Beneficial Effects of Acetyl-DL-Leucine (ADLL) in a Mouse Model of Sandhoff Disease

 , , ,
, , ,
Abstract
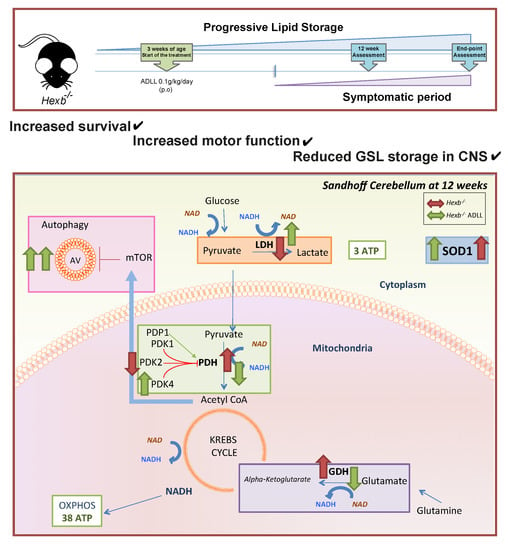
1. Introduction
2. Experimental Section
2.1. Mice
2.2. Drug Treatments
2.3. Behavioural Analysis
2.4. Tissue Handling
2.5. Glycosphingolipid Measurements
2.6. Western Blotting
2.7. Image Acquisition and Quantification
2.8. Statistical ANALYSES
3. Results
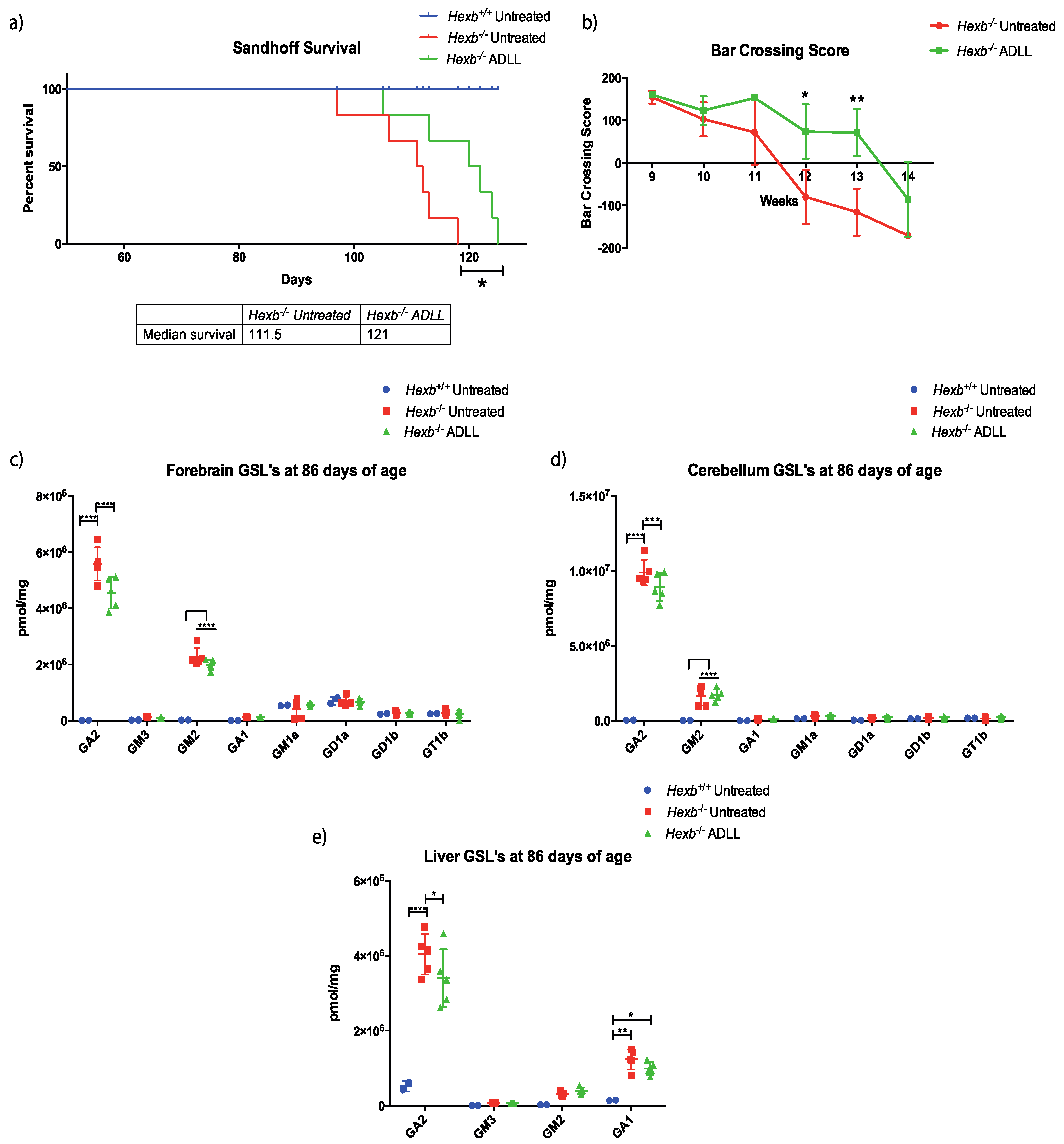
3.1. The Effects of ADLL in Hexb-/- Mice on Motor Function and Lipid Storage
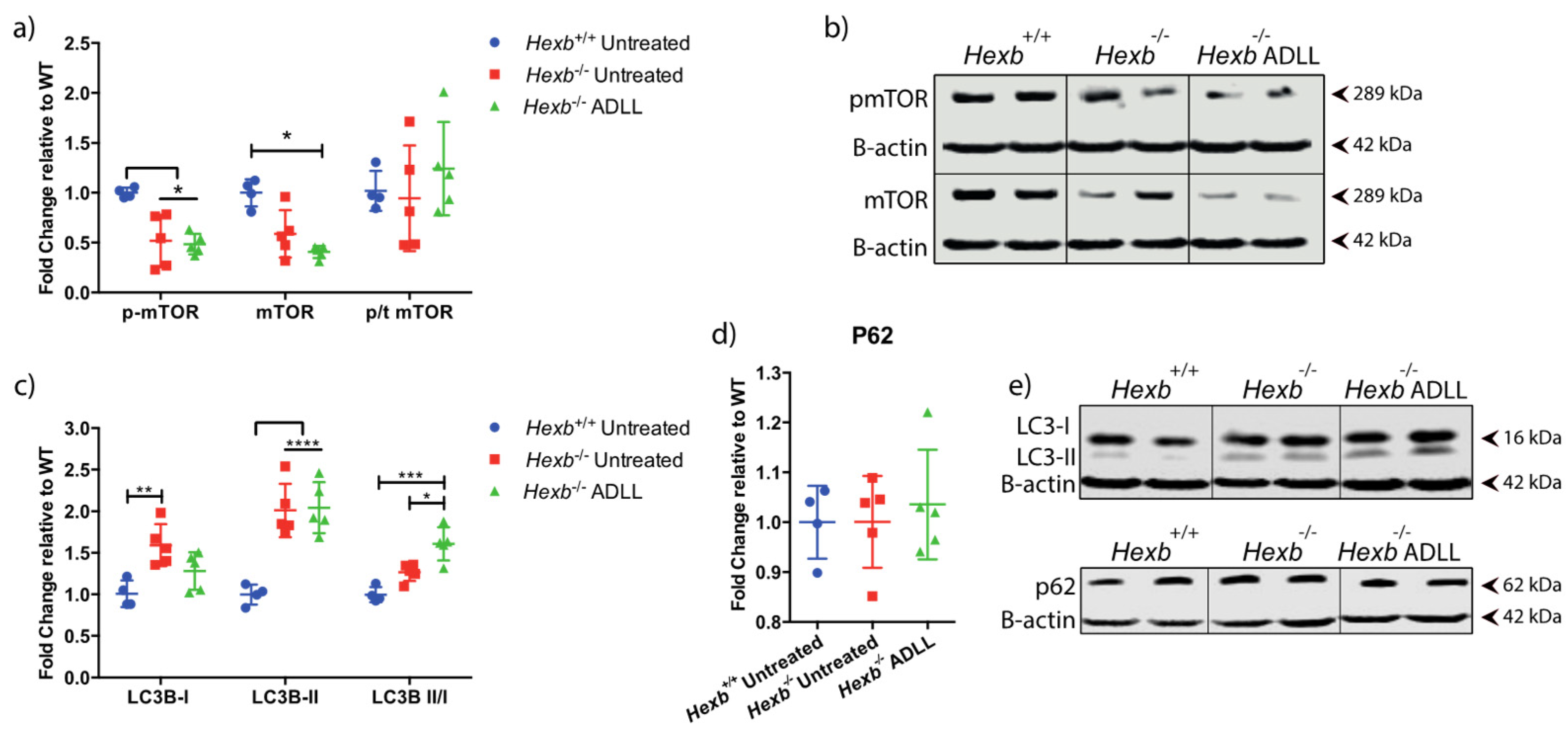
3.2. ADLL Increases Autophagy in Hexb-/- Mice Cerebellum
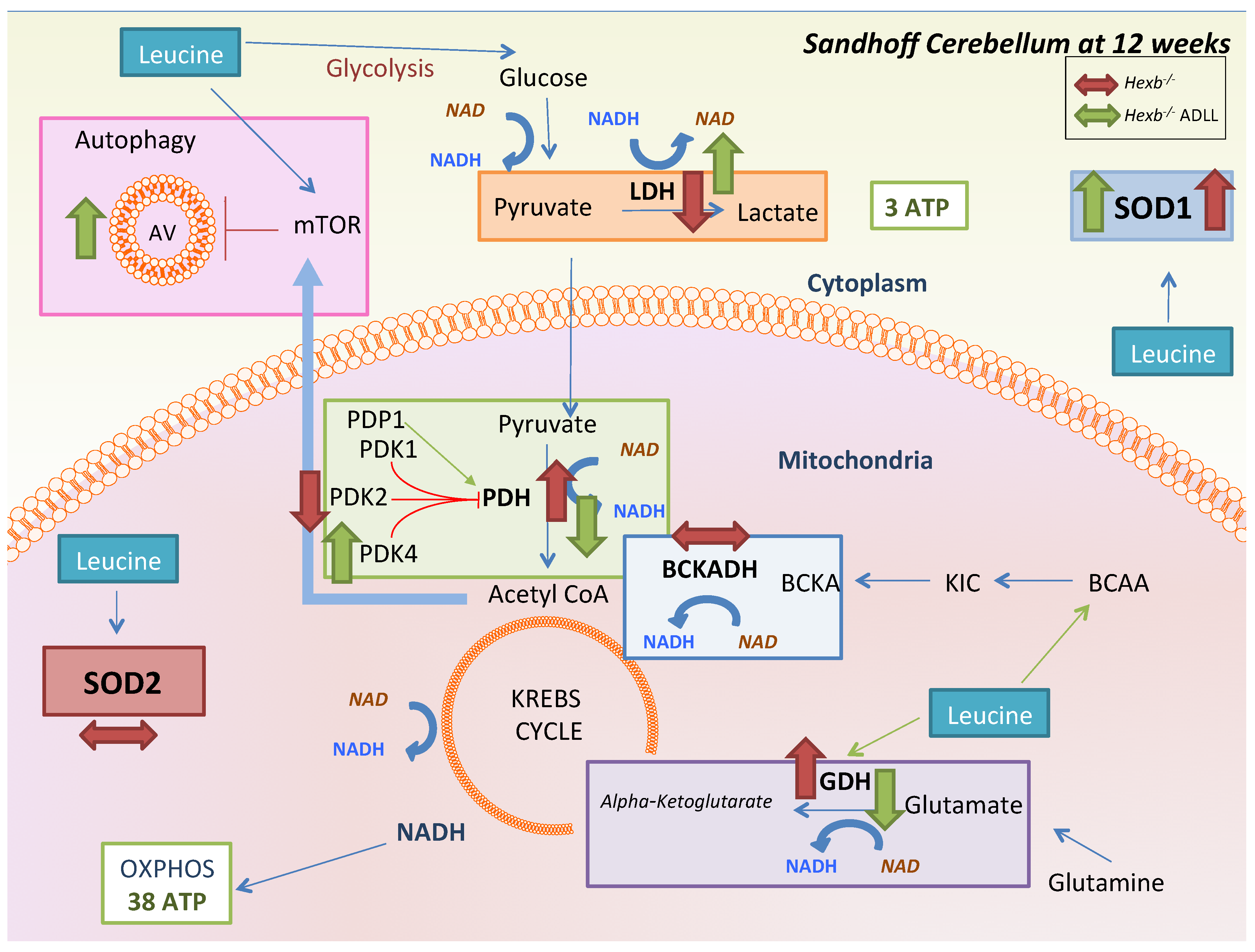
3.3. ADLL Normalizes Metabolic Abnormalities in Sandhoff Mouse Cerebellum
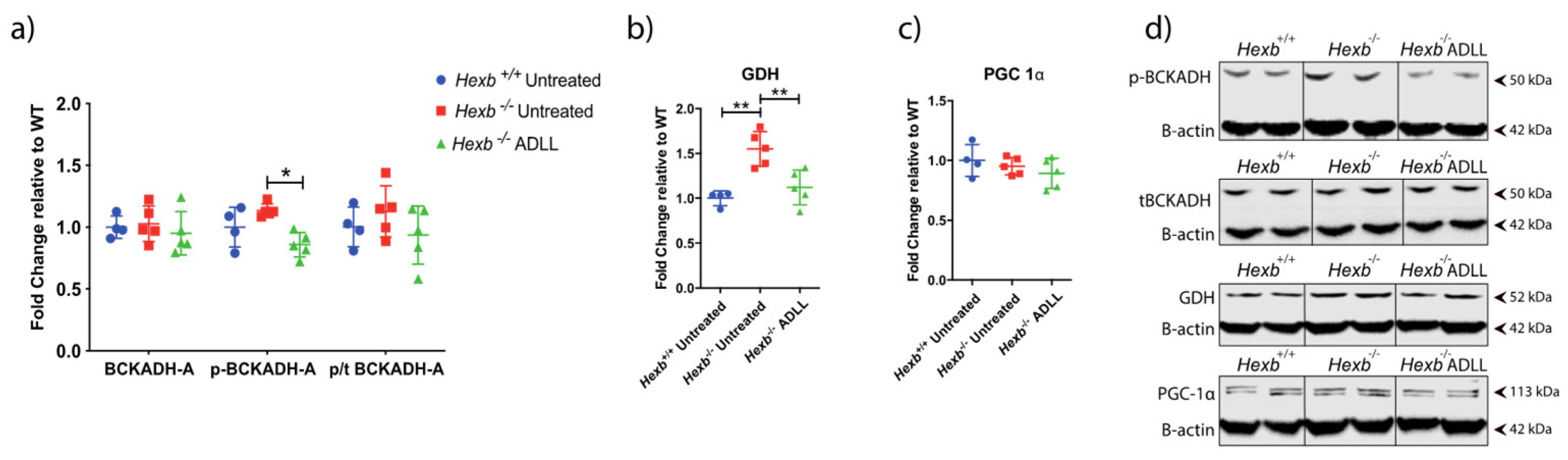
3.3.1. ADLL’s Effect on Branched Chain Amino Acid and Glutamate Metabolism
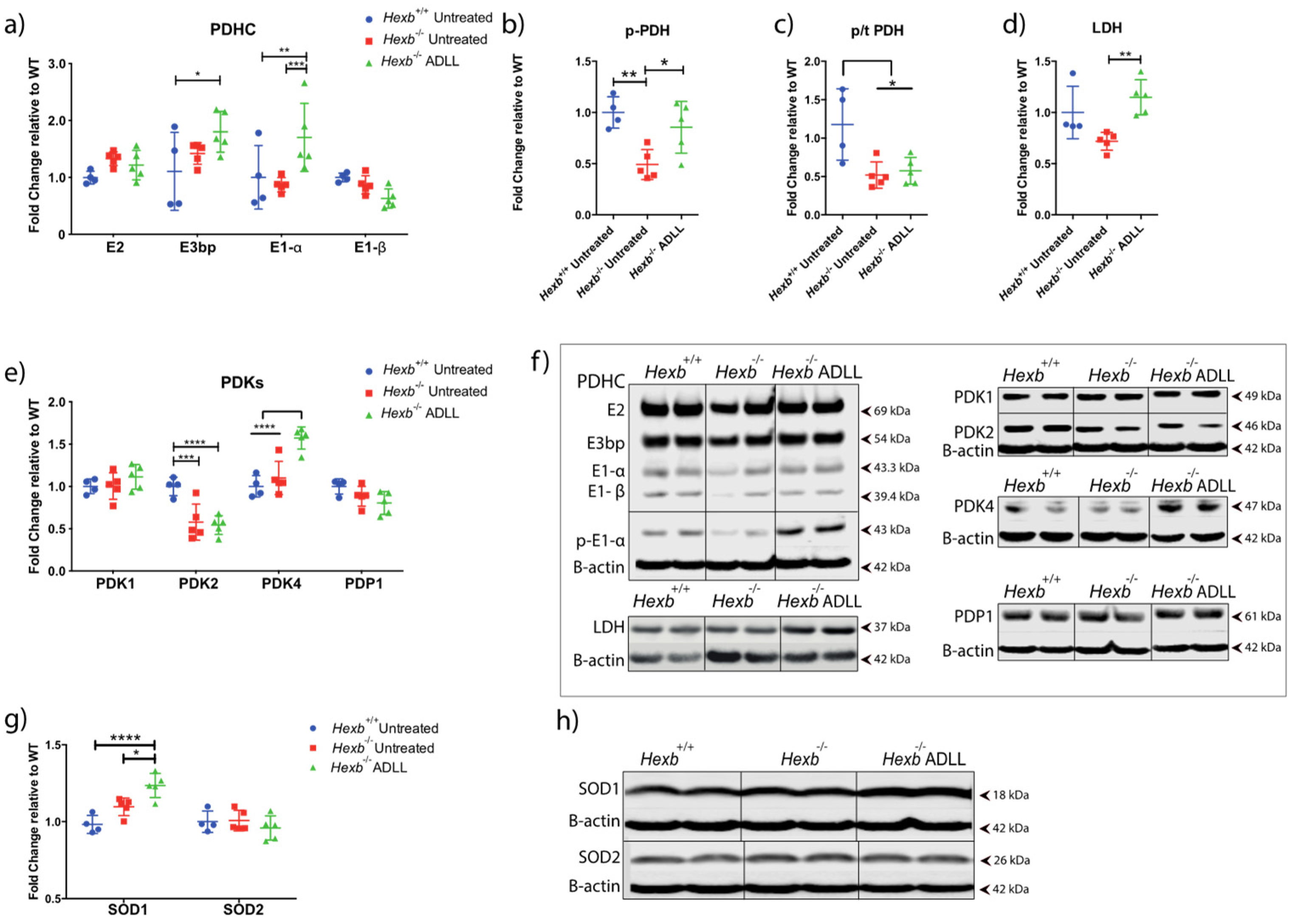
3.3.2. ADLL Promotes Anaerobic Glycolysis via PDK4 Up-Regulation
3.3.3. ADLL Elevates Antioxidant SOD1 Levels
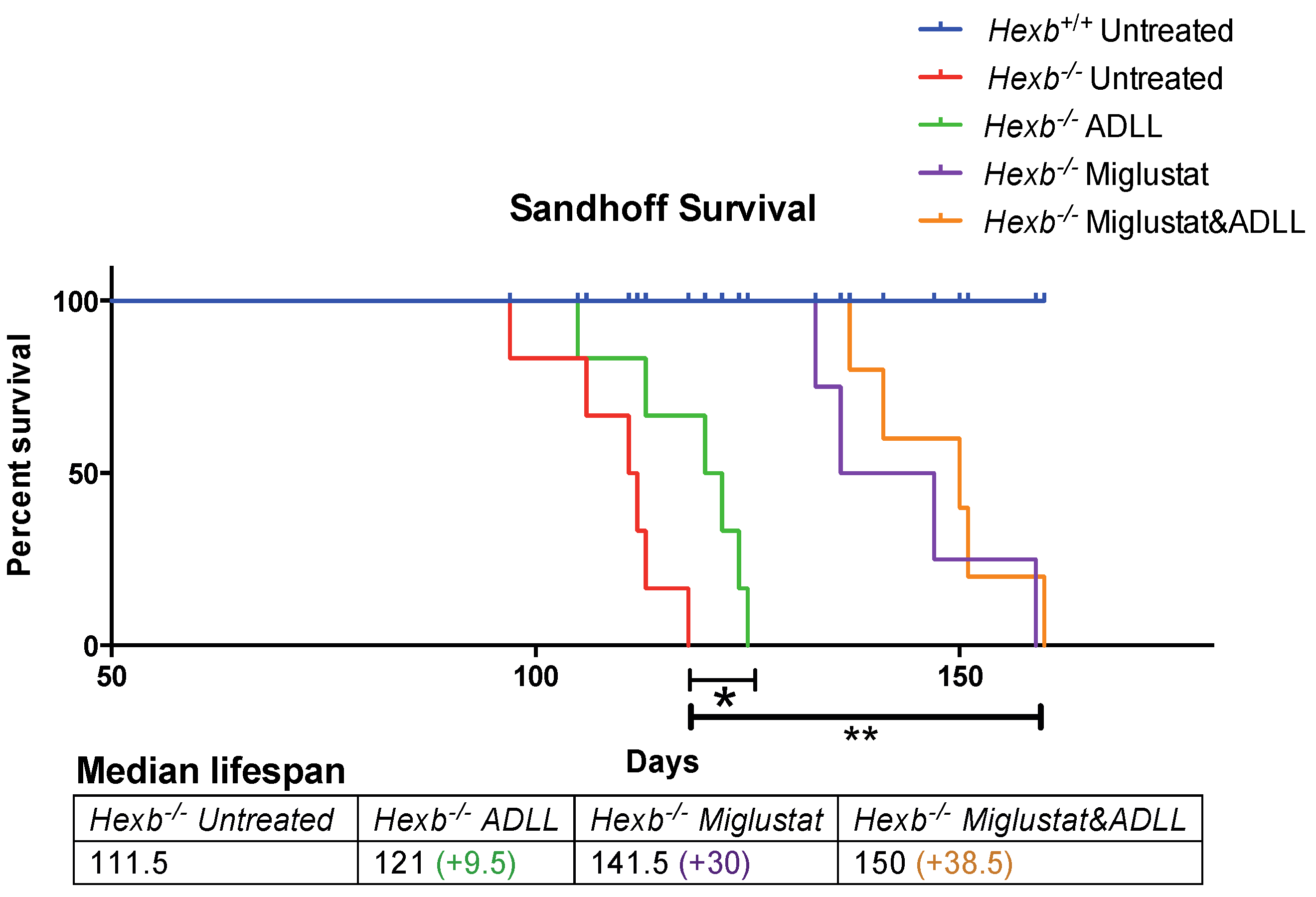
3.4. Combination Treatment of Miglustat and ADLL Results in an Additive Extension of Lifespan in Hexb-/- Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Wiederschain, G.Y. The metabolic and molecular bases of inherited disease. Biochem. 2002, 67, 611. [Google Scholar]
- Maegawa, G.H.B.; Stockley, T.; Tropak, M.; Banwell, B.; Blaser, S.; Kok, F.; Giugliani, R.; Mahuran, D.; Clarke, J.T.R. The Natural History of Juvenile or Subacute GM2 Gangliosidosis: 21 New Cases and Literature Review of 134 Previously Reported. Pediatrics 2006, 5, e1550-62. [Google Scholar] [CrossRef] [PubMed]
- Butters, T.D.; Dwek, R.A.; Platt, F.M. Imino sugar inhibitors for treating the lysosomal glycosphingolipidoses. Glycobiology 2005, 10, 43R–52R. [Google Scholar] [CrossRef] [PubMed]
