Early Phylogenetic Diversification of SARS-CoV-2: Determination of Variants and the Effect on Epidemiology, Immunology, and Diagnostics


Abstract
1. Introduction
2. Experimental Section
3. Results
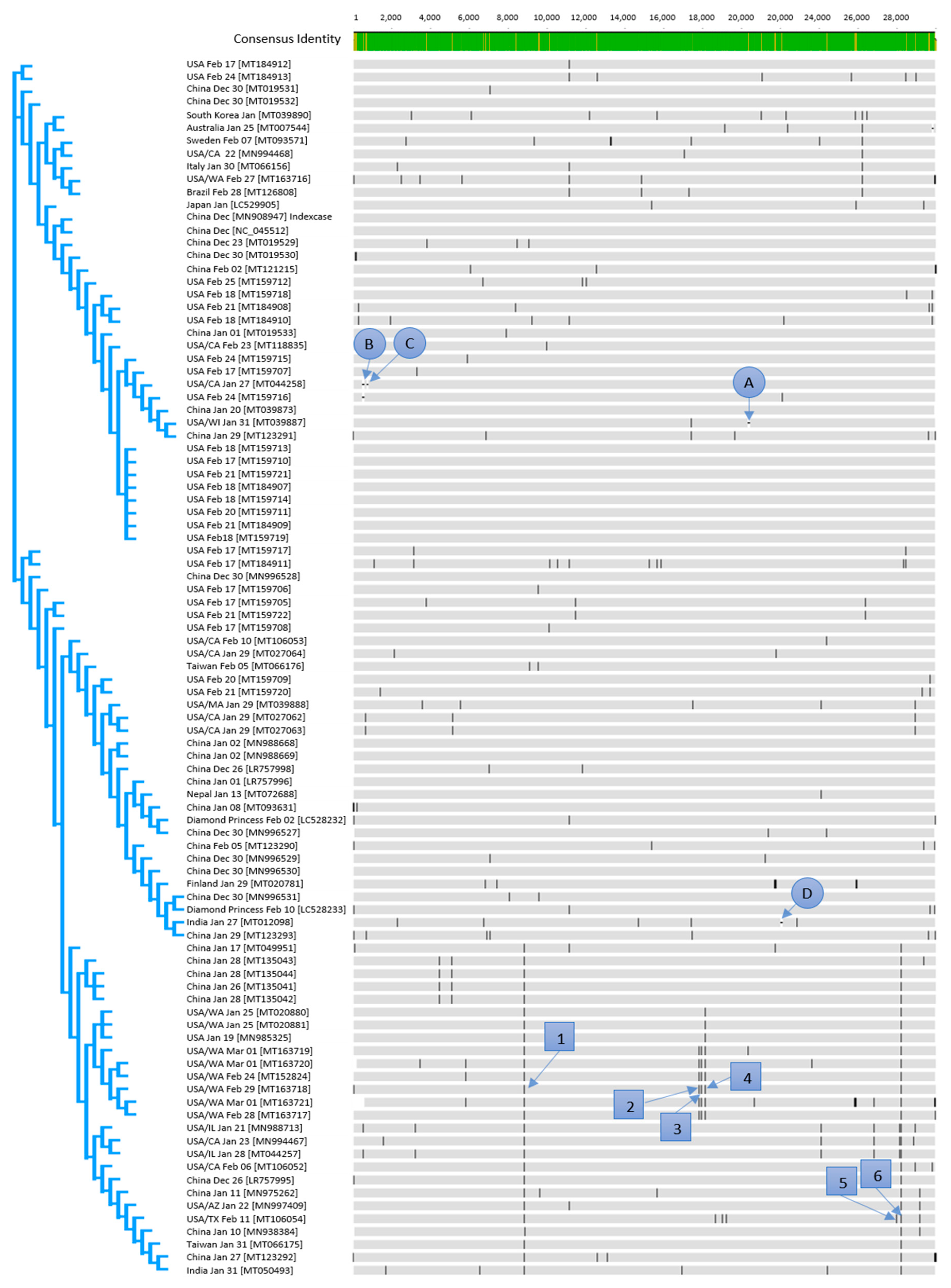
3.1. Phylogeny and Early Clusters
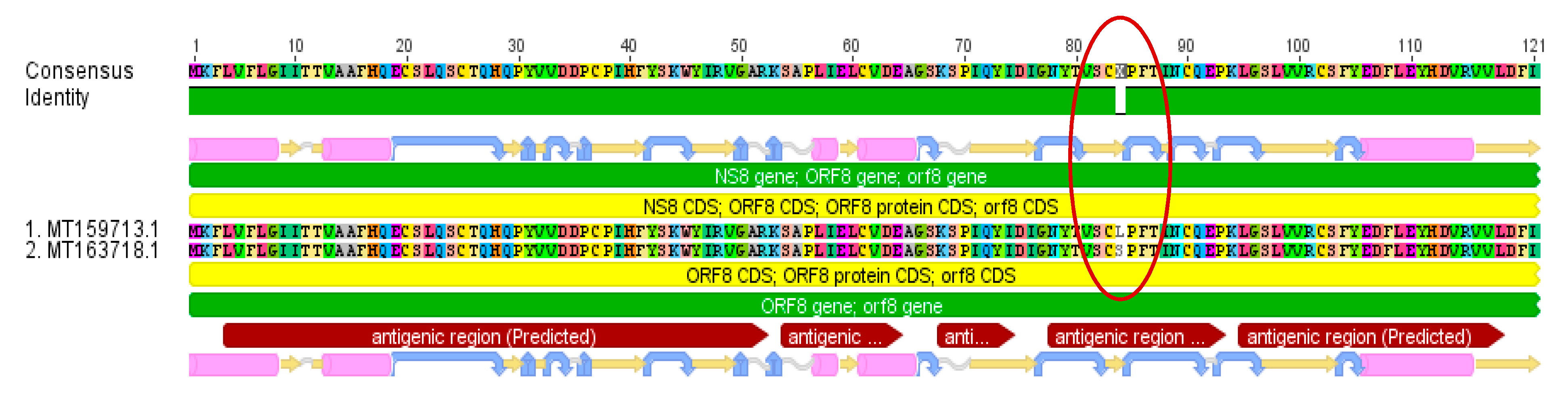
3.2. Genetic Variants
3.3. Diagnostics
4. Discussion
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Coronavirus Disease (COVID-2019) Situation Reports. Situation Report—84. Available online: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200413-sitrep-84-covid-19.pdf?sfvrsn=44f511ab_2 (accessed on 14 April 2020).
