Molecular and Metabolic Subtypes Correspondence for Pancreatic Ductal Adenocarcinoma Classification


Abstract
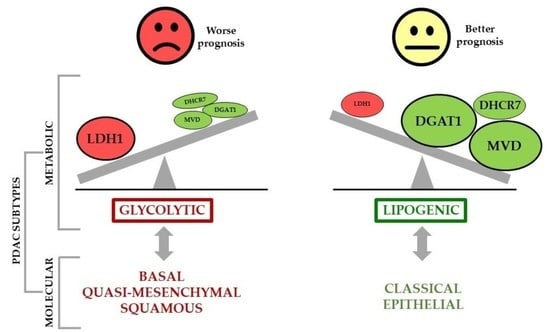
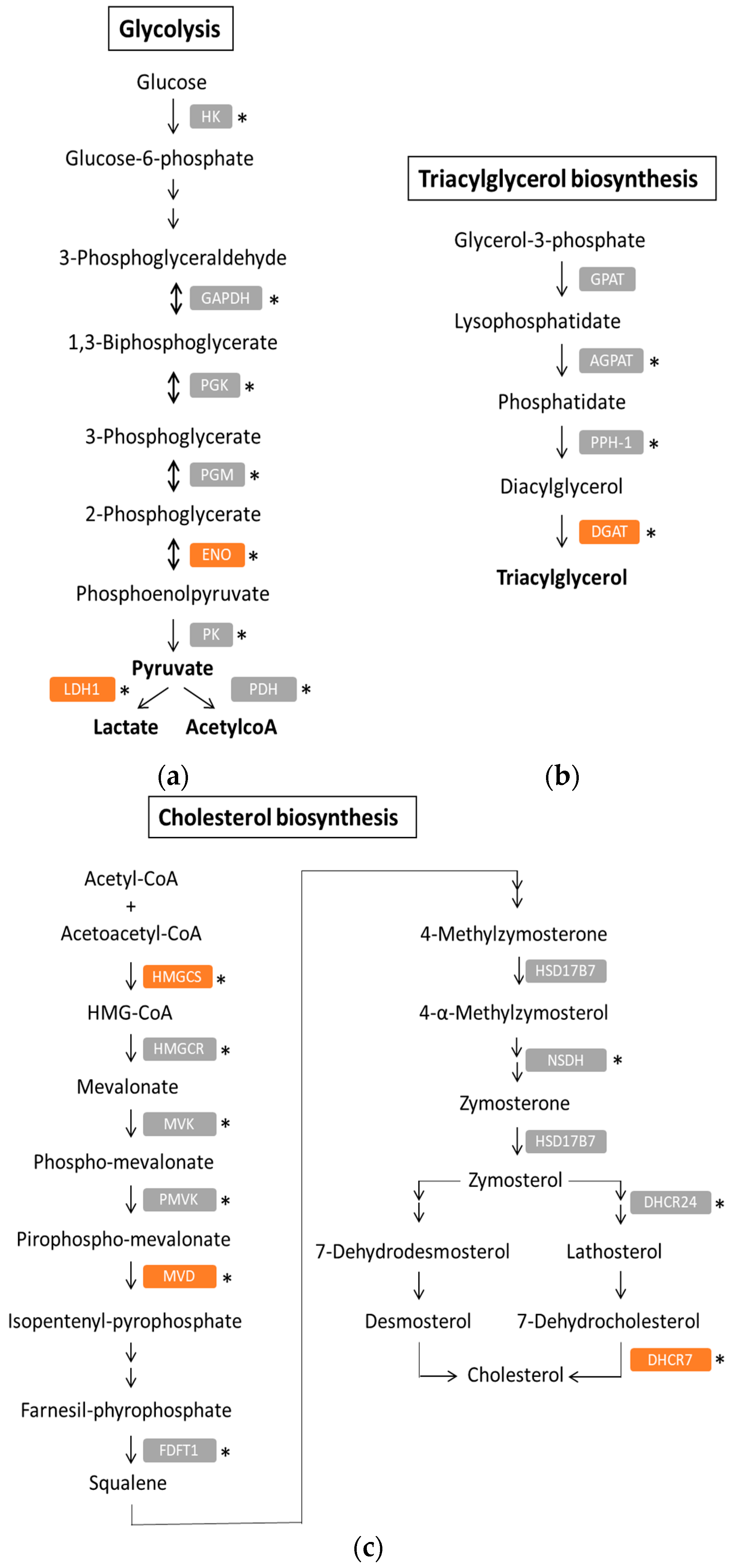
1. Introduction
2. PDAC Mutational Profile
2.1. K-RAS
2.2. p16/CDKN2A
2.3. TP53
2.4. SMAD4
3. PDAC Molecular Signatures
4. Metabolic Reprogramming in PDAC
4.1. Warburg Phenotype
4.2. Lipid Metabolism in PDAC
4.3. Amino Acid Metabolism in PDAC
5. PDAC Metabolic Signatures
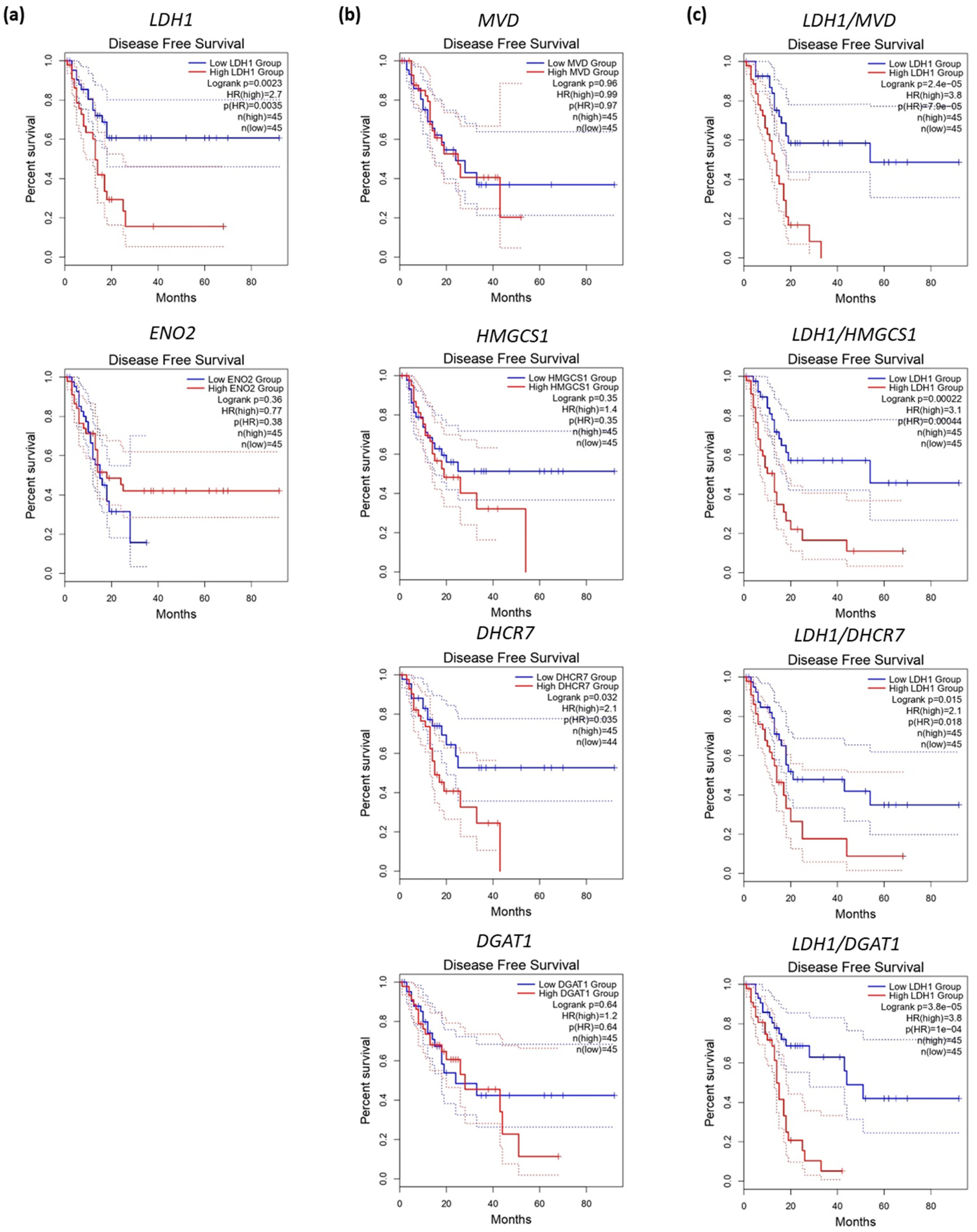
6. Metabolic Phenotypes and Survival in PDAC
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Foucher, E.D.; Ghigo, C.; Chouaib, S.; Galon, J.; Iovanna, J.; Olive, D. Pancreatic Ductal Adenocarcinoma: A Strong Imbalance of Good and Bad Immunological Cops in the Tumor Microenvironment. Front. Immunol. 2018, 9, 1044. [Google Scholar] [CrossRef]
- Oppong, K.W.; Bekkali, N.L.H. Pancreatic ductal adenocarcinoma epidemiology and risk assessment: Could we prevent? Possibility for an early diagnosis. Endosc. Ultrasound 2017, 6, 58–S61. [Google Scholar] [CrossRef] [PubMed]
- Hur, C.; Tramontano, A.C.; Dowling, E.C.; Brooks, G.A.; Jeon, A.; Brugge, W.R.; Gazelle, G.S.; Kong, C.Y.; Pandharipande, P.V. Early Pancreatic Ductal Adenocarcinoma Survival Is Dependent on Size. Pancreas 2016, 45, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, A.; Choudhuri, J.; Mukherjee, I. The subtypes of pancreatic ductal adenocarcinomas. SANAMED 2016, 11, 239. [Google Scholar] [CrossRef]
- Le, N.; Sund, M.; Vinci, A.; Beyer, G.; Javed, M.A.; Krug, S.; Neessee, A.; Schober, M.; Information, P.E.K.F.C. Prognostic and predictive markers in pancreatic adenocarcinoma. Dig. Liver Dis. 2016, 48, 223–230. [Google Scholar] [CrossRef]
- Zeitouni, D.; Pylayeva-Gupta, Y.; Der, C.J.; Bryant, K.L. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers 2016, 8, 45. [Google Scholar] [CrossRef]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 1–20. [Google Scholar] [CrossRef]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Pu, N.; Chen, Q.; Gao, S.; Liu, G.; Zhu, Y.; Yin, L.; Hu, H.; Wei, L.; Wu, Y.; Maeda, S.; et al. Genetic landscape of prognostic value in pancreatic ductal adenocarcinoma microenvironment. Ann. Transl. Med. 2019, 7, 645. [Google Scholar] [CrossRef]
