MSI and EBV Positive Gastric Cancer’s Subgroups and Their Link with Novel Immunotherapy

 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Molecular Landscape and Classification of Gastric Cancer
3. EBV Positive GCs
4. MSI Positive GCs
5. Immunotherapy in EBV Positive and MSI-H GCs
6. How to Better Select Patients for Immunotherapy: Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Chia, N.-Y.; Tan, P. Molecular classification of gastric cancer. Ann. Oncol. 2016, 27, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Kankeu Fonkoua, L.; Yee, N.S. Molecular Characterization of Gastric Carcinoma: Therapeutic Implications for Biomarkers and Targets. Biomedicines 2018, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Sethi, N.; Sepulveda, A.R.; Bass, A.J.; Wang, T.C. Oesophageal adenocarcinoma and gastric cancer: Should we mind the gap? Nat. Rev. Cancer 2016, 16, 305–318. [Google Scholar] [CrossRef]
- Li, X.; Wu, W.K.; Xing, R.; Wong, S.H.; Liu, Y.; Fang, X.; Zhang, Y.L.; Wang, M.; Wang, J.; Li, L.; et al. Distinct Subtypes of Gastric Cancer Defined by Molecular Characterization Include Novel Mutational Signatures with Prognostic Capability. Cancer Res. 2016, 76, 1724–1732. [Google Scholar] [CrossRef]
- Sunakawa, Y.; Lenz, H.-J. Molecular classification of gastric adenocarcinoma: Translating new insights from the cancer genome atlas research network. Curr. Treat. Options Oncol. 2015, 16, 331. [Google Scholar] [CrossRef]
- Tang, W.; Morgan, D.R.; Meyers, M.O.; Dominguez, R.L.; Martínez, E.; Kakudo, K.; Kuan, P.F.; Banet, N.; Muallem, H.; Woodward, K.; et al. Epstein-barr virus infected gastric adenocarcinoma expresses latent and lytic viral transcripts and has a distinct human gene expression profile. Infect. Agents Cancer 2012, 7, 21. [Google Scholar] [CrossRef]
- Murphy, G.; Pfeiffer, R.; Camargo, M.C.; Rabkin, C.S. Meta-analysis shows that prevalence of Epstein-Barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology 2009, 137, 824–833. [Google Scholar] [CrossRef]
- Geddert, H.; Zur Hausen, A.; Gabbert, H.E.; Sarbia, M. EBV-infection in cardiac and non-cardiac gastric adenocarcinomas is associated with promoter methylation of p16, p14 and APC, but not hMLH1. Anal. Cell. Pathol. 2010, 33, 143–149. [Google Scholar] [CrossRef]
- Camargo, M.C.; Kim, W.H.; Chiaravalli, A.M.; Kim, K.M.; Corvalan, A.H.; Matsuo, K.; Yu, J.; Sung, J.J.; Herrera-Goepfert, R.; Meneses-Gonzalez, F.; et al. Improved survival of gastric cancer with tumour Epstein-Barr virus positivity: An international pooled analysis. Gut 2014, 63, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.; Oliveira, A.; Malta, M.; Oliveira, C.; Silva, F.; Galaghar, A.; Afonso, L.P.; Cassiano Neves, M.; Medeiros, R.; Pimentel-Nunes, P.; et al. Clinical and pathological characterization of Epstein-Barr virus-associated gastric carcinomas in Portugal. World J. Gastroenterol. 2017, 23, 7292–7302. [Google Scholar] [CrossRef] [PubMed]
