Initial Steps for the Development of a Phage-Mediated Gene Replacement Therapy Using CRISPR-Cas9 Technology


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
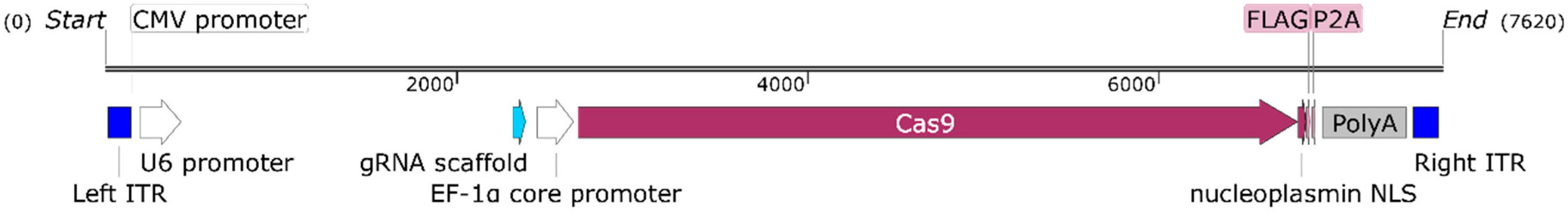
2.2. Cas9 Phage Plasmid Construction
2.3. M13 (Non-Targeted) and RGD4C (Targeted) Bacteriophage-Based Vector Production and Titration
2.4. Transfection of the Cas9 Phage Plasmid with Polyethyleneimine (PEI)
2.5. Cell Transduction by Phage Vectors
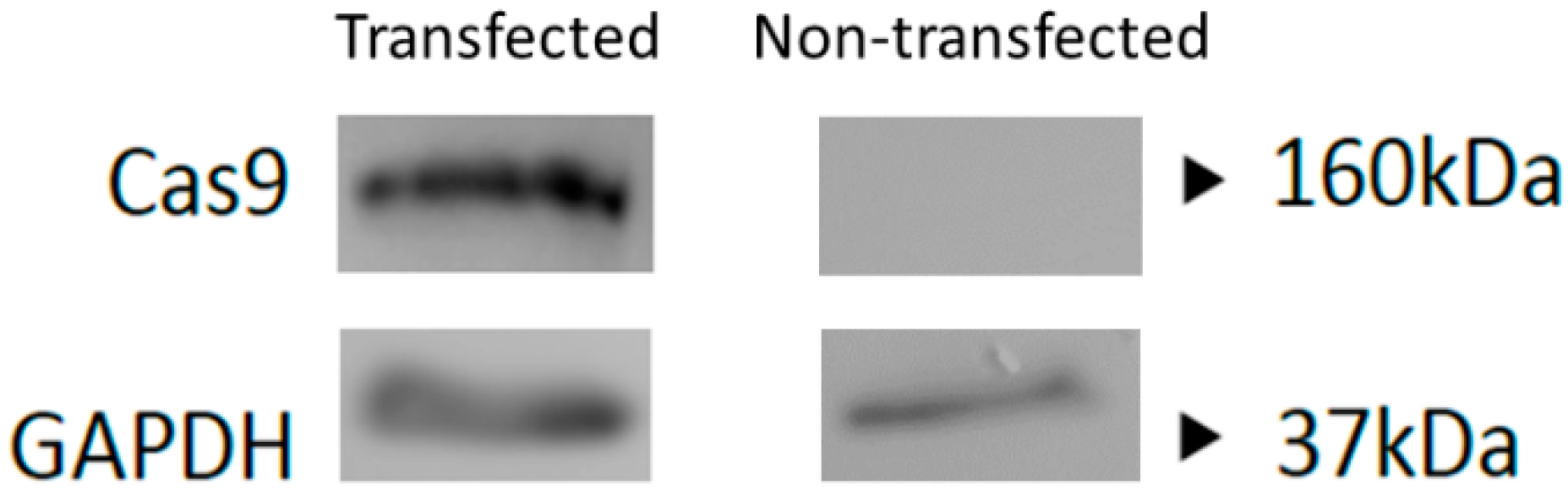
2.6. Western Blot
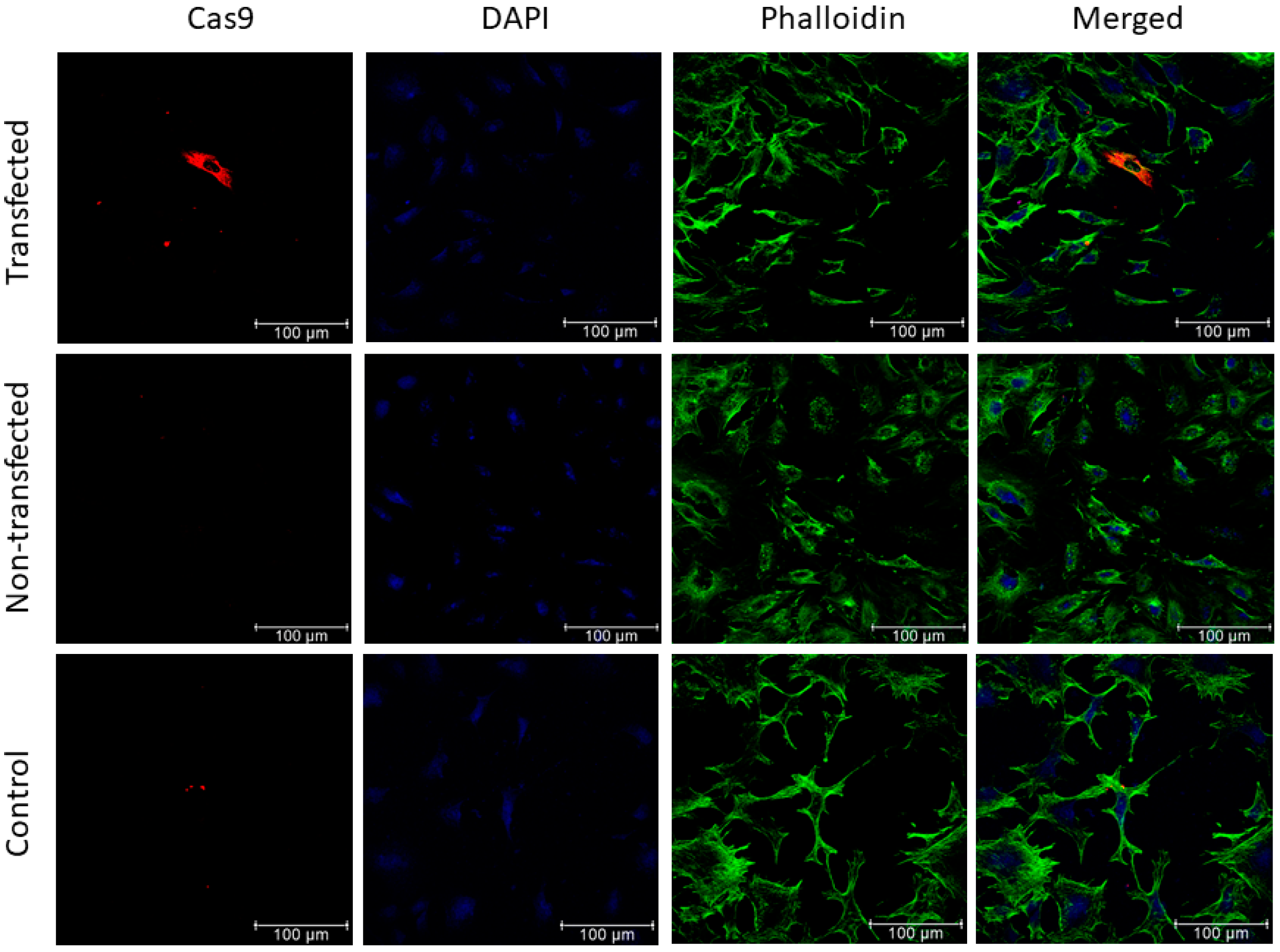
2.7. Immunofluorescence Staining
2.8. Statistical Analysis
3. Results
3.1. Positive Validation of the CRISPR-Cas9 Phage Plasmid
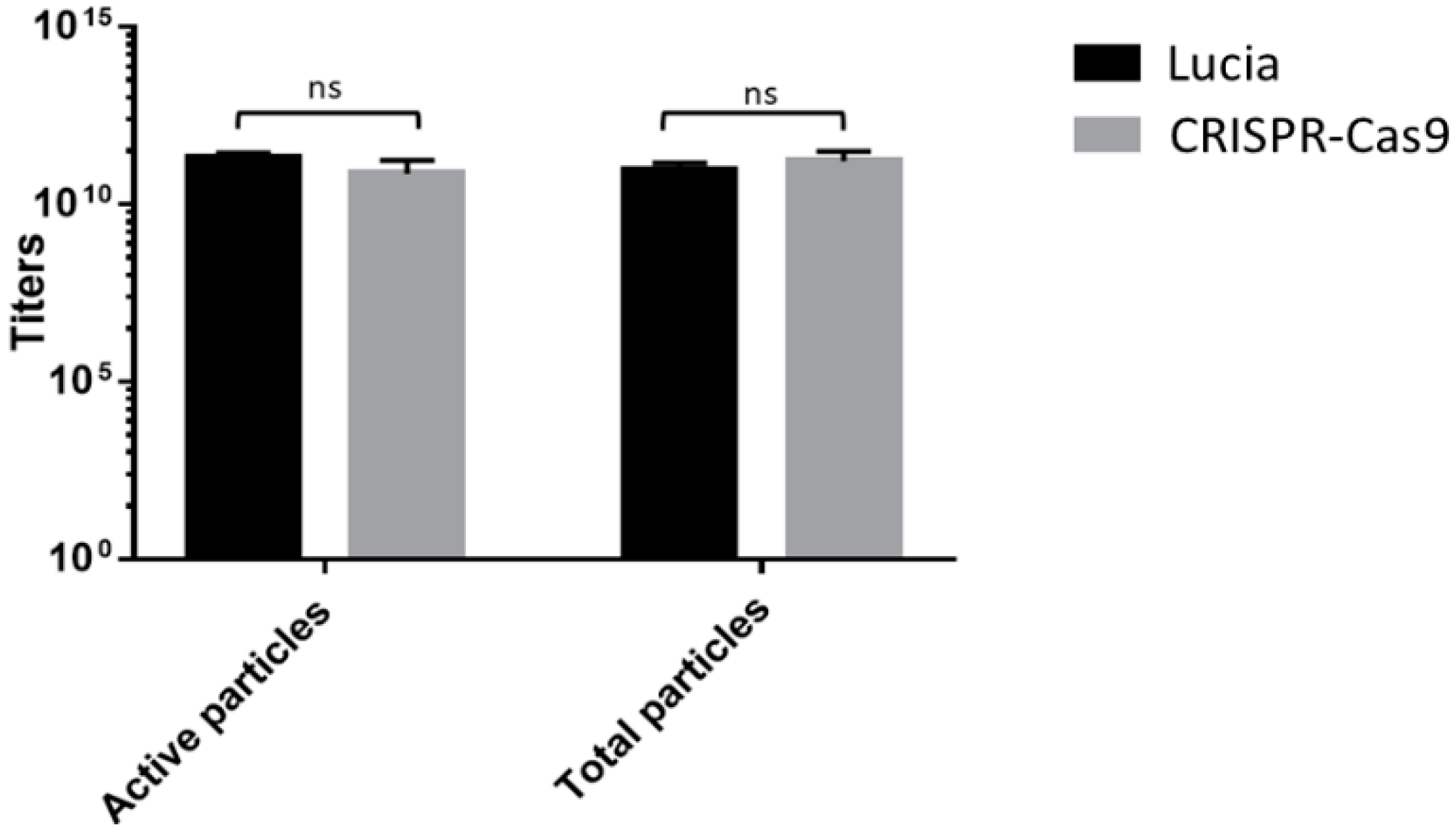
3.2. Analyzing the Impact of the CRISPR-Cas9 Transgene Cassette on Phage Vector Production and Titers Shows No Significant Alterations
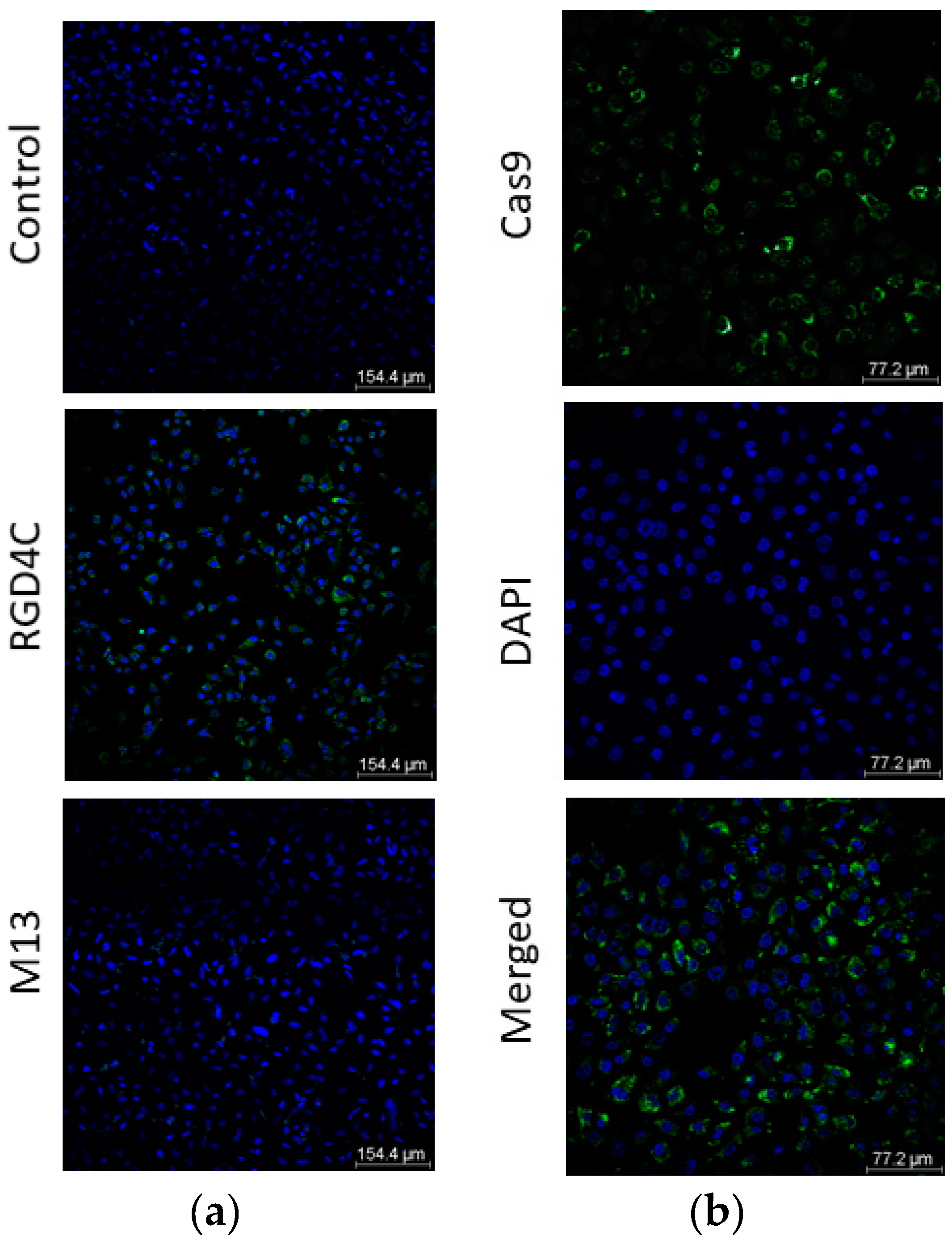
3.3. Evaluation of Cas9 Delivery to Lung Cancer Cells by Bacteriophage Vector Carrying the CRISPR-Cas9 Cassette Shows Efficient Transduction
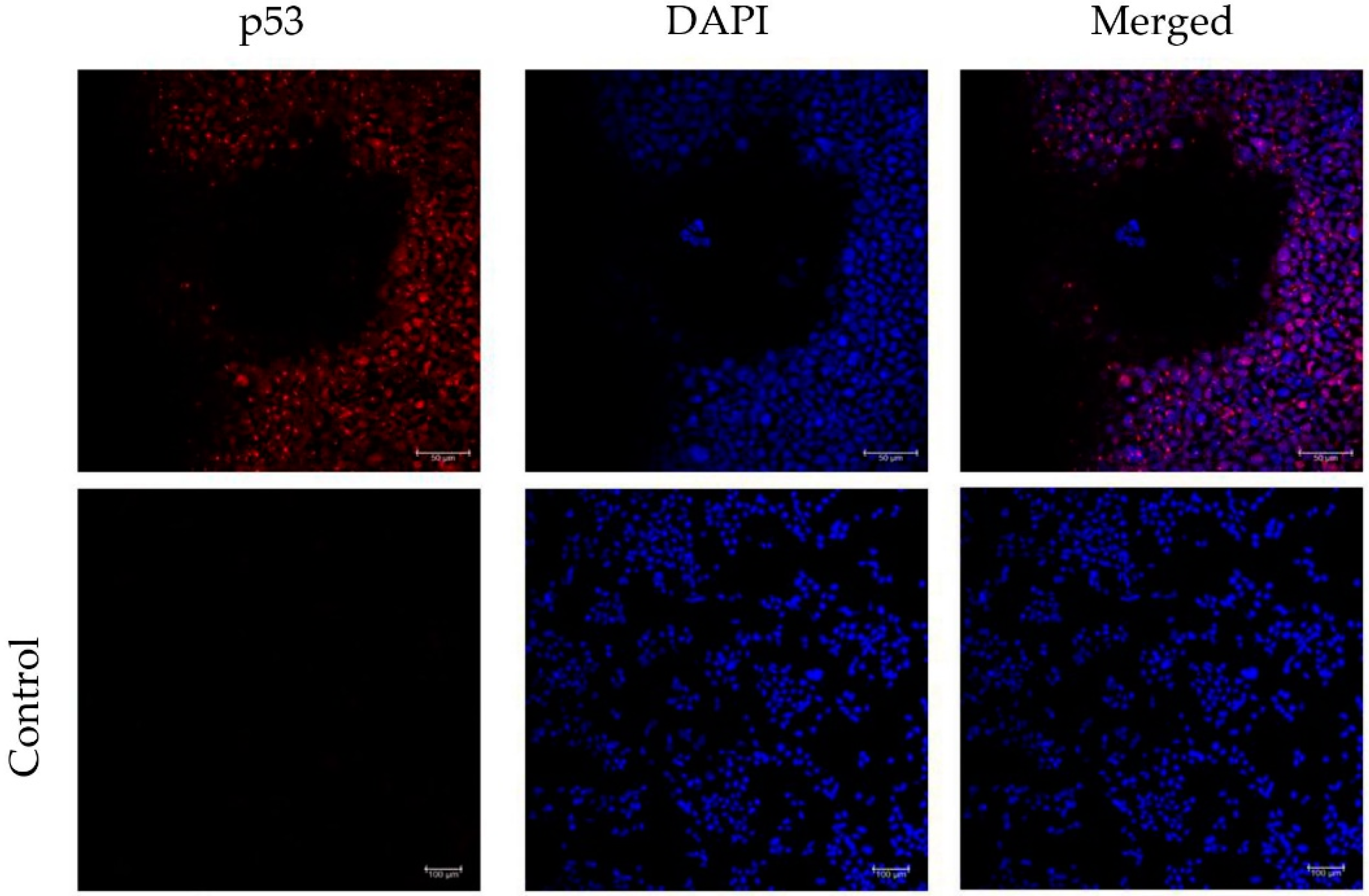
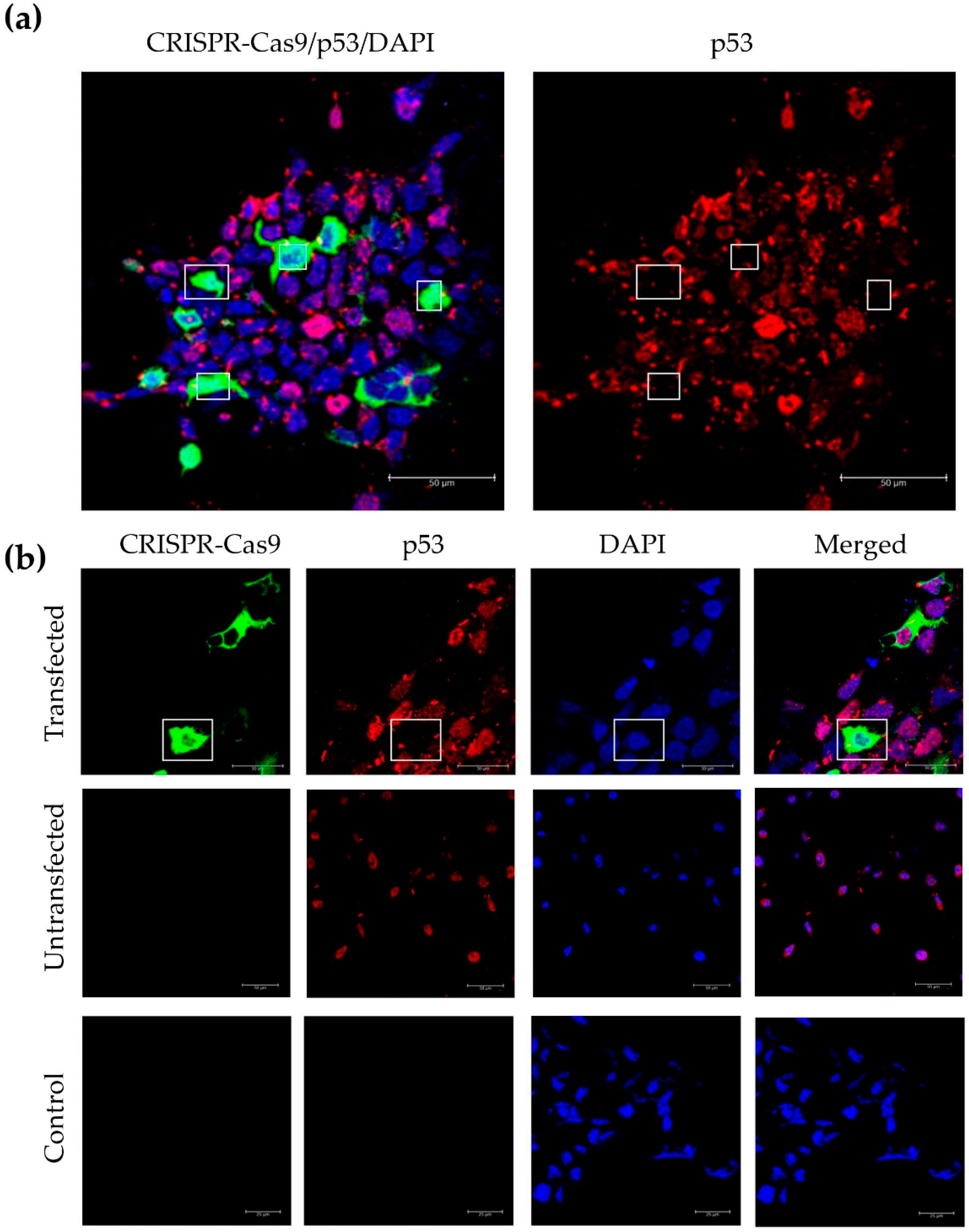