- Bailey, P.; Initiative, A.P.C.G.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Pihlak, R.; Weaver, J.M.J.; Valle, J.W.; McNamara, M.G. Advances in Molecular Profiling and Categorisation of Pancreatic Adenocarcinoma and the Implications for Therapy. Cancers 2018, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Bournet, B.; Muscari, F.; Buscail, C.; Assenat, E.; Barthet, M.; Hammel, P.; Selves, J.; Guimbaud, R.; Cordelier, P.; Buscail, L. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clin. Transl. Gastroenterol. 2016, 7, e157. [Google Scholar] [CrossRef]
- Sahin, I.H.; A Iacobuzio-Donahue, C.; O’Reilly, E.M. Molecular signature of pancreatic adenocarcinoma: An insight from genotype to phenotype and challenges for targeted therapy. Expert Opin. Ther. Targets 2015, 20, 341–359. [Google Scholar] [CrossRef]
- Roberts, N.J.; Norris, A.L.; Petersen, G.M.; Bondy, M.L.; E Brand, R.; Gallinger, S.; Roberts, N.J.; Olson, S.H.; Rustgi, A.K.; Schwartz, A.G.; et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov. 2016, 6, 166–175. [Google Scholar] [CrossRef]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.-B.; Brown, A.M.K.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef]
- Raphael, B.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef]
- Rice, A.; Hernández, A.E.D.R. The Mutational Landscape of Pancreatic and Liver Cancers, as Represented by Circulating Tumor DNA. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Iv, J.P.M.; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of Somatic Mutations in Most Early-Stage Pancreatic Intraepithelial Neoplasia. Gastroenterol. 2012, 142, 730–733.e9. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef] [PubMed]
- Eser, S.; Schnieke, A.; Schneider, G.; Saur, D. Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 2014, 111, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Öllinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nat. Cell Biol. 2018, 554, 62–68. [Google Scholar] [CrossRef]
- Grant, T.J.; Hua, K.; Singh, A. Molecular Pathogenesis of Pancreatic Cancer. In Progress in Molecular Biology and Translational Science; Elsevier BV: Amsterdam, The Netherlands, 2016; Volume 144, pp. 241–275. [Google Scholar]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.; Wagenblast, E.; Davis, C.A.; Moon, S.-H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 Drives Pancreatic Cancer Metastasis through Cell-Autonomous PDGF Receptor β Signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Dijke, P.T. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- David, C.J.; Huang, Y.