Genetic, Epigenetic, and Steroidogenic Modulation Mechanisms in Endometriosis


Abstract
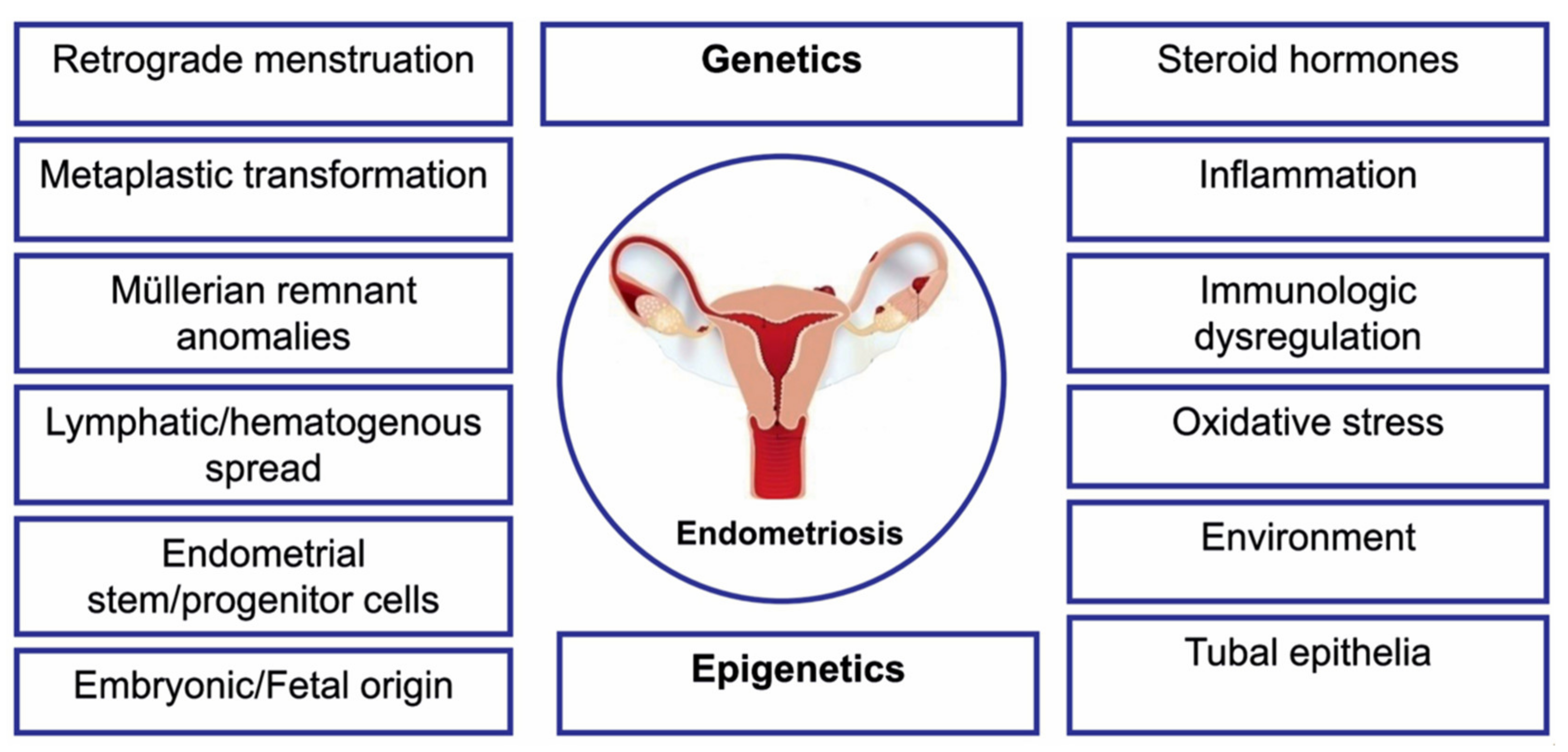
1. Introduction
2. Genetic Profile of Endometriosis
2.1. Familial Studies in Endometriosis
2.2. Genetic Studies in Endometriosis
2.3. Genome-Wide Association Studies in Endometriosis
2.4. Genes Associated with Endometriosis
3. Steroidogenic Pathway
4. Steroidogenic Factor-1
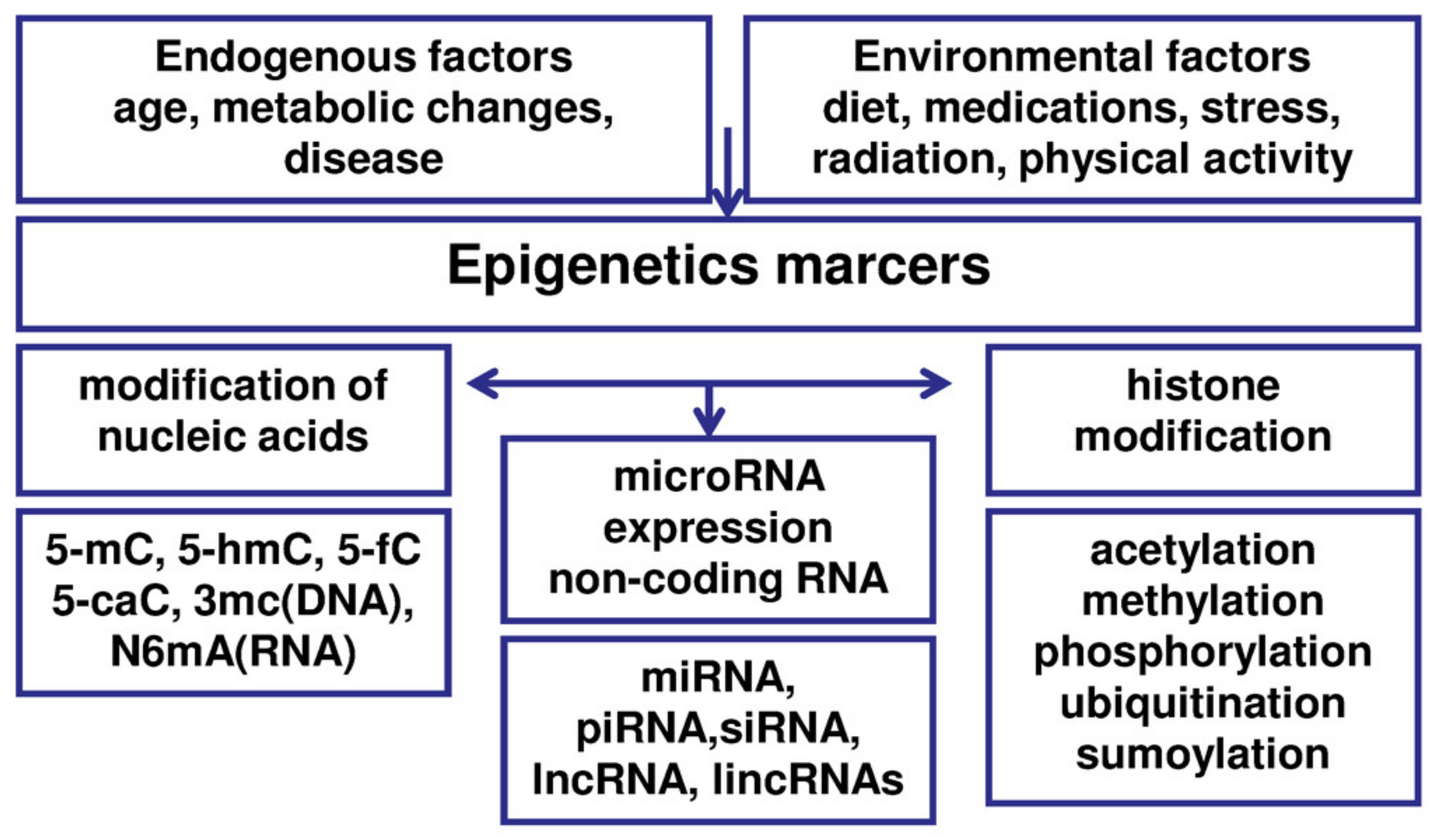
5. Epigenetic Processes in Endometriosis
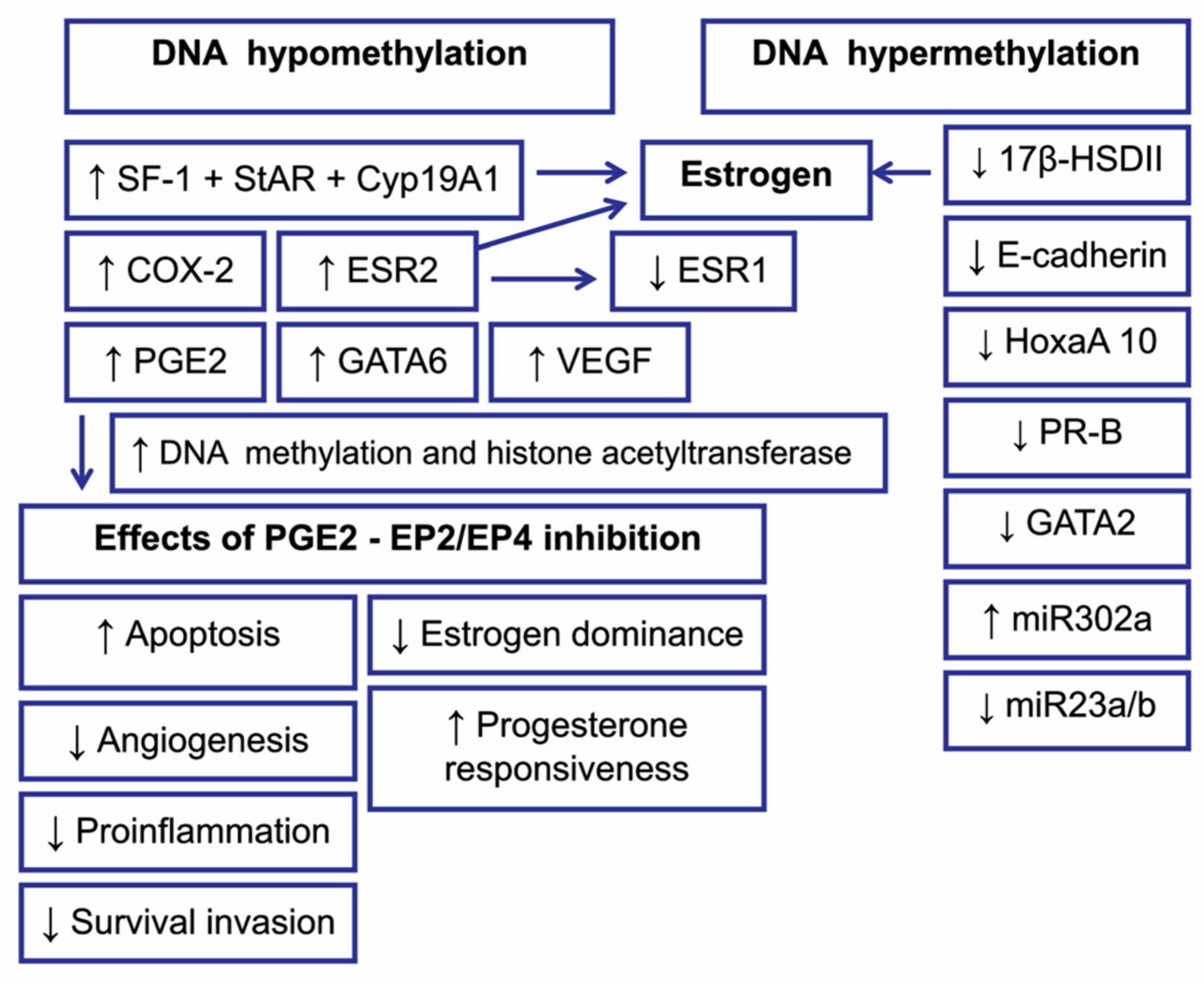
5.1. DNA Methylation in Endometriosis
5.2. Epigenetic Histone Modifications
5.3. Role of Non-Coding RNA in the Detection of Endometriosis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 17β-HSD | 17β-hydroxysteroid dehydrogenase |
| Bcl-2 | Antiapoptotic protein B cell lymphoma 2 |
| CDK | Cyclin-dependent kinase |
| COX-2 | Cyclo-oxygenase type 2 |
| CpG | Cytosine that precede a guanosine |
| CYP17 | 17, 20-lyase gene |
| CYP19 | Aromatase cytochrome P450 gene |
| DNMTs | DNA methyltransferase enzymes |
| EP2 | Prostaglandin E receptor 2 |
| EP4 | Prostaglandin E receptor 4 |
| ER | Estrogen receptor |
| ESR1 | Estrogen receptors type 1 (alpha) |
| ESR2 | Estrogen receptors type 2 (beta) |
| FUMA | Functional mapping and annotation |
| GREB1 | Growth regulating estrogen receptor binding 1 |
| GWA | Genome-wide association |
| HAT | Histone acetyltransferases |
| HDAC | Histone deacetylase |
| HDAC 1 | Histone deacetylase 1 |
| HDACI | Histone deacetylase inhibitors |
| HMT | Histone methyltransferases |
| HOXA10 | Homeobox A transcription factor |
| HSD17B2 | 17β-hydroxysteroid dehydrogenase type II |
| IL | Interleukin |
| MAGMA | Multi-marker Analysis of GenoMic Annotation |
| MBD2 | Methyl-CpG-binding domain protein 2 |
| miRNA | MicroRNA |
| NR5A1 | Nuclear receptor subfamily 5, group A, member 1 (SF-1) |
| PG | Prostaglandin |
| PGE2 | Prostaglandin E2 |
| PGR | Progesterone receptor gene |
| piRNA | Piwi interacting RNA |
| PR | Progesterone receptor |
| siRNA | Small interfering RNA |
| SF-1 | Steroidogenic factor-1 |
| SF-2 | Steroidogenic factor-2 |
| SNP | Single nucleotide polymorphisms |
| SR | Steroid/nuclear receptor |
| StAR | Steroid acute regulatory |
| SUMO | Small ubiquitin-like modifier |
| TGF-β | Transforming growth factor-β |
| TNF | Tumor necrosis factor |
| WNT4 | Wingless-type mouse mammary tumor virus integration site family 4 |
| VEGF | Vascular endothelial growth factor |
References
- Giudice, L.C.; Kao, L.C. Endometriosis. Lancet 2004, 364, 1789–1799. [Google Scholar] [CrossRef]
- Eisenberg, V.H.; Weil, C.; Chodick, G.; Shalev, V. Epidemiology of endometriosis: A large population-based database study from a healthcare provider with 2 million members. BJOG An. Int. J. Obstet. Gynaecol. 2018, 125, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Johnstone, E.B.; Bloom, M.S.; Huddleston, H.G.; Fujimoto, V.Y. A higher prevalence of endometriosis among Asian women does not contribute to poorer IVF outcomes. J. Assist. Reprod. Genet. 2017, 34, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Hickey, M.; Ballard, K.; Farquhar, C. Endometriosis. BMJ 2014, 348, g1752. [Google Scholar] [CrossRef]
- Kobayashi, H.; Higashiura, Y.; Shigetomi, H.; Kajihara, H. Pathogenesis of endometriosis: The role of initial infection and subsequent sterile inflammation (Review). Mol. Med. Rep. 2014, 9, 9–15. [Google Scholar] [CrossRef]
- Giudice, L.C. Endometriosis. N. Engl. J. Med. 2010, 362, 2389–2398. [Google Scholar] [CrossRef]
- Tomassetti, C.; D’Hooghe, T. Endometriosis and infertility: Insights into the causal link and management strategies. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 51, 25–33. [Google Scholar] [CrossRef]
- Indrielle-Kelly, T.; Frühauf, F.; Fanta, M.; Burgetova, A.; Lavu, D.; Dundr, P.; Cibula, D.; Fischerova, D. Application of the International Deep Endometriosis Analysis (IDEA) group consensus in preoperative ultrasound and MR imaging of deep pelvic endometriosis. Ultrasound Obstet. Gynecol. 2019. [Google Scholar] [CrossRef]
- Domínguez, F. Search for new molecular biomarkers to diagnose endometriosis continues. Fertil. Steril. 2018, 109, 615–616. [Google Scholar] [CrossRef]
- Agrawal, S.; Tapmeier, T.T.; Rahmioglu, N.; Kirtley, S.; Zondervan, K.T.; Becker, C.M. The miRNA mirage: How close are we to finding a non-invasive diagnostic biomarker in endometriosis? A systematic review. Int. J. Mol. Sci. 2018, 19, 599. [Google Scholar] [CrossRef]
- Zubrzycka, A.; Zubrzycki, M.; Janecka, A.; Zubrzycka, M. New Horizons in the Etiopathogenesis and Non-Invasive Diagnosis of Endometriosis. Curr. Mol. Med. 2015, 15, 697–713. [Google Scholar]
- Yuan, Z.; Wang, L.; Wang, Y.; Zhang, T.; Li, L.; Cragun, J.M.; Chambers, S.K.; Kong, B.; Zheng, W. Tubal origin of ovarian endometriosis. Mod. Pathol. 2014, 27, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulou, L.; Matalliotakis, M.; Zervou, M.I.; Matalliotaki, C.; Krithinakis, K.; Matalliotakis, I.; Spandidos, D.A.; Goulielmos, G.N. Defining the genetic profile of endometriosis. Exp. Ther. Med. 2019, 17, 3267–3281. [Google Scholar] [PubMed]