3.4. Incorporation of a p53 gRNA in the CRISPR-Cas9 Phage Plasmid Generates Knockout Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Byers, L.A.; Kurie, J.M. Smoking, p53 Mutation, and Lung Cancer. Mol. Cancer Res. 2014, 12, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Lieschke, E.; Wang, Z.; Kelly, G.L.; Strasser, A. Discussion of some “knowns” and some “unknowns” about the tumour suppressor p53. J. Mol. Cell Biol. 2019, 11, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; San Luis, B.; Lane, D.P. Intratumour heterogeneity of p53 expression; causes and consequences. J. Pathol. 2019, 249, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Tazawa, H.; Kagawa, S.; Fujiwara, T. p53 Replacement Therapy for Cancer. Recent Results Cancer Res. 2016, 209, 1–15. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2016, 8, 8921–8946. [Google Scholar] [CrossRef]
- Oren, M.; Rotter, V. Mutant p53 Gain-of-Function in Cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Goswami, R.; Subramanian, G.; Silayeva, L.; Newkirk, I.; Doctor, D.; Chawla, K.; Chattopadhyay, S.; Chandra, D.; Chilukuri, N.; Betapudi, V. Gene Therapy Leaves a Vicious Cycle. Front. Oncol. 2019, 9, 297. [Google Scholar] [CrossRef]