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.-K.; Sun, J.; Cao, G.; Liu, Y.; Lin, D.; Gao, Y.-Z.; Ren, W.-H.; Long, X.-D.; Zhang, H.; Ma, X.-P.; et al. Genetic variants in STAT4 and HLA-DQ genes confer risk of hepatitis B virus–related hepatocellular carcinoma. Nat Genet 2013, 45, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Pung, R.; Chiew, C.J.; Young, B.E.; Chin, S.; Chen, M.I.C.; Clapham, H.E.; Cook, A.R.; Maurer-Stroh, S.; Toh, M.P.H.S.; Poh, C.; et al. Investigation of three clusters of COVID-19 in Singapore: Implications for surveillance and response measures. Lancet 2020, 395, 1039–1046. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Yuan, S.; Kok, K.-H.; To, K.K.-W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.-Y.; Poon, R.W.-S.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Pan, Y.; Cheng, S.M.S.; Hui, K.P.Y.; Krishnan, P.; Liu, Y.; Ng, D.Y.M.; Wan, C.K.C.; Yang, P.; Wang, Q.; et al. Molecular Diagnosis of a Novel Coronavirus (2019-nCoV) Causing an Outbreak of Pneumonia. Clin. Chem. 2020, 66, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, Z.; Chen, Z.; Huang, X.; Xu, M.; He, T.; Zhang, Z. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J. Med. Virol. 2020, 92, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Salipante, S.J.; SenGupta, D.J.; Cummings, L.A.; Land, T.A.; Hoogestraat, D.R.; Cookson, B.T. Application of Whole-Genome Sequencing for Bacterial Strain Typing in Molecular Epidemiology. J. Clin. Microbiol. 2015, 53, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Díez, J.; Martínez, M.A.; Hernández, J.; Holguín, A.; Borrego, B.; Mateu, M.G. New observations on antigenic diversification of RNA viruses. Antigenic variation is not dependent on immune selection. J. Gen. Virol. 1993, 74, 2039–2045. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.J.; Gandhi, J.; Clewley, J.P. Genetic variants of parvovirus B19 identified in the United Kingdom: Implications for diagnostic testing. J. Clin. Virol. 2006, 36, 152–155. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Selected Region | 1 | 2 | 3 | 4 | 5 | 6 |
| Variant Sequence | MT163718 | MT163718 | MT163718 | MT163718 | LR757995 | MT163718 |
| Total number sequences with variation | 26 | 6 | 6 | 9 | 2 | 26 |
| Gene | orf 1 ab gene, nsp4 peptide | orf 1 ab gene, nsp13 peptide | orf 1 ab gene, nsp13 peptide | orf 1 ab gene, nsp14 peptide | NS8 gene | NS8 gene |
| Function | contains transmembrane domain | helicase and others | helicase and others | exonuclease | unknown | unknown |
| Variant Nucleotide | C8517T | C17483T | A17593G | C17795T | C32T | T251C |
| Variant Amino Acid | S2839S | P5828L | Y5865C | L5932L | T11I | L84S |
| Consequence | silent mutation | effective variant | effective variant | silent mutation | effective variant | effective variant |
| Substitution in antigenic region | not relevant | no | no | not relevant | yes | yes |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaden, R. Early Phylogenetic Diversification of SARS-CoV-2: Determination of Variants and the Effect on Epidemiology, Immunology, and Diagnostics. J. Clin. Med. 2020, 9, 1615. https://doi.org/10.3390/jcm9061615
Kaden R. Early Phylogenetic Diversification of SARS-CoV-2: Determination of Variants and the Effect on Epidemiology, Immunology, and Diagnostics. Journal of Clinical Medicine. 2020; 9(6):1615. https://doi.org/10.3390/jcm9061615
Chicago/Turabian StyleKaden, Rene. 2020. "Early Phylogenetic Diversification of SARS-CoV-2: Determination of Variants and the Effect on Epidemiology, Immunology, and Diagnostics" Journal of Clinical Medicine 9, no. 6: 1615. https://doi.org/10.3390/jcm9061615
APA StyleKaden, R. (2020). Early Phylogenetic Diversification of SARS-CoV-2: Determination of Variants and the Effect on Epidemiology, Immunology, and Diagnostics. Journal of Clinical Medicine, 9(6), 1615. https://doi.org/10.3390/jcm9061615