- Ito, C.; Nishizyka, S.S.; Ishida, K.; Uesugi, N.; Sugai, T.; Tamura, G.; Koeda, K.; Sasaki, A. Analysis of PIK3CA mutations and PI3K pathway proteins in advanced gastric cancer. J. Surg. Res. 2017, 212, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.H.; Ryu, M.-H.; Park, Y.S.; Lee, H.J.; Lee, C.; Ryoo, B.-Y.; Lee, J.-L.; Chang, H.-M.; Kim, T.W.; Kang, Y.-K. Phase II study of everolimus with biomarker exploration in patients with advanced gastric cancer refractory to chemotherapy including fluoropyrimidine and platinum. Br. J. Cancer 2012, 106, 1039–1044. [Google Scholar] [CrossRef]
- Doi, T.; Muro, K.; Boku, N.; Yamada, T.; Nishina, T.; Takiuchi, H.; Komatsu, Y.; Hamamoto, Y.; Ohno, N.; Fujita, Y.; et al. Multicenter Phase II Study of Everolimus in Patients Previously Treated Metastatic Gastric Cancer. J. Clin. Oncol. 2010, 28, 1904–1910. [Google Scholar] [CrossRef]
- Ohtsu, A.; Ajani, J.A.; Bai, Y.-X.; Bang, Y.-J.; Chung, H.C.; Pan, H.-M.; Sahmoud, T.; Shen, L.; Yeh, K.-H.; Chin, K.; et al. Everolimus for Previously Treated Advanced Gastric Cancer: Results of the Randomized, Double-Blind, Phase III GRANITE-1 Study. J. Clin. Oncol. 2013, 31, 3935–3943. [Google Scholar] [CrossRef]
- Chen, D.; Lin, X.; Zhang, C.; Liu, Z.; Chen, Z.; Li, Z.; Wang, J.; Li, B.; Hu, Y.; Dong, B.; et al. Dual PI3K/mTOR inhibitor BEZ235 as a promising therapeutic strategy against paclitaxel-resistant gastric cancer via targeting PI3K/Akt/mTOR pathway. Cell Death Dis. 2018, 9, 123. [Google Scholar] [CrossRef]
- Panda, A.; Mehnert, J.M.; Hirshfield, K.M.; Riedlinger, G.; Damare, S.; Saunders, T.; Kane, M.; Sokol, L.; Stein, M.N.; Poplin, E.; et al. Immune Activation and Benefit From Avelumab in EBV-Positive Gastric Cancer. J. Natl. Cancer Inst. 2017, 110, 316–320. [Google Scholar] [CrossRef]
- Kenney, S.C.; Mertz, J.E. Regulation of the latent-lytic switch in Epstein-Barr virus. Semin. Cancer Boil. 2014, 26, 60–68. [Google Scholar] [CrossRef]
- Liu, X.; Meltzer, S.J. Gastric Cancer in the Era of Precision Medicine. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.Y.; Yuen, S.T.; Chung, L.P.; Chu, K.M.; Chan, A.S.; Ho, J.C. hMLH1 promoter methylation and lack of hMLH1 expression in sporadic gastric carcinomas with high frequency microsatellite instability. Cancer Res. 1999, 59, 159–164. [Google Scholar] [PubMed]