- Hajitou, A.; Trepel, M.; Lilley, C.E.; Soghomonyan, S.; Alauddin, M.M.; Marini, F.C.; Restel, B.H.; Ozawa, M.G.; Moya, C.A.; Rangel, R.; et al. A hybrid vector for ligand-directed tumor targeting and molecular imaging. Cell 2006, 125, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Stoneham, C.A.; Hollinshead, M.; Hajitou, A. Clathrin-mediated endocytosis and subsequent endo-lysosomal trafficking of adeno-associated virus/phage. J. Biol. Chem. 2012, 287, 35849–35859. [Google Scholar] [CrossRef] [PubMed]
- Suwan, K.; Yata, T.; Waramit, S.; Przystal, J.M.; Stoneham, C.A.; Bentayebi, K.; Asavarut, P.; Chongchai, A.; Pothachaeron, P.; Lee, K.-Y.; et al. Next-generation of targeted AAVP vectors for systemic transgene delivery against cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 18571–18577. [Google Scholar] [CrossRef] [PubMed]
- Tsafa, E.; Al-Bahrani, M.; Bentayebi, K.; Przystal, J.; Suwan, K.; Hajitou, A. The natural dietary genistein boosts bacteriophage-mediated cancer cell killing by improving phage-targeted tumor cell transduction. Oncotarget 2016, 7, 52135–52149. [Google Scholar] [CrossRef][Green Version]