- Zondervan, K.T.; Rahmioglu, N.; Morris, A.P.; Nyholt, D.R.; Montgomery, G.W.; Becker, C.M.; Missmer, S.A. Beyond Endometriosis Genome-Wide Association Study: From Genomics to Phenomics to the Patient. Semin. Reprod. Med. 2016, 34, 242–254. [Google Scholar]
- Sapkota, Y.; Low, S.-K.; Attia, J.; Gordon, S.D.; Henders, A.K.; Holliday, E.G.; MacGregor, S.; Martin, N.G.; McEvoy, M.; Morris, A.P.; et al. Association between endometriosis and the interleukin 1A (IL1A) locus. Hum. Reprod. 2015, 30, 239–248. [Google Scholar] [CrossRef]
- Rahmioglu, N.; Banasik, K.; Christofidou, P.; Danning, R.; Galarneau, G.; Giri, A.; MacGregor, S.; Mortlock, S.; Sapkota, Y.; Schork, J.A.; et al. Large-scale genome-wide association meta-analysis of endometriosis reveals 13 novel loci and genetically-associated comorbidity with other pain conditions. bioRxiv 2018. [Google Scholar] [CrossRef]
- Sapkota, Y.; Steinthorsdottir, V.; Morris, A.P.; Fassbender, A.; Rahmioglu, N.; De Vivo, I.; Buring, J.E.; Zhang, F.; Edwards, T.L.; Jones, S.; et al. Meta-analysis identifies five novel loci associated with endometriosis highlighting key genes involved in hormone metabolism. Nat. Commun. 2017, 8, 15539. [Google Scholar] [CrossRef]
- Uimari, O.; Rahmioglu, N.; Nyholt, D.R.; Vincent, K.; Missmer, S.A.; Becker, C.; Morris, A.P.; Montgomery, G.W.; Zondervan, K.T. Genome-wide genetic analyses highlight mitogen-activated protein kinase (MAPK) signaling in the pathogenesis of endometriosis. Hum. Reprod. 2017, 32, 780–793. [Google Scholar] [CrossRef]
- Nyholt, D.R.; Low, S.-K.; Anderson, C.A.; Painter, J.N.; Uno, S.; Morris, A.P.; MacGregor, S.; Gordon, S.D.; Henders, A.K.; Martin, N.G.; et al. Genome-wide association meta-analysis identifies new endometriosis risk loci. Nat. Genet. 2012, 44, 1355–1359. [Google Scholar] [CrossRef]
- Saare, M.; Rekker, K.; Laisk-Podar, T.; Rahmioglu, N.; Zondervan, K.; Salumets, A.; Gotte, M.; Peters, M. Challenges in endometriosis miRNA studies—From tissue heterogeneity to disease specific miRNAs. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 2282–2292. [Google Scholar] [CrossRef]
- Krishnamoorthy, K.; Decherney, A.H. Genetics of Endometriosis. Clin. Obstet. Gynecol. 2017, 60, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-X.; Gao, S.-Y.; Wang, P.-Y.; Zhou, X.; Li, Y.-J.; Yu, Y.; Yan, Y.-F.; Zhang, H.H.; Lv, C.-J.; Zhou, H.-H.; et al. Reduced expression levels of let-7c in human breast cancer patients. Oncol. Lett. 2015, 9, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Koukoura, O.; Sifakis, S.; Spandidos, D.A. DNA methylation in endometriosis (Review). Mol. Med. Rep. 2016, 13, 2939–2948. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-L.; Zhao, M. A PubMed-wide study of endometriosis. Genomics 2016, 108, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Grimstad, F.W.; Decherney, A. A Review of the Epigenetic Contributions to Endometriosis. Clin. Obstet. Gynecol. 2017, 60, 467–476. [Google Scholar] [CrossRef]
- Akter, S.; Wilshire, G.; Davis, J.W.; Bromfield, J.; Crowder, S.; Joshi, T.; Pelch, K.; Schust, D.J.; Meng, A.; Barrier, B.; et al. A multi-omics informatics approach for identifying molecular mechanisms and biomarkers in clinical patients with endometriosis. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine, (BIBM), Kansas City, MO, USA, 13–16 November 2017; pp. 2221–2223. [Google Scholar]
- Daraï, E.; Ploteau, S.; Ballester, M.; Bendifallah, S. Endométriose: Physiopathologie, facteurs génétiques et diagnostic clinique. Press. Medicale 2017, 46, 1156–1165. [Google Scholar] [CrossRef]
- Aghajanova, L.; Burney, R.O.; Tran, N.D.; Giudice, L.C. mRNA and miRNA biomarkers for endometriosis. In Biomarkers for Endometriosis: State of the Art; Springer International Publishing AG: Berlin, Germany, 2017; pp. 165–183. ISBN 9783319598567. [Google Scholar]
- Simpson, J.L.; Elias, S.; Malinak, L.R.; Buttram, V.C.J. Heritable aspects of endometriosis. I. Genetic studies. Am. J. Obstet. Gynecol. 1980, 137, 327–331. [Google Scholar] [CrossRef]
- Goodall, J.R. A Study of Endometriosis; J.B. Lippinett Co.: Philadelphia, PA, USA, 1943. [Google Scholar]
- Buggio, L.; Pagliardini, L.; Gentilini, D.; De Braud, L.; Viganò, P.; Vercellini, P. A rare familial case of endometriosis with very severe gynecological and obstetric complications: Novel genetic variants at a glance. Gynecol. Obstet. Invest. 2014, 77, 201–204. [Google Scholar] [CrossRef]
- Kennedy, S. The genetics of endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol. 1999, 82, 129–133. [Google Scholar] [CrossRef]
- Coxhead, D.; Thomas, E.J. Familial inheritance of endometriosis in a british population. A case control study. J. Obstet. Gynaecol. (Lahore) 1993, 13, 42–44. [Google Scholar] [CrossRef]
- Nouri, K.; Ott, J.; Krupitz, B.; Huber, J.C.; Wenzl, R. Family incidence of endometriosis in first-, second-, and third-degree relatives: Case-control study. Reprod. Biol. Endocrinol. 2010, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Kashima, K.; Ishimaru, T.; Okamura, H.; Suginami, H.; Ikuma, K.; Murakami, T.; Iwashita, M.; Tanaka, K. Familial risk among Japanese patients with endometriosis. Int. J. Gynecol. Obstet. 2004, 84, 61–64. [Google Scholar] [CrossRef]