- Mathiak, M.; Warneke, V.S.; Behrens, H.-M.; Haag, J.; Böger, C.; Krüger, S.; Röcken, C. Clinicopathologic Characteristics of Microsatellite Instable Gastric Carcinomas Revisited: Urgent need for standardization. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Baniak, N.; Senger, J.-L.; Ahmed, S.; Kanthan, R. Gastric biomarkers: A global review. World J. Surg. Oncol. 2016, 14, 212. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.Y.; Kim, H.; Shin, S.-J.; Kim, H.Y.; Lee, J.; Yang, H.-K.; Kim, W.H.; Kim, H.K.; Kook, M.-C.; Park, Y.K.; et al. Microsatellite Instability and Programmed Cell Death-Ligand 1 Expression in Stage II/III Gastric Cancer: Post Hoc Analysis of the CLASSIC Randomized Controlled study. Ann. Surg. 2019, 270, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Wotherspoon, A.; Peckitt, C.; González, D.; Hulkki-Wilson, S.; Eltahir, Z.; Fassan, M.; Rugge, M.; Valeri, N.; Okines, A.; et al. Mismatch Repair Deficiency, Microsatellite Instability, and Survival: An Exploratory Analysis of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) Trial. JAMA Oncol. 2017, 3, 1197–1203. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Miceli, R.; Raimondi, A.; Kim, Y.W.; Kang, W.K.; Langley, R.E.; Choi, Y.Y.; Kim, K.-M.; Nankivell, M.G.; Morano, F.; et al. Individual Patient Data Meta-Analysis of the Value of Microsatellite Instability As a Biomarker in Gastric Cancer. J. Clin. Oncol. 2019, 37, 3392–3400. [Google Scholar] [CrossRef]
- Polom, K.; Marrelli, D.; Smyth, E.C.; Voglino, C.; Roviello, G.; Pascale, V.; Varas, J.; Vindigni, C.; Roviello, F. The Role of Microsatellite Instability in Positive Margin Gastric Cancer Patients. Surg. Innov. 2018, 25, 99–104. [Google Scholar] [CrossRef]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.N.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef]
- Willett, C.G.; Chang, D.T.; Czito, B.G.; Meyer, J.; Wo, J. The Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumors: Impact on clnical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J. Immunotherapy for Esophageal and Gastric Cancer. Am. Soc. Clin. Oncol. Educ. Book 2018, 37, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Derks, S.; Liao, X.; Chiaravalli, A.M.; Xu, X.; Camargo, M.C.; Solcia, E.; Sessa, F.; Fleitas, T.; Freeman, G.J.; Rodig, S.J.; et al. Abundant PD-L1 expression in Epstein-Barr Virus-infected gastric cancer. Oncotarget 2016, 7, 32925–32932. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Spaapen, R.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-Regulation of PD-L1, IDO, and T(regs) in the Melanoma Tumor Microenvironment Is Driven by CD8(+) T Cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef] [PubMed]
- Liosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment or microsatellite instable colon cancer patients receiving first-line chemotherapy. Gastric Cancer 2017, 20, 156–163. [Google Scholar]
- Muro, K.; Chung, H.C.; Shankaran, V.; Geva, R.; Catenacci, D.; Gupta, S.; Eder, J.P.; Golan, T.; Le, D.T.; Burtness, B.; et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): A multicentre, open-label, phase 1b trial. Lancet Oncol. 2016, 17, 717–726. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Bendell, J.; Calvo, E.; Kim, J.W.; Ascierto, P.A.; Sharma, P.; Ott, P.A.; Peltola, K.; Jaeger, D.; Evans, J.; et al. CheckMate-032 Study: Efficacy and Safety of Nivolumab and Nivolumab Plus Ipilimumab in Patients With Metastatic Esophagogastric Cancer. J. Clin. Oncol. 2018, 36, 2836–2844. [Google Scholar] [CrossRef]