- Hajitou, A. Targeted systemic gene therapy and molecular imaging of cancer contribution of the vascular-targeted AAVP vector. Adv. Genet. 2010, 69, 65–82. [Google Scholar]
- Pranjol, M.Z.I.; Hajitou, A. Bacteriophage-Derived Vectors for Targeted Cancer Gene Therapy. Viruses 2015, 7, 268–284. [Google Scholar] [CrossRef]
- Hajitou, A.; Rangel, R.; Trepel, M.; Soghomonyan, S.; Gelovani, J.G.; Alauddin, M.M.; Pasqualini, R.; Arap, W. Design and construction of targeted AAVP vectors for mammalian cell transduction. Nat. Protoc. 2007, 2, 523–531. [Google Scholar] [CrossRef]
- Paoloni, M.C.; Tandle, A.; Mazcko, C.; Hanna, E.; Kachala, S.; Leblanc, A.; Newman, S.; Vail, D.; Henry, C.; Thamm, D.; et al. Launching a novel preclinical infrastructure: Comparative oncology trials consortium directed therapeutic targeting of TNFalpha to cancer vasculature. PLoS ONE 2009, 4, e4972. [Google Scholar] [CrossRef]
- Tandle, A.; Hanna, E.; Lorang, D.; Hajitou, A.; Moya, C.A.; Pasqualini, R.; Arap, W.; Adem, A.; Starker, E.; Hewitt, S.; et al. Tumor vasculature-targeted delivery of tumor necrosis factor-α*. Cancer 2009, 115, 128–139. [Google Scholar] [CrossRef]