- Wada, R.; Tifft, C.J.; Proia, R.L. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc. Natl. Acad. Sci. USA 2000, 97, 10954–10959. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, M.; Norflus, F.; Tiff, C.J.; Cortina-Borja, M.; Butters, T.D.; Proia, R.L.; Perry, V.H.; Dwek, R.A.; Platt, F.M. Enhanced survival in Sandhoff disease mice receiving a combination of substrate deprivation therapy and bone marrow transplantation. Blood 2001, 97, 327–329. [Google Scholar] [CrossRef]
- Begoacachon-González, M.; Wang, S.Z.; McNair, R.; Bradley, J.; Lunn, D.; Ziegler, R.; Cheng, S.H.; Cox, T.M. Gene transfer corrects acute GM2 gangliosidosispotential therapeutic contribution of perivascular enzyme flow. Mol. Ther. 2012, 8, 1489–1500. [Google Scholar] [CrossRef]
- Jeyakumar, M.; Butters, T.D.; Cortina-Borja, M.; Hunnam, V.; Proia, R.L.; Perry, V.H.; Dwek, R.A.; Platt, F.M. Delayed symptom onset and increased life expectancy in Sandhoff disease mice treated with N-butyldeoxynojirimycin. Proc. Natl. Acad. Sci. USA 1999, 96, 6388–6393. [Google Scholar] [CrossRef]
- Tallaksen, C.M.E.; Berg, J.E. Miglustat therapy in juvenile Sandhoff disease. J. Inherit. Metab. Dis. 2009, 32, 289–293. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Lefeber, D.J.; Dekomien, G.; Willemsen, M.A.A.P.; Wevers, R.A.; Morava, E. Substrate deprivation therapy in juvenile Sandhoff disease. J. Inherit. Metab. Dis. 2009, 32, 307–311. [Google Scholar] [CrossRef]
- Benard, P.; Cousse, H.; Bengonei, T.; Germaini, C. Autoradiography in Brain of Macaca Iesciculeris Monkeys after injection of Acetyl-DL-Leucine [ 2 _ 14C ] ( Tanganil®). Eur. J. Drug Metab. Pharmacokinet. 2001, 26, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Vibert, N.; Vidal, P.-P. In vitro effects of acetyl-DL-leucine (Tanganil®) on central vestibular neurons and vestibulo-ocular networks of the guinea-pig. Eur. J. Neurosci. 2001, 13, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Schniepp, R.; Strupp, M.; Wuehr, M.; Jahn, K.; Dieterich, M.; Brandt, T.; Feil, K. Acetyl-DL-leucine improves gait variability in patients with cerebellar ataxia-a case series. Cerebellum & ataxias 2016, 3, 8. [Google Scholar]