- Moehler, M.H.; Ryu, M.-H.; Lee, K.-W.; Coskun, H.S.; Wong, R.; Ozguroglu, M.; Chung, H.-C.; Poltoratsky, A.; Tsuji, A.; Yen, C.-J.; et al. Maintenance therapy with avelumab (MSB0010718C; an -PD-L1) vs connuation of rst-line chemotherapy in patients with unresectable, locally advanced or metastatic gastric cancer: The phase 3 JAVELIN Gastric 100 trial. J. Clin. Oncol. 2018, 36, TPS195. [Google Scholar] [CrossRef]
- Kang, Y.-K.; Boku, N.; Satoh, T.; Ryu, M.-H.; Chao, Y.; Kato, K.; Chung, H.C.; Chen, J.-S.; Muro, K.; Kang, W.K.; et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 2461–2471. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef]
- Shitara, K.; Özgüroğlu, M.; Bang, Y.J.; Di Bartolomeo, M.; Mandalà, M.; Ryu, M.H.; Fornaro, L.; Olesiński, T.; Caglevic, C.; Chung, H.C.; et al. KEYNOTE-061 investigators. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): A randomised, open-label, controlled, phase 3 trial. Lancet 2018, 392, 123–133. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Banks, K.; Pectasides, E.; Kim, K.; Lanman, R.; Talasaz, A.; An, J.; Choi, M.; Lee, J.; Sohn, T.; et al. Impact of genomic alterations on lapatinib treatment outcome and cellfree genomic landscape during HER2 therapy in HER2+ gastric cancer patients. Ann. Oncol. 2018, 29, 1037–1048. [Google Scholar] [CrossRef]
- Kato, S.; Okamura, R.; Baumgartner, J.M.; Patel, H.; Leichman, L.; Kelly, K.; Sicklick, J.K.; Fanta, P.T.; Lippman, S.M.; Kurzrock, R. Analysis of Circulating Tumor DNA and Clinical Correlates in Patients with Esophageal, Gastroesophageal Junction, and Gastric Adenocarcinoma. Clin. Cancer Res. 2018, 24, 6248–6256. [Google Scholar] [CrossRef] [PubMed]
- Garlan, F.; Laurent-Puig, P.; Sefrioui, D.; Siauve, N.; Didelot, A.; Perkins, G.; Mulot, C.; Zaanan, A.; Sarafan-Vasseur, N.; Michel, P.; et al. Early Evaluation of Circulating Tumor DNA as Marker of Therapeutic Efficacy in Metastatic Colorectal Cancer Patients (PLACOL Study). Clin. Cancer Res. 2017, 23, 5416–5425. [Google Scholar] [CrossRef]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.-M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef]
- Rubinstein, J.C.; Nicolson, N.G.; Ahuja, N. Next-generation Sequencing in the Management of Gastric and Esophageal Cancers. Surg. Clin. N. Am. 2019, 99, 511–527. [Google Scholar] [CrossRef]
- Harada, K.; Kaya, D.M.; Shimodaira, Y.; Song, S.; Baba, H.; Ajani, J. Translating genomic profiling to gastrointestinal cancer treatment. Future Oncol. 2017, 13, 919–934. [Google Scholar] [CrossRef]