- Kia, A.; Przystal, J.M.; Nianiaris, N.; Mazarakis, N.D.; Mintz, P.J.; Hajitou, A. Dual systemic tumor targeting with ligand-directed phage and Grp78 promoter induces tumor regression. Mol. Cancer Ther. 2012, 11, 2566–2577. [Google Scholar] [CrossRef]
- Przystal, J.M.; Umukoro, E.; Stoneham, C.A.; Yata, T.; O’Neill, K.; Syed, N.; Hajitou, A. Proteasome inhibition in cancer is associated with enhanced tumor targeting by the adeno-associated virus/phage. Mol. Oncol. 2013, 7, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Dobroff, A.S.; D’Angelo, S.; Eckhardt, B.L.; Ferrara, F.; Staquicini, D.I.; Cardó-Vila, M.; Staquicini, F.I.; Nunes, D.N.; Kim, K.; Driessen, W.H.P.; et al. Towards a transcriptome-based theranostic platform for unfavorable breast cancer phenotypes. Proc. Natl. Acad. Sci. USA 2016, 113, 12780–12785. [Google Scholar] [CrossRef] [PubMed]
- Przystal, J.M.; Waramit, S.; Pranjol, M.Z.I.; Yan, W.; Chu, G.; Chongchai, A.; Samarth, G.; Olaciregui, N.G.; Tabatabai, G.; Carcaboso, A.M.; et al. Efficacy of systemic temozolomide-activated phage-targeted gene therapy in human glioblastoma. EMBO Mol. Med. 2019, 11, e8492. [Google Scholar] [CrossRef] [PubMed]