- Cortina-Borja, M.; Te Vruchte, D.; Mengel, E.; Amraoui, Y.; Imrie, J.; Jones, S.A.; I Dali, C.; Fineran, P.; Kirkegaard, T.; Runz, H.; et al. Annual severity increment score as a tool for stratifying patients with Niemann-Pick disease type C and for recruitment to clinical trials. Orphanet J. Rare Dis. 2018, 13, 1–16. [Google Scholar] [CrossRef]
- Pelz, J.O.; Fricke, C.; Saur, D.; Classen, J. Failure to confirm benefit of acetyl-dl-leucine in degenerative cerebellar ataxia: a case series. J. Neurol. 2015, 262, 1373–1375. [Google Scholar] [CrossRef]
- Bremova, T.; Malinova, V.; Amraoui, Y.; Mengel, E.; Reinke, J.; Kolnikova, M.; Strupp, M. Acetyl-DL-leucine in Niemann-Pick type C: A case series. Neurology 2015, 85, 1368–1375. [Google Scholar] [CrossRef]
- Colaco, A.; Kaya, E.; Adriaenssens, E.; Davis, L.C.; Zampieri, S.; Fernández-Suárez, M.E.; Tan, C.Y.; Deegan, P.; Porter, F.D.; Galione, A.; et al. Mechanistic convergence and shared therapeutic targets in Niemann-Pick disease type C and Tangier disease. J. Inherit. Metab. Dis. 2019, 1–12. [Google Scholar]
- Te-Vruchte, D.; Galione, A.; Strupp, M.; Mann, M. Effects of N-Acetyl-Leucine and its enantiomers in Niemann-Pick disease type C cells Danielle. bioRxiv Prepr. 2019. [Google Scholar] [CrossRef]
- Churchill, G.C.; Strupp, M.; Galione, A.; Platt, F.M. Unexpected differences in the pharmacokinetics of N-acetyl-DL-leucine enantiomers after oral dosing and their clinical relevance. PLoS One 2020, 15, e0229585. [Google Scholar] [CrossRef]