-H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massagué, J. TGF-β Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Veenstra, V.L.; Garcia-Garijo, A.; Van Laarhoven, H.W.M.; Bijlsma, M.F. Extracellular Influences: Molecular Subclasses and the Microenvironment in Pancreatic Cancer. Cancers 2018, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhao, H.; Yan, H. Gene expression profiling of 1200 pancreatic ductal adenocarcinoma reveals novel subtypes. BMC Cancer 2018, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Maurer, H.C.; Holmstrom, S.R.; He, J.; Laise, P.; Su, T.; Ahmed, A.; Hibshoosh, H.; A Chabot, J.; E Oberstein, P.; Sepulveda, A.R.; et al. Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes. Gut 2019, 68, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Dijk, F.; Veenstra, V.L.; Soer, E.C.; Dings, M.P.; Zhao, L.; Halfwerk, J.B.; Hooijer, G.K.; Damhofer, H.; Marzano, M.; Steins, A.; et al. Unsupervised class discovery in pancreatic ductal adenocarcinoma reveals cell-intrinsic mesenchymal features and high concordance between existing classification systems. Sci. Rep. 2020, 10, 337. [Google Scholar] [CrossRef] [PubMed]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef]
- Nicolle, R.; Blum, Y.; Duconseil, P.; Vanbrugghe, C.; Brandone, N.; Poizat, F.; Roques, J.; Bigonnet, M.; Gayet, O.; Rubis, M.; et al. Establishment of a pancreatic adenocarcinoma molecular gradient (PAMG) that predicts the clinical outcome of pancreatic cancer. EBioMedicine 2020, 57, 102858. [Google Scholar] [CrossRef]
- Juiz, N.; Elkaoutari, A.; Bigonnet, M.; Gayet, O.; Roques, J.; Nicolle, R.; Iovanna, J.; Dusetti, N. Basal-like and classical cells coexist in pancreatic cancer revealed by single-cell analysis on biopsy-derived pancreatic cancer organoids from the classical subtype. FASEB J. 2020, 34, 12214–12228. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; De Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Q.; Russo, J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim. et Biophys. Acta (BBA) Bioenerg. 2012, 1826, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Heiden, M.G.V.; Kroemer, G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. 2013, 12, 829–846. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. THE METABOLISM OF TUMORS IN THE BODY. J. Gen. Physiol. 2004, 8, 519–530. [Google Scholar] [CrossRef]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U.E. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Kheir, T.B.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef]
- Koong, A.; Mehta, V.K.; Le, Q.T.; Fisher, G.A.; Terris, D.J.; Brown, J.; Bastidas, A.J.; Vierra, M. Pancreatic tumors show high levels of hypoxia. Int. J. Radiat. Oncol. 2000, 48, 919–922. [Google Scholar] [CrossRef]