- Kennedy, S.; Mardon, H.; Barlow, D. Familial endometriosis. J. Assist. Reprod. Genet. 1995, 12, 32–34. [Google Scholar] [CrossRef]
- Tulandi, T.; Redwine, D.B. Endometriosis: Advances and Controversies; Marcel Dekker: New York, NY, USA, 2004; Volume 3, pp. 55–56. [Google Scholar]
- Matalliotakis, M.; Goulielmos, G.N.; Zervou, M.I.; Matalliotaki, C.; Koumantakis, G.; Matalliotakis, I. The Familial Risk of Endometriosis among the Female Relatives of Patients with Endometriosis in Greece. J. Endometr. Pelvic Pain Disord. 2017, 9, 184–187. [Google Scholar] [CrossRef]
- Treloar, S.A.; O’Connor, D.T.; O’Connor, V.M.; Martin, N.G. Genetic influences on endometriosis in an Australian twin sample. sueT@qimar.edu.au. Fertil. Steril. 1999, 71, 701–710. [Google Scholar] [CrossRef]
- Hadfield, R.M.; Mardon, H.J.; Barlow, D.H.; Kennedy, S.H. Endometriosis in monozygotic twins. Fertil. Steril. 1997, 68, 941–942. [Google Scholar] [CrossRef]
- Moen, M.H. Endometriosis in monozygotic twins. Acta Obstet. Gynecol. Scand. 1994, 73, 59–62. [Google Scholar] [CrossRef]
- Matalliotakis, M.; Zervou, M.I.; Matalliotaki, C.; Rahmioglu, N.; Koumantakis, G.; Kalogiannidis, I.; Prapas, I.; Zondervan, K.; Spandidos, D.A.; Matalliotakis, I.; et al. The role of gene polymorphisms in endometriosis. Mol. Med. Rep. 2017, 16, 5881–5886. [Google Scholar] [CrossRef]
- Pagliardini, L.; Gentilini, D.; Vigano’, P.; Panina-Bordignon, P.; Busacca, M.; Candiani, M.; Di Blasio, A.M. An Italian association study and meta-analysis with previous GWAS confirm WNT4, CDKN2BAS and FN1 as the first identified susceptibility loci for endometriosis. J. Med. Genet. 2013, 50, 43–46. [Google Scholar] [CrossRef]
- Treloar, S.; Hadfield, R.; Montgomery, G.; Lambert, A.; Wicks, J.; Barlow, D.H.; O’Connor, D.T.; Kennedy, S. The International Endogene Study: A collection of families for genetic research in endometriosis. Fertil. Steril. 2002, 78, 679–685. [Google Scholar] [CrossRef]
- Uno, S.; Zembutsu, H.; Hirasawa, A.; Takahashi, A.; Kubo, M.; Akahane, T.; Aoki, D.; Kamatani, N.; Hirata, K.; Nakamura, Y. A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat. Genet. 2010, 42, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Painter, J.N.; Zondervan, K.T.; Montgomery, G.W. Understanding the Pathogenesis of Endometriosis: Gene Mapping Studies. In Endometriosis: Science and Practicr 6; Wiley-Blackwell: Oxford, UK, 2012; pp. 54–64. [Google Scholar]
- Treloar, S.A.; Wicks, J.; Nyholt, D.R.; Montgomery, G.W.; Bahlo, M.; Smith, V.; Dawson, G.; Mackay, I.J.; Weeks, D.E.; Bennett, S.T.; et al. Genomewide linkage study in 1,176 affected sister pair families identifies a significant susceptibility locus for endometriosis on chromosome 10q26. Am. J. Hum. Genet. 2005, 77, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Zondervan, K.T.; Treloar, S.A.; Lin, J.; Weeks, D.E.; Nyholt, D.R.; Mangion, J.; MacKay, I.J.; Cardon, L.R.; Martin, N.G.; Kennedy, S.H.; et al. Significant evidence of one or more susceptibility loci for endometriosis with Near-Mendelian inheritance on chromosome 7p13–15. Hum. Reprod. 2007, 22, 717–728. [Google Scholar] [CrossRef]
- Painter, J.N.; Anderson, C.A.; Nyholt, D.R.; Macgregor, S.; Lin, J.; Lee, S.H.; Lambert, A.; Zhao, Z.Z.; Roseman, F.; Guo, Q.; et al. Genome-wide association study identifies a locus at 7p15.2 associated with endometriosis. Nat. Genet. 2011, 43, 51–54. [Google Scholar] [CrossRef]
- Painter, J.N.; Nyholt, D.R.; Morris, A.; Zhao, Z.Z.; Henders, A.K.; Lambert, A.; Wallace, L.; Martin, N.G.; Kennedy, S.H.; Treloar, S.A.; et al. High-density fine-mapping of a chromosome 10q26 linkage peak suggests association between endometriosis and variants close to CYP2C19. Fertil. Steril. 2011, 95, 2236–2240. [Google Scholar] [CrossRef]
- Rahmioglu, N.; Missmer, S.A.; Montgomery, G.W.; Zondervan, K.T. Insights into Assessing the Genetics of Endometriosis. Curr. Obstet. Gynecol. Rep. 2012, 1, 124–137. [Google Scholar] [CrossRef]
- Fung, J.N.; Montgomery, G.W. Genetics of endometriosis: State of the art on genetic risk factors for endometriosis. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 50, 61–71. [Google Scholar] [CrossRef]
- Bush, W.S.; Moore, J.H. Chapter 11: Genome-wide association studies. PLoS Comput. Biol. 2012, 8, e1002822. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef]
- Vigano, P.; Somigliana, E.; Vignali, M.; Busacca, M.; Blasio, A.M. Di Genetics of endometriosis: Current status and prospects. Front. Biosci. 2007, 12, 3247–3255. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.A.; Eyster, K.M. Genetics and genomics of endometriosis. Clin. Obstet. Gynecol. 2010, 53, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Darabi, H.; McCue, K.; Beesley, J.; Michailidou, K.; Nord, S.; Kar, S.; Humphreys, K.; Thompson, D.; Ghoussaini, M.; Bolla, M.K.; et al. Polymorphisms in a Putative Enhancer at the 10q21.2 Breast Cancer Risk Locus Regulate NRBF2 Expression. Am. J. Hum. Genet. 2015, 97, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Dunning, A.M.; Michailidou, K.; Kuchenbaecker, K.B.; Thompson, D.; French, J.D.; Beesley, J.; Healey, C.S.; Kar, S.; Pooley, K.A.; Lopez-Knowles, E.; et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat. Genet. 