- Namdee, K.; Khongkow, M.; Boonrungsiman, S.; Nittayasut, N.; Asavarut, P.; Temisak, S.; Saengkrit, N.; Puttipipatkhachorn, S.; Hajitou, A.; Ruxrungtham, K.; et al. Thermoresponsive Bacteriophage Nanocarrier as a Gene Delivery Vector Targeted to the Gastrointestinal Tract. Mol. Ther. Nucleic Acids 2018, 12, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Aurnhammer, C.; Haase, M.; Muether, N.; Hausl, M.; Rauschhuber, C.; Huber, I.; Nitschko, H.; Busch, U.; Sing, A.; Ehrhardt, A.; et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum. Gene Ther. Methods 2012, 23, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated in Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef]
- Lau, C.-H.; Suh, Y. In vivo genome editing in animals using AAV-CRISPR system: Applications to translational research of human disease. F1000Res 2017, 6, 2153. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef]
- VandenDriessche, T.; Chuah, M.K. Moving Forward Toward a Cure for Hemophilia B. Mol. Ther. 2015, 23, 809–811. [Google Scholar] [CrossRef][Green Version]
- Meliani, A.; Boisgerault, F.; Hardet, R.; Marmier, S.; Collaud, F.; Ronzitti, G.; Leborgne, C.; Verdera, H.C.; Sola, M.S.; Charles, S.; et al. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat. Commun. 2018, 9, 4098. [Google Scholar] [CrossRef]