- Spivak-Kroizman, T.R.; Hostetter, G.; Posner, R.; Aziz, M.; Hu, C.; Demeure, M.J.; Von Hoff, D.; Hingorani, S.R.; Palculict, T.B.; Izzo, J.; et al. Hypoxia Triggers Hedgehog-Mediated Tumor-Stromal Interactions in Pancreatic Cancer. Cancer Res. 2013, 73, 3235–3247. [Google Scholar] [CrossRef]
- Cao, X.; Cao, Y.; Li, W.; Zhang, H.; Zhu, Z. P4HA1/HIF1α feedback loop drives the glycolytic and malignant phenotypes of pancreatic cancer. Biochem. Biophys. Res. Commun. 2019, 516, 606–612. [Google Scholar] [CrossRef]
- Chaika, N.V.; Yu, F.; Purohit, V.; Mehla, K.; Lazenby, A.J.; DiMaio, D.; Anderson, J.M.; Yeh, J.J.; Johnson, K.R.; Hollingsworth, M.A.; et al. Differential Expression of Metabolic Genes in Tumor and Stromal Components of Primary and Metastatic Loci in Pancreatic Adenocarcinoma. PLoS ONE 2012, 7, e32996. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hancock, S.E.; Sultani, G.; Wilkins, B.P.; Ding, E.; Osborne, B.; Quek, L.-E.; Turner, N. Snail-Overexpression Induces Epithelial-mesenchymal Transition and Metabolic Reprogramming in Human Pancreatic Ductal Adenocarcinoma and Non-tumorigenic Ductal Cells. J. Clin. Med. 2019, 8, 822. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.E.; Yakovenko, A.; Trevino, J.G. Glucose and Lactate Transport in Pancreatic Cancer: Glycolytic Metabolism Revisited. J. Oncol. 2018, 2018, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Basturk, O.; Singh, R.; Kaygusuz, E.; Balci, S.; Dursun, N.; Culhaci, N.; Adsay, N.V. GLUT-1 Expression in Pancreatic Neoplasia. Pancreas 2011, 40, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.C.; Nøhr-Nielsen, A.; Zeeberg, K.; Reshkin, S.J.; Hoffmann, E.K.; Novak, I.; Pedersen, S.F. Monocarboxylate Transporters MCT1 and MCT4 Regulate Migration and Invasion of Pancreatic Ductal Adenocarcinoma Cells. Pancreas 2016, 45, 1036–1047. [Google Scholar] [CrossRef]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Tian, G.; Li, G.; Liu, P.; Wang, Z.; Li, N. Glycolysis-Based Genes Associated with the Clinical Outcome of Pancreatic Ductal Adenocarcinoma Identified by The Cancer Genome Atlas Data Analysis. DNA Cell Biol. 2020, 39, 417–427. [Google Scholar] [CrossRef]
- Liang, C.; Qin, Y.; Zhang, B.; Ji, S.; Shi, S.; Xu, W.; Liu, J.; Xiang, J.; Liang, D.; Hu, Q.; et al. Energy sources identify metabolic phenotypes in pancreatic cancer. Acta Biochim. et Biophys. Sin. 2016, 48, 969–979. [Google Scholar] [CrossRef]