2016, 48, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Ghoussaini, M.; French, J.D.; Michailidou, K.; Nord, S.; Beesley, J.; Canisus, S.; Hillman, K.M.; Kaufmann, S.; Sivakumaran, H.; Moradi Marjaneh, M.; et al. Evidence that the 5p12 Variant rs10941679 Confers Susceptibility to Estrogen-Receptor-Positive Breast Cancer through FGF10 and MRPS30 Regulation. Am. J. Hum. Genet. 2016, 99, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.E.; Fung, J.N.; Shakhbazov, K.; Sapkota, Y.; Cloonan, N.; Hemani, G.; Hillman, K.M.; Kaufmann, S.; Luong, H.T.; Bowdler, L.; et al. Endometriosis risk alleles at 1p36.12 act through inverse regulation of CDC42 and LINC00339. Hum. Mol. Genet. 2016, 25, 5046–5058. [Google Scholar]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Albertsen, H.M.; Chettier, R.; Farrington, P.; Ward, K. Genome-Wide Association Study Link Novel Loci to Endometriosis. PLoS ONE 2013, 8, 58257. [Google Scholar] [CrossRef]
- Steinthorsdottir, V.; Thorleifsson, G.; Aradottir, K.; Feenstra, B.; Sigurdsson, A.; Stefansdottir, L.; Kristinsdottir, A.M.; Zink, F.; Halldorsson, G.H.; Munk Nielsen, N.; et al. Common variants upstream of KDR encoding VEGFR2 and in TTC39B associate with endometriosis. Nat. Commun. 2016, 7, 12350. [Google Scholar] [CrossRef]
- Borghese, B.; Tost, J.; De Surville, M.; Busato, F.; Letourneur, F.; Mondon, F.; Vaiman, D.; Chapron, C. Identification of susceptibility genes for peritoneal, ovarian, and deep infiltrating endometriosis using a pooled sample-based genome-wide association study. Biomed. Res. Int. 2015, 2015, 461024. [Google Scholar] [CrossRef]
- Matalliotakis, M.; Zervou, M.I.; Eliopoulos, E.; Matalliotaki, C.; Rahmioglu, N.; Kalogiannidis, I.; Zondervan, K.; Spandidos, D.A.; Matalliotakis, I.; Goulielmos, G.N. The role of IL-16 gene polymorphisms in endometriosis. Int. J. Mol. Med. 2018, 41, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Taskesen, E.; Van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef] [PubMed]
- Mwinyi, J.; Cavaco, I.; Pedersen, R.S.; Persson, A.; Burkhardt, S.; Mkrtchian, S.; Ingelman-Sundberg, M. Regulation of CYP2C19 expression by estrogen receptor alpha: Implications for estrogen-dependent inhibition of drug metabolism. Mol. Pharmacol. 2010, 78, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, K.; Forrest, L.A.; Vuong, N.; Garson, K.; Djordjevic, B.; Vanderhyden, B.C. GREB1 is an estrogen receptor-regulated tumour promoter that is frequently expressed in ovarian cancer. Oncogene 2018, 37, 5873–5886. [Google Scholar] [CrossRef]
- Veeraraghavan, J.; Tan, Y.; Cao, X.-X.; Kim, J.A.; Wang, X.; Chamness, G.C.; Maiti, S.N.; Cooper, L.J.N.; Edwards, D.P.; Contreras, A.; et al. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat. Commun. 2014, 5, 4577. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y.; Zhao, L.; Wang, L.; Wu, Z.; Mei, Q.; Nie, J.; Li, X.; Li, Y.; Fu, X.; et al. Whole-exome sequencing of endometriosis identifies frequent alterations in genes involved in cell adhesion and chromatin-remodeling complexes. Hum. Mol. Genet. 2014, 23, 6008–6021. [Google Scholar] [CrossRef]
- Rahmioglu, N.; Nyholt, D.R.; Morris, A.P.; Missmer, S.A.; Montgomery, G.W.; Zondervan, K.T. Genetic variants underlying risk of endometriosis: Insights from meta-analysis of eight genome-wide association and replication datasets. Hum. Reprod. Update 2014, 20, 702–716. [Google Scholar] [CrossRef]
- Hsiao, K.-Y.; Wu, M.-H.; Tsai, S.-J. Epigenetic regulation of the pathological process in endometriosis. Reprod. Med. Biol. 2017, 16, 314–319. [Google Scholar] [CrossRef]
- Bulun, S.E.; Yilmaz, B.D.; Sison, C.; Miyazaki, K.; Bernardi, L.; Liu, S.; Kohlmeier, A.; Yin, P.; Milad, M.; Wei, J. Endometriosis. Endocr. Rev. 2019, 40, 1048–1079. [Google Scholar] [CrossRef]
- Shao, R.; Cao, S.; Wang, X.; Feng, Y.; Billig, H. The elusive and controversial roles of estrogen and progesterone receptors in human endometriosis. Am. J. Transl. Res. 2014, 6, 104–113. [Google Scholar]
- Yilmaz, B.D.; Bulun, S.E. Endometriosis and nuclear receptors. Hum. Reprod. Update 2019, 25, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Ryan, I.P.; Taylor, R.N. Endometriosis and infertility: New concepts. Obstet. Gynecol. Surv. 1997, 52, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Lin, Z.; Yin, P.; Milad, M.P.; Cheng, Y.-H.; Confino, E.; Reierstad, S.; Bulun, S.E. Transcriptional activation of steroidogenic factor-1 by hypomethylation of the 5′ CpG island in endometriosis. J. Clin. Endocrinol. Metab. 2007, 92, 3261–3267. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; O’Malley, B.W. The dynamics of nuclear receptors and nuclear receptor coregulators in the pathogenesis of endometriosis. Hum. Reprod. Update 2014, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Pavone, M.E.; Reierstad, S.; Sun, H.; Milad, M.; Bulun, S.E.; Cheng, Y.-H. Altered retinoid uptake and action contributes to cell survival in endometriosis. J. Clin. Endocrinol. Metab. 2010, 95, E300–E309. [Google Scholar] [CrossRef] [PubMed]