- Yuan, Z.; Syrkin, G.; Adem, A.; Geha, R.; Pastoriza, J.; Vrikshajanani, C.; Smith, T.; Quinn, T.J.; Alemu, G.; Cho, H.; et al. Blockade of inhibitors of apoptosis (IAPs) in combination with tumor-targeted delivery of tumor necrosis factor-α leads to synergistic antitumor activity. Cancer Gene Ther. 2013, 20, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.L.; Yuan, Z.; Cardó-Vila, M.; Sanchez Claros, C.; Adem, A.; Cui, M.-H.; Branch, C.A.; Gelovani, J.G.; Libutti, S.K.; Sidman, R.L.; et al. AAVP displaying octreotide for ligand-directed therapeutic transgene delivery in neuroendocrine tumors of the pancreas. Proc. Natl. Acad. Sci. USA 2016, 113, 2466–2471. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, F.; Staquicini, D.I.; Driessen, W.H.P.; D’Angelo, S.; Dobroff, A.S.; Barry, M.; Lomo, L.C.; Staquicini, F.I.; Cardó-Vila, M.; Soghomonyan, S.; et al. Targeted molecular-genetic imaging and ligand-directed therapy in aggressive variant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 12786–12791. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.L.; Souza, G.R.; Sidman, R.L.; Arap, W.; Pasqualini, R. An AAVP-based solid-phase transducing matrix for transgene delivery: Potential for translational applications. Cancer Gene Ther. 2017, 24, 358–360. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang Zhou, J.; Suwan, K.; Hajitou, A. Initial Steps for the Development of a Phage-Mediated Gene Replacement Therapy Using CRISPR-Cas9 Technology. J. Clin. Med. 2020, 9, 1498. https://doi.org/10.3390/jcm9051498
Yang Zhou J, Suwan K, Hajitou A. Initial Steps for the Development of a Phage-Mediated Gene Replacement Therapy Using CRISPR-Cas9 Technology. Journal of Clinical Medicine. 2020; 9(5):1498. https://doi.org/10.3390/jcm9051498
Chicago/Turabian StyleYang Zhou, Jordi, Keittisak Suwan, and Amin Hajitou. 2020. "Initial Steps for the Development of a Phage-Mediated Gene Replacement Therapy Using CRISPR-Cas9 Technology" Journal of Clinical Medicine 9, no. 5: 1498. https://doi.org/10.3390/jcm9051498
APA StyleYang Zhou, J., Suwan, K., & Hajitou, A. (2020). Initial Steps for the Development of a Phage-Mediated Gene Replacement Therapy Using CRISPR-Cas9 Technology. Journal of Clinical Medicine, 9(5), 1498. https://doi.org/10.3390/jcm9051498