | 28.0 | 26.0 | 24.0 | 26.0 | 16.0 | 13.5 | 11.0 | 13.5 | 12.0 | 12.5 | 13.0 | 12.5 |
| Total | 229.0 | 106.0 | 96.2 | 123.0 | 132.8 | 61.0 | 50.8 | 60.0 | 70.2 | 45.0 | 45.3 | 63.0 | 62.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Gaetano, A.; Gibellini, L.; Bianchini, E.; Borella, R.; De Biasi, S.; Nasi, M.; Boraldi, F.; Cossarizza, A.; Pinti, M. Impaired Mitochondrial Morphology and Functionality in Lonp1wt/− Mice. J. Clin. Med. 2020, 9, 1783. https://doi.org/10.3390/jcm9061783
De Gaetano A, Gibellini L, Bianchini E, Borella R, De Biasi S, Nasi M, Boraldi F, Cossarizza A, Pinti M. Impaired Mitochondrial Morphology and Functionality in Lonp1wt/− Mice. Journal of Clinical Medicine. 2020; 9(6):1783. https://doi.org/10.3390/jcm9061783
Chicago/Turabian StyleDe Gaetano, Anna, Lara Gibellini, Elena Bianchini, Rebecca Borella, Sara De Biasi, Milena Nasi, Federica Boraldi, Andrea Cossarizza, and Marcello Pinti. 2020. "Impaired Mitochondrial Morphology and Functionality in Lonp1wt/− Mice" Journal of Clinical Medicine 9, no. 6: 1783. https://doi.org/10.3390/jcm9061783
APA StyleDe Gaetano, A., Gibellini, L., Bianchini, E., Borella, R., De Biasi, S., Nasi, M., Boraldi, F., Cossarizza, A., & Pinti, M. (2020). Impaired Mitochondrial Morphology and Functionality in Lonp1wt/− Mice. Journal of Clinical Medicine, 9(6), 1783. https://doi.org/10.3390/jcm9061783