- Mustonen, M.V.; Isomaa, V.V.; Vaskivuo, T.; Tapanainen, J.; Poutanen, M.H.; Stenback, F.; Vihko, R.K.; Vihko, P.T. Human 17beta-hydroxysteroid dehydrogenase type 2 messenger ribonucleic acid expression and localization in term placenta and in endometrium during the menstrual cycle. J. Clin. Endocrinol. Metab. 1998, 83, 1319–1324. [Google Scholar] [PubMed]
- Cheng, Y.H.; Yin, P.; Xue, Q.; Yilmaz, B.; Dawson, M.I.; Bulun, S.E. Retinoic Acid (RA) regulates 17β-hydroxysteroid dehydrogenase type 2 expression in endometrium: Interaction of RA receptors with Specificity Protein (SP) 1/SP3 for estradiol metabolism. J. Clin. Endocrinol. Metab. 2008, 93, 1915–1923. [Google Scholar] [CrossRef]
- Cheng, Y.H.; Imir, A.; Fenkci, V.; Yilmaz, M.B.; Bulun, S.E. Stromal cells of endometriosis fail to produce paracrine factors that induce epithelial 17β-hydroxysteroid dehydrogenase type 2 gene and its transcriptional regulator Sp1: A mechanism for defective estradiol metabolism. Am. J. Obstet. Gynecol. 2007, 196, 391.e1–391.e8. [Google Scholar] [CrossRef]
- Zeitoun, K.; Takayama, K.; Sasano, H.; Suzuki, T.; Moghrabi, N.; Andersson, S.; Johns, A.; Meng, L.; Putman, M.; Carr, B.; et al. Deficient 17beta-hydroxysteroid dehydrogenase type 2 expression in endometriosis: Failure to metabolize 17beta-estradiol. J. Clin. Endocrinol. Metab. 1998, 83, 4474–4480. [Google Scholar]
- Zhao, H.; Chen, Z.-J. Genetic association studies in female reproduction: From candidate-gene approaches to genome-wide mapping. Mol. Hum. Reprod. 2013, 19, 644–654. [Google Scholar] [CrossRef]
- Herynk, M.H.; Fuqua, S.A.W. Estrogen receptor mutations in human disease. Endocr. Rev. 2004, 25, 869–898. [Google Scholar] [CrossRef] [PubMed]
- Deschênes, J.; Bourdeau, V.; White, J.H.; Mader, S. Regulation of GREB1 transcription by estrogen receptor α through a multipartite enhancer spread over 20 kb of upstream flanking sequences. J. Biol. Chem. 2007, 282, 17335–17339. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.; D’Santos, C.; Serandour, A.A.; Ali, H.R.; Brown, G.D.; Atkins, A.; Rueda, O.M.; Holmes, K.A.; Theodorou, V.; Robinson, J.L.L.; et al. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013, 3, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Izawa, M.; Taniguchi, F.; Terakawa, N.; Harada, T. Epigenetic aberration of gene expression in endometriosis. Front. Biosci. (Elite Ed.) 2013, 5, 900–910. [Google Scholar] [CrossRef]
- Meinsohn, M.-C.; Smith, O.E.; Bertolin, K.; Murphy, B.D. The orphan nuclear receptors steroidogenic factor-1 and liver receptor homolog-1: Structure, regulation, and essential roles in mammalian reproduction. Physiol. Rev. 2019, 99, 1249–1279. [Google Scholar] [CrossRef]
- Vasquez, Y.M.; Wu, S.P.; Anderson, M.L.; Hawkins, S.M.; Creighton, C.J.; Ray, M.; Tsai, S.Y.; Tsai, M.J.; Lydon, J.P.; De Mayo, F.J. Endometrial expression of steroidogenic factor 1 promotes cystic glandular morphogenesis. Mol. Endocrinol. 2016, 30, 518–532. [Google Scholar] [CrossRef]
- Bulun, S.E.; Utsunomiya, H.; Lin, Z.; Yin, P.; Cheng, Y.H.; Pavone, M.E.; Tokunaga, H.; Trukhacheva, E.; Attar, E.; Gurates, B.; et al. Steroidogenic factor-1 and endometriosis. Moll. Cell. Endocrinol. 2009, 300, 104–108. [Google Scholar] [CrossRef]
- Utsunomiya, H.; Cheng, Y.-H.; Lin, Z.; Reierstad, S.; Yin, P.; Attar, E.; Xue, Q.; Imir, G.; Thung, S.; Trukhacheva, E.; et al. Upstream stimulatory factor-2 regulates steroidogenic factor-1 expression in endometriosis. Mol. Endocrinol. 2008, 22, 904–914. [Google Scholar] [CrossRef]
- Xue, Q.; Xu, Y.; Yang, H.; Zhang, L.; Shang, J.; Zeng, C.; Yin, P.; Bulun, S.E. Methylation of a novel CpG island of intron 1 is associated with stereogenic factor 1 expression in endometriotic stromal cells. Reprod. Sci. 2014, 21, 395–400. [Google Scholar] [CrossRef]
- Hu, Z.; Mamillapalli, R.; Taylor, H. Increased circulating mi-R370-3p regulates steroidogenic factor 1 in endometriosis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, 373–382. [Google Scholar] [CrossRef]
- Shen, L.; Yang, S.; Huang, W.; Xu, W.; Wang, Q.; Song, Y.; Liu, Y. MicroRNA23a and microRNA23b deregulation derepresses SF-1 and upregulates estrogen signaling in ovarian endometriosis. Endocrinol. Metab. 2013, 98, 1575–1582. [Google Scholar] [CrossRef] [PubMed]
- Koninckx, P.R.; Ussia, A.; Adamyan, L.; Wattiez, A.; Gomel, V.; Martin, D.C. Pathogenesis of endometriosis: The genetic/epigenetic theory. Fertil. Steril. 2019, 111, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.M.; Fry, R.C. Environmental Influences on the Epigenome: Exposure- Associated DNA Methylation in Human Populations. Annu. Rev. Public Health 2018, 39, 309–333. [Google Scholar] [CrossRef] [PubMed]
- Dyson, M.T.; Roqueiro, D.; Monsivais, D.; Ercan, C.M.; Pavone, M.E.; Brooks, D.C.; Kakinuma, T.; Ono, M.; Jafari, N.; Dai, Y.; et al. Genome-Wide DNA Methylation Analysis Predicts an Epigenetic Switch for GATA Factor Expression in Endometriosis. PLoS Genet. 2014, 10, e1004158. [Google Scholar] [CrossRef] [PubMed]
- Dyson, M.T.; Kakinuma, T.; Pavone, M.E.; Monsivais, D.; Navarro, A.; Malpani, S.S.; Ono, M.; Bulun, S.E. Aberrant expression and localization of deoxyribonucleic acid methyltransferase 3B in endometriotic stromal cells. Fertil. Steril. 2015, 104, 953–963. [Google Scholar] [CrossRef][Green Version]