- Sango, K.; Yamanaka, S.; Hoffmann, A.; Okuda, Y.; Grinberg, A.; Westphal, H.; McDonald, M.P.; Crawley, J.N.; Sandhoff, K.; Suzuki, K.; et al. Mouse models of Tay–Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat. Genet. 1995, 2, 170–176. [Google Scholar] [CrossRef]
- Barclay, L.L.; Gibson, G.E.; Blass, J.P. The string test: An early behavioral change in thiamine deficiency. Pharmacol. Biochem. Behav. 1981, 2, 153–157. [Google Scholar] [CrossRef]
- Neville, D.C.A.; Coquard, V.; Priestman, D.A.; Te Vruchte, D.J.M.; Sillence, D.J.; Dwek, R.A.; Platt, F.M.; Butters, T.D. Analysis of fluorescently labeled glycosphingolipid-derived oligosaccharides following ceramide glycanase digestion and anthranilic acid labeling. Anal. Biochem. 2004, 331, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. MTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, H.; Ishii, T.; Endo, K.; Kawakami, E.; Nagao, K.; Miyashita, T.; Akiyama, K.; Watabe, K.; Komatsu, M.; Yamamoto, D.; et al. L-leucine and SPNS1 coordinately ameliorate dysfunction of autophagy in mouse and human Niemann-Pick type C disease. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, K.; Leblanc, R.E.; Loh, D.; Schwartz, G.J.; Yu, Y.H. Increasing Dietary Leucine Intake Reduces Diet-Induced Obesity and Improves Glucose and Cholesterol Metabolism in Mice via Multimechanisms. Diabetes 2007, 56, 1647–1654. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Yamamoto, A.; Cremona, M.L.; Rothman, J.E. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J. Cell Biol. 2006, 172, 719–731. [Google Scholar] [CrossRef]
- Yudkoff, M. Brain metabolism of branched-chain amino acids. Glia 1997, 21, 92–98. [Google Scholar] [CrossRef]
- Kennedy, B.E.; Hundert, A.S.; Goguen, D.; Weaver, I.C.G.; Karten, B. Presymptomatic Alterations in Amino Acid Metabolism and DNA Methylation in the Cerebellum of a Murine Model of Niemann-Pick Type C Disease. Am. J. Pathol. 2016, 186, 1–18. [Google Scholar] [CrossRef]