- Lee, H.S.; Kim, W.H.; Kwak, Y.; Koh, J.; Bae, J.M.; Kim, K.M.; Chang, M.S.; Han, H.S.; Kim, J.M.; Kim, H.W.; et al. Molecular Testing for Gastrointestinal Cancer. J. Pathol. Transl. Med. 2017, 51, 103–121. [Google Scholar] [CrossRef][Green Version]


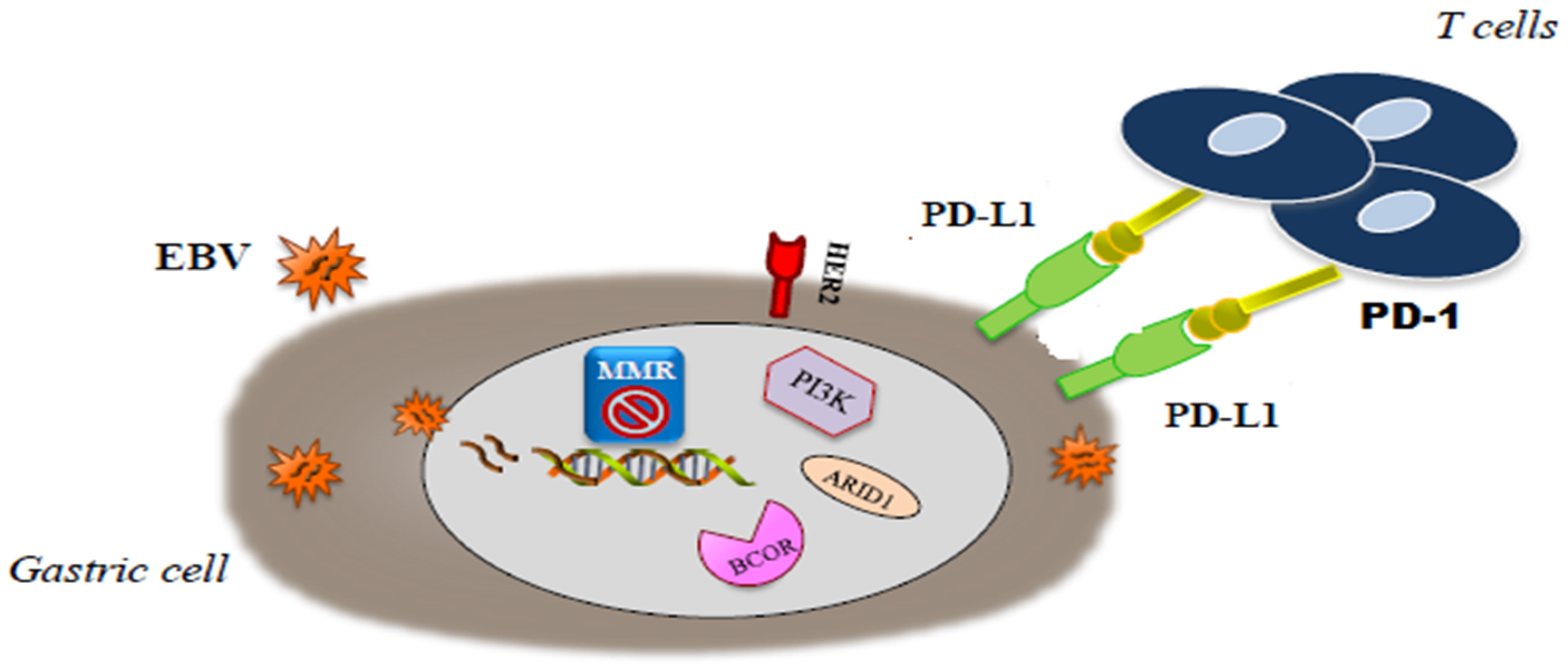{kind=link}
| SUBTYPES | MOLECULAR FEATURES | |
|---|---|---|
| TCGA (The Cancer Genome Atlas) | EBV (9%) | - DNA hypermethylation, including CDKN2A (p16) but not MLH1 promoters |
| - PIK3CA mutations | ||
| - JAK2 gene amplification | ||
| - PDL1/PDL2 overexpression | ||
| MSI (22%) | - high mutation rate | |
| - DNA methylation with epigenetic silencing of MLH1 | ||
| - Hypermutation of many genes including HLA class 1 factors | ||
| GS (20%) | - molecular alterations in cell adhesion/ cell migration pathways | |
| - ARID1 and BCOR mutations | ||
| CIN (50%) | - chromosomal instability (CIN) | |
| - amplification of genes (most encoding tyrosine kinase receptors) | ||
| ACRG (Asian Cancer Research Group) | MSS/TP53 + (26%) | - frequent EBV positivity |
| - intermediate mutation rate | ||
| MSI (23%) | - high mutation rate | |
| EMT (15%) | - low mutation rate | |
| - loss of epithelial markers | ||
| MSS/TP53- (36%) | - TP53 mutations | |
| - genomic instability | ||
| Li et al. Classification | REGULAR C1 | - 2.4 mutations /megabase; range, 0–8.3 |
| - TP53, XIRP2, APC mutations | ||
| REGULAR C2 | - 2.4 mutations /megabase; range, 0–8.3 | |
| - ARID1A, CDH1, PIK3CA, ERBB2, RHOA mutations | ||
| - | ||
| HYPERMUTATED | - 20.5 mutations/megabase; range, 9.6–200.2) |
| Trial/Author | Target | Agent | Line | Control | Endpoint | Result | Difference mOS (m) (HR) |
|---|---|---|---|---|---|---|---|
| Keynote061 | PD1 | Pembrolizumab | 2nd | Paclitaxel | OS | Negative | +0.8 (HR 0.82) |
| JAVELIN300 | PD1 | Avelumab | 3rd | Irinotecan/taxanes | OS | Negative | −0.4 (HR 1.1) |
| ATTRACTION-2 | PD1 | Nivolumab | 3rd | Placebo | OS | Positive | +1.2 (HR 0.63) |
| AVAGAST | VEGF-A | Bevacizumab | 1st | Placebo (+chemo) | OS | Negative | +2 (HR 0.87) |
| RAINFALL | VEGFR2 | Ramucirumab | 1st | Placebo (+chemo) | OS | Negative | +0.4 (HR 0.96) |
| REGARD | VEGFR2 | Ramucirumab | 2nd | Placebo | OS | Positive | +1.4 (HR 0.776) |
| RAINBOW | VEGFR2 | Ramucirumab | 2nd | Placebo (+chemo) | OS | Positive | +2.2 (HR 0.807) |
| Li. et al. | VEGFR2 | Apatinib | 3rd | Placebo | OS | Positive | +1.8 (HR 0.71) |
| REAL-3 | EGFR | Panitumumab | 1st | (+chemo) | OS | Negative | −2.5 (HR 1.37) |
| EXPAND | EGFR | Cetuximab | 1st | Placebo (+chemo) | PFS | Negative | −1.3 (HR 1.0) |
| ToGA | HER2 | Trastuzumab | 1st | (+chemo) | OS | Positive | +2.7 (HR 0.74) |
| Logic | HER2 | Lapatinib | 1st | Placebo (+chemo) | OS | Negative | +1.7 (HR 0.91) |
| JACOB | HER2 | Pertuzumab | 1st | Placebo (+chemo + Tmab) | OS | Negative | +3.3 (HR 0.84) |