- Xu, F.; Mao, C.; Ding, Y.; Rui, C.; Wu, L.; Shi, A.; Zhang, H.; Zhang, L.; Xu, Z. Molecular and enzymatic profiles of mammalian DNA methyltransferases: Structures and targets for drugs. Curr. Med. Chem. 2010, 17, 4052–4071. [Google Scholar] [CrossRef]
- Jeltsch, A. Molecular enzymology of mammalian DNA methyltransferases. Curr. Top. Microbiol. Immunol. 2006, 301, 203–225. [Google Scholar]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Yun, J.; Johnson, J.L.; Hanigan, C.L.; Locasale, J.W. Interactions between epigenetics and metabolism in cancers. Front. Oncol. 2012, 2, 163. [Google Scholar] [CrossRef]
- Wu, Y.; Strawn, E.; Basir, Z.; Halverson, G.; Guo, S.-W. Aberrant expression of deoxyribonucleic acid methyltransferases DNMT1, DNMT3A, and DNMT3B in women with endometriosis. Fertil. Steril. 2007, 87, 24–32. [Google Scholar] [CrossRef]
- Szczepanska, M.; Wirstlein, P.; Skrzypczak, J.; Jagodzinski, P.P. Expression of HOXA11 in the mid-luteal endometrium from women with endometriosis-associated infertility. Reprod. Biol. Endocrinol. 2012, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Van Kaam, K.J.A.F.; Delvoux, B.; Romano, A.; D’Hooghe, T.; Dunselman, G.A.J.; Groothuis, P.G. Deoxyribonucleic acid methyltransferases and methyl-CpG-binding domain proteins in human endometrium and endometriosis. Fertil. Steril. 2011, 95, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.-Y.; Wu, M.-H.; Chang, N.; Yang, S.-H.; Wu, C.-W.; Sun, H.S.; Tsai, S.-J. Coordination of AUF1 and miR-148a destabilizes DNA methyltransferase 1 mRNA under hypoxia in endometriosis. Mol. Hum. Reprod. 2015, 21, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, J.; Li, Y.; Wang, Z.; Kang, S. Genome-wide analysis of DNA methylation in endometriosis using Illumina Human Methylation 450 K Beadchips. Mol. Dev. 2019, 86, 491–501. [Google Scholar] [CrossRef]
- Nasu, K.; Kawano, Y.; Tsukamoto, Y.; Takano, M.; Takai, N.; Li, H.; Furukawa, Y.; Abe, W.; Moriyama, M.; Narahara, H. Aberrant DNA methylation status of endometriosis: Epigenetics as the pathogenesis, biomarker and therapeutic target. J. Obstet. Gynaecol. Res. 2011, 37, 683–695. [Google Scholar] [CrossRef]
- Romani, M.; Pistillo, M.P.; Banelli, B. Environmental Epigenetics: Crossroad between Public Health, Lifestyle, and Cancer Prevention. Biomed. Res. Int. 2015, 2015, 587983. [Google Scholar] [CrossRef]
- Arosh, J.A.; Lee, J.H.; Starzinski-Powitz, A.; Banu, S.K. Selective inhibition of prostaglandin E2 receptors EP2 and EP4 modulates DNA methylation and histone modification machinery proteins in human endometriotic cells. Mol. Cell. Endocrinol. 2015, 409, 51–58. [Google Scholar] [CrossRef]
- Senthong, A.; Kitkumthorn, N.; Rattanatanyong, P.; Khemapech, N.; Triratanachart, S.; Mutirangura, A. Differences in LINE-1 methylation between endometriotic ovarian cyst and endometriosis-associated ovarian cancer. Int. J. Gynecol. Cancer 2014, 24, 36–42. [Google Scholar] [CrossRef]
- Bernardi, L.A.; Dyson, M.T.; Tokunaga, H.; Sison, C.; Oral, M.; Robins, J.C.; Bulun, S.E. The Essential Role of GATA6 in the Activation of Estrogen Synthesis in Endometriosis. Reprod. Sci. 2019, 26, 60–69. [Google Scholar] [CrossRef]
- Barragan, F.; Irwin, J.C.; Balayan, S.; Erikson, D.W.; Chen, J.C.; Houshdaran, S.; Piltonen, T.T.; Spitzer, T.L.B.; George, A.; Rabban, J.T.; et al. Human Endometrial Fibroblasts Derived from Mesenchymal Progenitors Inherit Progesterone Resistance and Acquire an Inflammatory Phenotype in the Endometrial Niche in Endometriosis1. Biol. Reprod. 2016, 94, 118. [Google Scholar] [CrossRef]
- Naqvi, H.; Ilagan, Y.; Krikun, G.; Taylor, H.S. Altered genome-wide methylation in endometriosis. Reprod. Sci. 2014, 21, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Koike, N.; Higashiura, Y.; Akasaka, J.; Uekuri, C.; Ito, F.; Kobayashi, H. Epigenetic dysregulation of endometriosis susceptibility genes (Review). Mol. Med. Rep. 2015, 12, 1611–1616. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Turinsky, A.L.; Turner, B.; Borja, R.C.; Gleeson, J.A.; Heath, M.; Pu, S.; Switzer, T.; Dong, D.; Gong, Y.; On, T.; et al. DAnCER: Disease-annotated chromatin epigenetics resource. Nucleic Acids Res. 2011, 39, D889–D894. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, Y.Z. Altered histone modifications in gliomas. Brain Tumor Res. Treat. 2014, 2, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, 1–26. [Google Scholar] [CrossRef]
- Nasu, K.; Kawano, Y.; Kai, K.; Aoyagi, Y.; Abe, W.; Okamoto, M.; Narahara, H. Aberrant histone modification in endometriosis. Front. Biosci. Landmark Ed. 2014, 19, 1202–1214. [Google Scholar] [CrossRef]
- Hatzimichael, E.; Crook, T. Cancer Epigenetics: New Therapies and New Challenges. J. Drug Deliv. 2013, 2013, 529312. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef]
- Shiio, Y.; Eisenman, R.N. Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 2003, 100, 13225–13230. [Google Scholar] [CrossRef]
- Gill, G. SUMO and ubiquitin in the nucleus: Different functions, similar mechanisms? Genes Dev. 2004, 18, 2046–2059. [Google Scholar] [CrossRef]