- Harris, A.; Paxton, R.; Powell, S.M.; Gillim, S.E. Physiological covalent regulation of rat liver Branched-Chain a-Ketoacid Dehydrogenase. Arch. Biochem. Biophys. 1985, 243, 542–555. [Google Scholar] [CrossRef]
- Harris, R.A.; Joshi, M.; Jeoung, N.H.; Obayashi, M. Overview of the molecular and biochemical basis of branched-chain amino acid catabolism. J. Nutr. 2005, 135, 1527S–1530S. [Google Scholar] [CrossRef] [PubMed]
- Erecińska, M.; Nelson, D. Activation of Glutamate Dehydrogenase by Leucine and its nonmetabolizable analogue in rat brain synaptosomes. J. Neurochem. 1990, 54, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Pedroso, J.A.B.; Zampieri, T.T.; Donato, J. Reviewing the effects of l-leucine supplementation in the regulation of food intake, energy balance, and glucose homeostasis. Nutrients 2015, 7, 3914–3937. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Jeon, J.H.; Min, B.K.; Ha, C.M.; Thoudam, T.; Park, B.Y.; Lee, I.K. Role of the pyruvate dehydrogenase complex in metabolic remodeling: Differential pyruvate dehydrogenase complex functions in metabolism. Diabetes Metab. J. 2018, 42, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hulver, M.W.; McMillan, R.P.; Cline, M.A.; Gilbert, E.R. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. 2014, 11, 1–9. [Google Scholar] [CrossRef]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci. 2014, 71, 2577–2604. [Google Scholar] [CrossRef]
- Hu, J.; Nie, Y.; Chen, S.; Xie, C.; Fan, Q.; Wang, Z.; Long, B.; Yan, G.; Zhong, Q.; Yan, X. Leucine reduces reactive oxygen species levels via an energy metabolism switch by activation of the mTOR-HIF-1α pathway in porcine intestinal epithelial cells. Int. J. Biochem. Cell Biol. 2017, 89, 42–56. [Google Scholar] [CrossRef]