- Swierczynski, J.; Hebanowska, A.; Sledzinski, T. Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World J. Gastroenterol. 2014, 20, 2279–2303. [Google Scholar] [CrossRef]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Liang, Y.; Xing, C.; Wang, H.; Hu, P.; Li, J.; Huang, H.; Wang, W.; Jiang, C. Cancer-associated adipocytes exhibit distinct phenotypes and facilitate tumor progression in pancreatic cancer. Oncol. Rep. 2019, 42, 2537–2549. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Gotoh, K.; Eguchi, H.; Kobayashi, S.; Iwagami, Y.; Tomimaru, Y.; Akita, H.; Asaoka, T.; Noda, T.; Takeda, Y.; et al. Impact of CD36 on Chemoresistance in Pancreatic Ductal Adenocarcinoma. Ann. Surg. Oncol. 2019, 27, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Eguchi, H. ASO Author Reflections: Regulation of Chemoresistance in Pancreatic Ductal Adenocarcinoma by Scavenger Receptor CD36. Ann. Surg. Oncol. 2019, 27, 620–621. [Google Scholar] [CrossRef] [PubMed]
- Tanase, C.; Gheorghisan-Galateanu, A.A.; Popescu, I.D.; Mihai, S.; Codrici, E.; Albulescu, R.; Hinescu, M.E. CD36 and CD97 in Pancreatic Cancer versus Other Malignancies. Int. J. Mol. Sci. 2020, 21, 5656. [Google Scholar] [CrossRef]
- Qin, C.; Yang, G.; Yang, J.; Ren, B.; Wang, H.; Chen, G.; Zhao, F.; You, L.; Wang, W.; Zhao, Y. Metabolism of pancreatic cancer: Paving the way to better anticancer strategies. Mol. Cancer 2020, 19, 1–19. [Google Scholar] [CrossRef]
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid Metabolism and Lipid Droplets in Pancreatic Cancer and Stellate Cells. Cancers 2017, 10, 3. [Google Scholar] [CrossRef]
- Cohen, R.; Neuzillet, C.; Tijeras-Raballand, A.; Faivre, S.; De Gramont, A.; Raymond, E. Targeting cancer cell metabolism in pancreatic adenocarcinoma. Oncotarget 2015, 6, 16832–16847. [Google Scholar] [CrossRef]
- Bian, Y.; Yu, Y.; Wang, S.; Li, L. Up-regulation of fatty acid synthase induced by EGFR/ERK activation promotes tumor growth in pancreatic cancer. Biochem. Biophys. Res. Commun. 2015, 463, 612–617. [Google Scholar] [CrossRef]
- Tadros, S.B.; Shukla, S.K.; King, R.J.; Gunda, V.; Vernucci, E.; Abrego, J.; Chaika, N.V.; Lyudmyla, B.; Lazenby, A.J.; Berim, L.; et al. De NovoLipid Synthesis Facilitates Gemcitabine Resistance through Endoplasmic Reticulum Stress in Pancreatic Cancer. Cancer Res. 2017, 77, 5503–5517. [Google Scholar] [CrossRef]