- Fung, J.N.; Mortlock, S.; Girling, J.E.; Holdsworth-Carson, S.J.; Teh, W.T.; Zhu, Z.; Lukowski, S.W.; McKinnon, B.D.; McRae, A.; Yang, J.; et al. Genetic regulation of disease risk and endometrial gene expression highlights potential target genes for endometriosis and polycystic ovarian syndrome. Sci. Rep. 2018, 8, 11424. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.K.; Farquhar, C.M.; Mitchell, M.D.; Ponnampalam, A.P. Epigenetic regulation of endometrium during the menstrual cycle. Mol. Hum. Reprod. 2010, 16, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Xiaomeng, X.; Ming, Z.; Jiezhi, M.; Xiaoling, F. Aberrant histone acetylation and methylation levels in woman with endometriosis. Arch. Gynecol. Obstet. 2013, 287, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, J.B.; Colon-Diaz, M.; Garcia, M.; Gutierrez, S.; Colon, M.; Seto, E.; Laboy, J.; Flores, I. Endometriosis is characterized by a distinct pattern of histone 3 and histone 4 lysine modifications. Reprod. Sci. 2014, 21, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Krusche, C.A.; Vloet, A.J.; Classen-Linke, I.; von Rango, U.; Beier, H.M.; Alfer, J. Class I histone deacetylase expression in the human cyclic endometrium and endometrial adenocarcinomas. Hum. Reprod. 2007, 22, 2956–2966. [Google Scholar] [CrossRef] [PubMed]
- Colón-Díaz, M.; Báez-Vega, P.; García, M.; Ruiz, A.; Monteiro, J.B.; Fourquet, J.; Bayona, M.; Alvarez-Garriga, C.; Achille, A.; Seto, E.; et al. HDAC1 and HDAC2 are differentially expressed in endometriosis. Reprod. Sci. 2012, 19, 483–492. [Google Scholar] [CrossRef]
- Sakai, N.; Maruyama, T.; Sakurai, R.; Masuda, H.; Yamamoto, Y.; Shimizu, A.; Kishi, I.; Asada, H.; Yamagoe, S.; Yoshimura, Y. Involvement of histone acetylation in ovarian steroid-induced decidualization of human endometrial stromal cells. J. Biol. Chem. 2003, 278, 16675–16682. [Google Scholar] [CrossRef]
- Uchida, H.; Maruyama, T.; Nagashima, T.; Ono, M.; Masuda, H.; Arase, T.; Sugiura, I.; Onouchi, M.; Kajitani, T.; Asada, H.; et al. Human endometrial cytodifferentiation by histone deacetylase inhibitors. Hum. Cell 2006, 19, 38–42. [Google Scholar] [CrossRef]
- Uchida, H.; Maruyama, T.; Ohta, K.; Ono, M.; Arase, T.; Kagami, M.; Oda, H.; Kajitani, T.; Asada, H.; Yoshimura, Y. Histone deacetylase inhibitor-induced glycodelin enhances the initial step of implantation. Hum. Reprod. 2007, 22, 2615–2622. [Google Scholar] [CrossRef]
- Kawano, Y.; Nasu, K.; Li, H.; Tsuno, A.; Abe, W.; Takai, N.; Narahara, H. Application of the histone deacetylase inhibitors for the treatment of endometriosis: Histone modifications as pathogenesis and novel therapeutic target. Hum. Reprod. 2011, 26, 2486–2498. [Google Scholar] [CrossRef]
- Samartzis, E.P.; Noske, A.; Samartzis, N.; Fink, D.; Imesch, P. The expression of histone deacetylase 1, but not other class I histone deacetylases, is significantly increased in endometriosis. Reprod. Sci. 2013, 20, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Altucci, L.; Rots, M.G. Epigenetic drugs: From chemistry via biology to medicine and back. Clin. Epigenet. 2016, 8, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Lagana, A.S.; Garzon, S.; Gotte, M.; Vigano, P.; Franchi, M.; Ghezzi, F.; Martin, D.C. The Pathogenesis of Endometriosis: Molecular and Cell Biology Insights. Int. J. Mol. Sci. 2019, 20, 5615. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.; Mattick, J.S. Non-coding RNAs: Regulators of disease. J. Pathol. 2010, 220, 126–139. [Google Scholar] [CrossRef]
- Mitra, S.A.; Mitra, A.P.; Triche, T.J. A central role for long non-coding RNA in cancer. Front. Genet. 2012, 3, 17. [Google Scholar] [CrossRef]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in control of gene expression: An overview of nuclear functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef]
- Petracco, R.; Dias, A.C.D.O.; Taylor, H.; Petracco, Á.; Badalotti, M.; Michelon, J.D.R.; Marinowic, D.R.; Hentschke, M.; Azevedo, P.N.D.; Zanirati, G.; et al. Evaluation of miR-135a/b expression in endometriosis lesions. Biomed. Rep. 2019, 11, 181–187. [Google Scholar] [CrossRef]
- Hull, M.L.; Nisenblat, V. Tissue and circulating microRNA influence reproductive function in endometrial disease. Reprod. Biomed. Online 2013, 27, 515–529. [Google Scholar] [CrossRef]
- Jia, S.-Z.; Yang, Y.; Lang, J.; Sun, P.; Leng, J. Plasma miR-17-5p, miR-20a and miR-22 are down-regulated in women with endometriosis. Hum. Reprod. 2013, 28, 322–330. [Google Scholar] [CrossRef]
- Wang, W.-T.; Zhao, Y.-N.; Han, B.-W.; Hong, S.-J.; Chen, Y.-Q. Circulating microRNAs identified in a genome-wide serum microRNA expression analysis as noninvasive biomarkers for endometriosis. J. Clin. Endocrinol. Metab. 2013, 98, 281–289. [Google Scholar] [CrossRef]
- Cho, S.; Mutlu, L.; Grechukhina, O.; Taylor, H.S. Circulating microRNAs as potential biomarkers for endometriosis. Fertil. Steril. 2015, 103, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Rekker, K.; Saare, M.; Roost, A.M.; Kaart, T.; Soritsa, D.; Karro, H.; Soritsa, A.; Simon, C.; Salumets, A.; Peters, M.R.K. Circulating miR-200-family microRNAs have altered plasma levels in patients with endometriosis and vary with blood collection time. Fertil. Steril. 2015, 104, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Mashayekhi, P.; Noruzinia, M.; Zeinali, S.; Khodaverdi, S. Endometriotic Mesenchymal Stem Cells Epigenetic Pathogenesis: Deregulation of miR-200b, miR-145, and let7b in A Functional Imblanced Epigenetic Disease. Cell. J. 2019, 21, 179–185. [Google Scholar] [PubMed]