- Cojocaru, E.; Filip, N.; Ungureanu, C.; Filip, C.; Danciu, M. Effects of Valine and Leucine on Some Antioxidant Enzymes in Hypercholesterolemic Rats. Health (Irvine. Calif). 2014, 06, 2313–2321. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef]
- Nagamori, S.; Wiriyasermkul, P.; Okuda, S.; Kojima, N.; Hari, Y.; Kiyonaka, S.; Mori, Y.; Tominaga, H.; Ohgaki, R.; Kanai, Y. Structure-activity relations of leucine derivatives reveal critical moieties for cellular uptake and activation of mTORC1-mediated signaling. Amino Acids 2016, 48, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science (80-. ). 2016, 351, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; Przybilla, M.J.; Whitley, C.B. Metabolomics profiling reveals profound metabolic impairments in mice and patients with Sandhoff disease. Mol. Genet. Metab. 2018, 126, 151–156. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, M.; Thomas, R.; Elliot-Smith, E.; Smith, D.A.; Van der Spoel, A.C.; D’Azzo, A.; Perry, V.H.; Butters, T.D.; Dwek, R.A.; Platt, F.M. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain 2003, 126, 974–987. [Google Scholar] [CrossRef]
- Liu, R.; Li, H.; Fan, W.; Jin, Q.; Chao, T.; Wu, Y.; Huang, J.; Hao, L.; Yang, X. Leucine supplementation differently modulates branched-chain amino acid catabolism, mitochondrial function and metabolic profiles at the different stage of insulin resistance in rats on high-fat diet. Nutrients 2017, 9, 565–575. [Google Scholar]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife 2016, 5, 1–25. [Google Scholar] [CrossRef]
- Vary, T.C. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: Effects on plasma lactate. Shock 1996, 2, 89–94. [Google Scholar] [CrossRef]
- Patel, M.S.; Korotchkina, L.G. Regulation of mammalian pyruvate dehydrogenase complex by phosphorylation: complexity of multiple phosphorylation sites and kinases. Exp. Mol. Med. 2001, 33, 191–197. [Google Scholar] [CrossRef]