- Jagust, P.; De Luxán-Delgado, B.; Parejo-Alonso, B.; Sancho, P. Metabolism-Based Therapeutic Strategies Targeting Cancer Stem Cells. Front. Pharmacol. 2019, 10, 203. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Lupien, L.; Kuemmerle, N.B.; Kinlaw, W.B.; Swinnen, J.V.; Smans, K. Lipogenesis and lipolysis: The pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid Res. 2013, 52, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Hong, Y.; Tao, X.; Wei, X.; Zhang, L.; Li, Q. An indispensable role of CPT-1a to survive cancer cells during energy stress through rewiring cancer metabolism. Tumor Biol. 2016, 37, 15795–15804. [Google Scholar] [CrossRef] [PubMed]
- Gabitova-Cornell, L.; Surumbayeva, A.; Peri, S.; Franco-Barraza, J.; Restifo, D.; Weitz, N.; Ogier, C.; Goldman, A.; Hartman, T.R.; Francescone, R.; et al. Cholesterol Pathway Inhibition Induces TGF-β Signaling to Promote Basal Differentiation in Pancreatic Cancer. Cancer Cell 2020, 38, 567–583.e11. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, S.; Guillaumond, F. LDL Receptor: An open route to feed pancreatic tumor cells. Mol. Cell. Oncol. 2015, 3, e1033586. [Google Scholar] [CrossRef]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef]
- Zhang, G.; He, P.; Tan, H.; Budhu, A.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Yfantis, H.G.; Lee, D.H.; Maitra, A.; et al. Integration of Metabolomics and Transcriptomics Revealed a Fatty Acid Network Exerting Growth Inhibitory Effects in Human Pancreatic Cancer. Clin. Cancer Res. 2013, 19, 4983–4993. [Google Scholar] [CrossRef]
- Lee, J.H.; Cho, Y.-R.; Kim, J.H.; Kim, J.; Nam, H.Y.; Kim, S.W.; Son, J. Branched-chain amino acids sustain pancreatic cancer growth by regulating lipid metabolism. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef]
- Mayers, J.R.; Wu, C.; Clish, C.B.; Kraft, P.; Torrence, M.E.; Fiske, B.P.; Yuan, C.; Bao, Y.; Townsend, M.K.; Tworoger, S.S.; et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med. 2014, 20, 1193–1198. [Google Scholar] [CrossRef]
- Maertin, S.; Elperin, J.M.; Lotshaw, E.; Sendler, M.; Speakman, S.D.; Takakura, K.; Reicher, B.M.; Mareninova, O.A.; Grippo, P.J.; Mayerle, J.; et al. Roles of autophagy and metabolism in pancreatic cancer cell adaptation to environmental challenges. Am. J. Physiol. Liver Physiol. 2017, 313, G524–G536. [Google Scholar] [CrossRef]
- Tumas, J.; Baskirova, I.; Petrenas, T.; Norkuniene, J.; Strupas, K.; Sileikis, A. Towards a Personalized Approach in Pancreatic Cancer Diagnostics Through Plasma Amino Acid Analysis. Anticancer. Res. 2019, 39, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, S.R.; Olive, K.P. Protein breakdown precedes pancreatic tumor development. Nat. Med. 2014, 20, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nat. Cell Biol. 