- Ohlsson Teague, E.M.C.; Van der Hoek, K.H.; Van der Hoek, M.B.; Perry, N.; Wagaarachchi, P.; Robertson, S.A.; Print, C.G.; Hull, L.M. MicroRNA-regulated pathways associated with endometriosis. Mol. Endocrinol. 2009, 23, 265–275. [Google Scholar] [CrossRef]
- Moustafa, S.; Burun, M.; Mamillapalli, R.; Nematian, S.; Flores, V.; Taylor, H.S. Accurate Diagnosis of Endometriosis Using Serum MicroRNAs. Am. J. Obstet. Gynecol. 2020. [Google Scholar] [CrossRef]
- Anastasiu, C.V.; Moga, M.A.; Neculau, A.E.; Balan, A.; Scarneciu, I.; Dragomir, R.M.; Dull, A.-M.; Chicea, L.-M. Biomarkers for the Noninvasive Diagnosis of Endometriosis: State of the Art and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 1750. [Google Scholar] [CrossRef]
- Nothnick, W.B. MicroRNAs and Endometriosis: Distinguishing Drivers from passengers in Disease Pathogenesis. Semin. Reprod. Med. 2017, 35, 173–180. [Google Scholar] [CrossRef]
- Zhang, L.; Li, H.; Yuan, M.; Li, D.; Sun, Ch.; Wang, G. Serum Exosomal MicroRNAs as Potential Circulating Biomarkers for Endometriosis. Dis. Markers 2020, 2020, 2456340. [Google Scholar] [CrossRef]
- Guil, S.; Esteller, M. DNA methylomes, histone codes and miRNAs: Tying it all together. Int. J. Biochem. Cell Biol. 2009, 41, 87–95. [Google Scholar] [CrossRef]
- Nelson, P.T.; Wang, W.-X.; Rajeev, B.W. MicroRNAs (miRNAs) in neurodegenerative diseases. Brain Pathol. 2008, 18, 130–138. [Google Scholar] [CrossRef]
- Attar, E.; Tokunaga, H.; Imir, G.; Yilmaz, M.B.; Redwine, D.; Putman, M.; Gurates, B.; Atter, R.; Yaegashi, N.; Hales, D.B.; et al. Prostaglandin E2 via stereoidogenic factor-1 coordinately regulates transcription of steroidogenic genes necessary for estrogen synthesis in endometriosis. J. Clin. Endocrinol. Metab. 2009, 94, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Vasudervan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: MicroRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Goulielmos, G.N.; Matalliotakis, M.; Matalliotaki, C.; Eliopoulos, E.; Matalliotakis, I.; Zervou, M.I. Endometriosis research in the-omics era (Review). Gene 2020, 741, 144545. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Chr | SNP | Associated Gene/Cytoband | References |
|---|---|---|---|
| 1 1 1 1 1 2 2 2 2 2 2 2 2 4 4 4 4 5 6 6 6 6 6 6 6 7 7 7 7 8 9 9 9 9 9 9 10 10 11 12 12 12 12 14 15 17 17 | rs12037376 rs7521902 rs16826658 rs1894692 rs495590 rs11674184 rs77294520 rs13394619 rs4141819 rs654324 rs10167914 rs1250247 rs1250241 rs10012589 rs1903068 rs17773813 rs2510770 rs13177597 rs6938760 rs760794 rs7759516 rs1971256 rs2206949 rs17803970 rs71575922 rs12700667 rs62468795 rs55909142 rs74491657 rs10090060 rs9987548 rs1537377 rs10757272 rs1448792 rs10965235 rs519664 rs1802669 rs796945 rs74485684 rs12320196 rs4762326 rs10859871 rs17727841 rs7151531 rs4923850 rs66683298 rs76731691 | WNT4/1p36.12 WNT4/1p36.12 WNT4/1p36 SLC19A2/1q24.2 DNM3/1q24.3 GREB1/2p25.1 GREB1/2p25.1 GREB1/2p25.1 ETAA1/2p14 ETAA1/2p14 IL1AI/2q13 FN1/2q35 FN1/2q35 KDR/4q12 KDR/4q12 VEGFR2/4q12 PDLIM5/4q22.3 ATP6AP1L/5q14.2 ID4/6p22.3 ID4/6p22.3 CCDC170/6q25.1 CCDC170/6q25.1 ESR1/6q25.1 SYNE1/6q25.1 SYNE1/6q25.1 7p15.2/7p15.2 IGF2BP3/7p15.3 7p12.3/7p12.3 7p12.3/7p12.3 GDAP1/8q21.11 CDKN2-BAS1/9p21.3 CDKN2-BAS1/9p21.3 CDKN2-BAS1/9p21.3 CDKN2-BAS1/9p21.3 CDKN2-BAS1/9p21 TTC39B/9p22 MLLT10/10p12.31 RNLS/10q23.31 FSHB/11p14.1 VEZT/12q22 VEZT/12q22 VEZT/12q22 IGF1/12q23.2 RIN3/14q32.12 BMF/15q15.1 SKAP1/17q21.32 CEP112/17q24.1 | [16,17] [19] [46] [16] [16] [16,17] [17] [19] [15,16,19] [17] [16,17] [16] [17] [16] [17] [64] [16] [16] [16] [17] [16] [17] [17] [17] [16,17] [15,16,17,19,50] [16] [16] [17] [16] [16] [15,17,19] [17] [17] [19,46] [64] [16] [16] [16,17] [16] [17] [19] [16] [16] [16] [16] [16] |
| Modification | Global Effect of Modification |
|---|---|
| Acetylation | Activation of Transcription Silencing of Telomeres DNA Repair |
| Methylation | Inactivation of transcription |
| Phosphorylation | DNA repair Mitosis |
| Ubiquitination | Activation of transcription |
| Sumoylation | Silencing of transcription |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zubrzycka, A.; Zubrzycki, M.; Perdas, E.; Zubrzycka, M. Genetic, Epigenetic, and Steroidogenic Modulation Mechanisms in Endometriosis. J. Clin. Med. 2020, 9, 1309. https://doi.org/10.3390/jcm9051309
Zubrzycka A, Zubrzycki M, Perdas E, Zubrzycka M. Genetic, Epigenetic, and Steroidogenic Modulation Mechanisms in Endometriosis. Journal of Clinical Medicine. 2020; 9(5):1309. https://doi.org/10.3390/jcm9051309
Chicago/Turabian StyleZubrzycka, Anna, Marek Zubrzycki, Ewelina Perdas, and Maria Zubrzycka. 2020. "Genetic, Epigenetic, and Steroidogenic Modulation Mechanisms in Endometriosis" Journal of Clinical Medicine 9, no. 5: 1309. https://doi.org/10.3390/jcm9051309
APA StyleZubrzycka, A., Zubrzycki, M., Perdas, E., & Zubrzycka, M. (2020). Genetic, Epigenetic, and Steroidogenic Modulation Mechanisms in Endometriosis. Journal of Clinical Medicine, 9(5), 1309. https://doi.org/10.3390/jcm9051309