- Mariño, G.; Pietrocola, F.; Eisenberg, T.; Kong, Y.; Malik, S.A.; Andryushkova, A.; Schroeder, S.; Pendl, T.; Harger, A.; Niso-Santano, M.; et al. Regulation of autophagy by cytosolic Acetyl-Coenzyme A. Mol. Cell 2014, 53, 710–725. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Sensing of energy and nutrients by AMP-activated protein kinase. Am. J. Clin. Nutr. 2011, 93, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.B.; Wei, Y.H. AMPK-mediated increase of glycolysis as an adaptive response to oxidative stress in human cells: Implication of the cell survival in mitochondrial diseases. Biochim. Biophys. Acta - Mol. Basis Dis. 2012, 1822, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Viollet, B.; Bhat, M.K. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1α-mediated mitochondrial biogenesis. Cell Death Discov. 2015, 15063. [Google Scholar] [CrossRef]
- Bruckbauer, A.; Zemel, M.B. Synergistic effects of metformin, resveratrol, and hydroxymethylbutyrate on insulin sensitivity. Diabetes, Metab. Syndr. Obes. Targets Ther. 2013, 6, 93–102. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein/Epitope | Antibody Type | Host | Source | Catalogue Number | Dilution |
|---|---|---|---|---|---|
| β-Actin (HRP conjugated) | 1o Monoclonal | Mouse | Invitrogen | MA5-15739-HRP | 1:15,000 |
| BCKADH-A | 1o Polyclonal | Rabbit | abcam | ab90691 | 1:1000 |
| p-BCKADH-A | 1o Polyclonal | Rabbit | abcam | ab200577 | 1:1000 |
| GDH | 1o Monoclonal | Rabbit | Cell Signalling | D9F7P | 1:1000 |
| LC3B | 1o Polyclonal | Rabbit | abcam | ab51520 | 1:2000 |
| LDHB | 1o Monoclonal | Mouse | abcam | ab85319 | 1:1500 |
| mTOR | 1o Polyclonal | Rabbit | Cell Signalling | 2972 | 1:500 |
| p-mTOR (Ser 2448) | 1o Polyclonal | Rabbit | Cell Signalling | 2971 | 1:500 |
| P62 | 1o Monoclonal | Mouse | abcam | ab56416 | 1:2000 |