| TyTAN | HER2 | Lapatinib | 2nd | (+chemo) | OS | Negative | +3 (HR 0.84) |
| GATSBY | HER2 | T-DM1 | 2nd | Taxanes | OS | Negative | −0.7 (HR 1.15) |
| GRANITE-1 | mTOR | Everolimus | 2nd/3rd | Placebo | OS | Negative | +1.05 (HR 0.9) |
| GRANITE-2 | mTOR | Everolimus | 2nd | Placebo (+chemo) | OS | Negative | +1.0 (HR 0.92) |
| RILOMET1 | HGF | Rilotumumab | 1st | Placebo (+chemo) | OS | Negative | −2.9 (HR 1.36) |
| METgastric | MET | Onartuzumab | 1st | Placebo (+chemo) | OS | Negative | −0.3 (HR 0.82) |
| GOLD | PARP | Olaparib | 2nd | Placebo (+chemo) | OS | Negative | +1.9 (HR 0.79) |
| BRIGHTER | STAT3 | Napabucasin | 2nd | Placebo (+chemo) | OS | Negative | −0.4 (HR 1.01) |
| Next-Generation Sequencing Techniques | |
|---|---|
| Whole-genome sequencing (WGS) | Single-nucleotide resolution of all genome bases |
| Whole-exome sequencing (WES) | Single-nucleotide resolution of protein-codon areas of the genome |
| Targeted sequencing | Covers limited subsets of candidate genes |
| RNA sequencing | Sequencing of each RNA transcript |
| Gene expression profiling | Evaluates the RNA level of a single gene with further functional associations; cell environment as potential bias |
| Esophageal Cancer | Gastroesophageal Cancer | ||
|---|---|---|---|
| Gene | Frequency (%) | Gene | Frequency (%) |
| TP53 | 60–93 | TP53 | 14–59 |
| CCND1 | 33–46 | PIK3CA | 7–36 |
| CDKN2A | 12–47 | CDH1 | 4–36 |
| KMT2D | 19–26 | HER2 | 2–32 |
| FAT1 | 14–27 | ARID1A | 8–27 |
| KRAS | 5–27 | KRAS | 0–27 |
| EGFR | 6–24 | PTEN | 0–27 |
| NOTCH | 9–19 | RHOA | 0–23 |
| PIK3CA | 4–10 | APC | 3–14 |
| ERBB3 | 0–10 | ||
| CTNNB1 | 2–9 | ||
| MET | 0–9 | ||
| SMAD4 | 4–6 | ||
| FBXW7 | 2–6 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriquenz, M.G.; Roviello, G.; D’Angelo, A.; Lavacchi, D.; Roviello, F.; Polom, K. MSI and EBV Positive Gastric Cancer’s Subgroups and Their Link with Novel Immunotherapy. J. Clin. Med. 2020, 9, 1427. https://doi.org/10.3390/jcm9051427
Rodriquenz MG, Roviello G, D’Angelo A, Lavacchi D, Roviello F, Polom K. MSI and EBV Positive Gastric Cancer’s Subgroups and Their Link with Novel Immunotherapy. Journal of Clinical Medicine. 2020; 9(5):1427. https://doi.org/10.3390/jcm9051427
Chicago/Turabian StyleRodriquenz, Maria Grazia, Giandomenico Roviello, Alberto D’Angelo, Daniele Lavacchi, Franco Roviello, and Karol Polom. 2020. "MSI and EBV Positive Gastric Cancer’s Subgroups and Their Link with Novel Immunotherapy" Journal of Clinical Medicine 9, no. 5: 1427. https://doi.org/10.3390/jcm9051427
APA StyleRodriquenz, M. G., Roviello, G., D’Angelo, A., Lavacchi, D., Roviello, F., & Polom, K. (2020). MSI and EBV Positive Gastric Cancer’s Subgroups and Their Link with Novel Immunotherapy. Journal of Clinical Medicine, 9(5), 1427. https://doi.org/10.3390/jcm9051427