| p-PDH (Ser 293) | 1o Polyclonal | Rabbit | abcam | ab92696 | 1:500 |
| PDH Complex (PDHC) | 1o Polyclonal | Mouse | abcam | ab110416 | 1:1000 |
| PDK1 | 1o Monoclonal | Mouse | abcam | ab110025 | 1:1000 |
| PDK2 | 1o Monoclonal | Rabbit | abcam | ab68164 | 1:1000 |
| PDK4 | 1o Monoclonal | Rabbit | abcam | ab214938 | 1:1000 |
| PDP1 | 1o Polyclonal | Rabbit | abcam | ab228578 | 1:1000 |
| PGC-1 α | 1o Monoclonal | Mouse | Merck Millipore | ST1202 | 1:1500 |
| SOD1 | 1o Polyclonal | Rabbit | abcam | ab13498 | 1:1500 |
| SOD2 | 1o Polyclonal | Rabbit | abcam | ab13533 | 1:1500 |
| Anti-Mouse IgG (H + L) | 2o Polyclonal | Goat | Licor-IRDye | 925-32210 | 1:10,000 |
| Anti-Rabbit IgG (H + L) | 2o Polyclonal | Goat | Licor-IRDye | 925-68071 | 1:10000 |
| Pierce ECL Substrate | - | - | Thermo Fisher | 32106 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaya, E.; Smith, D.A.; Smith, C.; Boland, B.; Strupp, M.; Platt, F.M. Beneficial Effects of Acetyl-DL-Leucine (ADLL) in a Mouse Model of Sandhoff Disease. J. Clin. Med. 2020, 9, 1050. https://doi.org/10.3390/jcm9041050
Kaya E, Smith DA, Smith C, Boland B, Strupp M, Platt FM. Beneficial Effects of Acetyl-DL-Leucine (ADLL) in a Mouse Model of Sandhoff Disease. Journal of Clinical Medicine. 2020; 9(4):1050. https://doi.org/10.3390/jcm9041050
Chicago/Turabian StyleKaya, Ecem, David A. Smith, Claire Smith, Barry Boland, Michael Strupp, and Frances M. Platt. 2020. "Beneficial Effects of Acetyl-DL-Leucine (ADLL) in a Mouse Model of Sandhoff Disease" Journal of Clinical Medicine 9, no. 4: 1050. https://doi.org/10.3390/jcm9041050
APA StyleKaya, E., Smith, D. A., Smith, C., Boland, B., Strupp, M., & Platt, F. M. (2020). Beneficial Effects of Acetyl-DL-Leucine (ADLL) in a Mouse Model of Sandhoff Disease. Journal of Clinical Medicine, 9(4), 1050. https://doi.org/10.3390/jcm9041050