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.J.; Amendola, C.R.; Hollinshead, K.E.R.; Yu, Q.; Yamamoto, K.; Encarnación-Rosado, J.; Rose, R.E.; LaRue, M.M.; Sohn, A.S.W.; Biancur, D.E.; et al. Selective Alanine Transporter Utilization Creates a Targetable Metabolic Niche in Pancreatic Cancer. Cancer Discov. 2020, 10, 1018–1037. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Achreja, A.; Meurs, N.; Animasahun, O.; Owen, S.; Mittal, A.; Parikh, P.; Lo, T.; Franco-Barraza, J.; Shi, J.; et al. Tumour-reprogrammed stromal BCAT1 fuels branched-chain ketoacid dependency in stromal-rich PDAC tumours. Nat. Metab. 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.-N.; Berthezène, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef]
- Altan, B.; Kaira, K.; Watanabe, A.; Kubo, N.; Bao, P.; Dolgormaa, G.; Bilguun, E.-O.; Araki, K.; Kanai, Y.; Yokobori, T.; et al. Relationship between LAT1 expression and resistance to chemotherapy in pancreatic ductal adenocarcinoma. Cancer Chemother. Pharmacol. 2018, 81, 141–153. [Google Scholar] [CrossRef]
- Daher, B.; Parks, S.K.; Durivault, J.; Cormerais, Y.; Baidarjad, H.; Tambutte, E.; Pouyssegur, J.; Vucetic, M. Genetic Ablation of the Cystine Transporter xCT in PDAC Cells Inhibits mTORC1, Growth, Survival, and Tumor Formation via Nutrient and Oxidative Stresses. Cancer Res. 2019, 79, 3877–3890. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nat. Cell Biol. 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Daemen, A.; Peterson, D.W.; Sahu, N.; Mccord, R.; Du, X.; Liu, B.; Kowanetz, K.; Hong, R.; Moffat, J.G.; Gao, M.; et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, E4410–E4417. [Google Scholar] [CrossRef] [PubMed]
- Karasinska, J.M.; Topham, J.T.; Kalloger, S.E.; Jang, G.H.; Denroche, R.E.; Culibrk, L.; Williamson, L.M.; Wong, H.-L.; Lee, M.K.-C.; O’Kane, G.M.; et al. Altered Gene Expression along the Glycolysis–Cholesterol Synthesis Axis Is Associated with Outcome in Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhao, B.; Liu, Z.; Ren, X.; Zhao, W.; Li, Z.; You, L.; Zhao, Y. Molecular Subtyping of Pancreatic Cancer: Translating Genomics and Transcriptomics into the Clinic. J. Cancer 2017, 8, 513–522. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Author | Type of Study | Type and Number of PDAC Samples | Dysregulated Pathways and Mutations | Outcome |
|---|---|---|---|---|
| Collison et al. [31] | Transcriptional | 1. Clinical samples microarray datasets Microdissected (n = 27) GSE15471 (n = 36) GSE11838 (n = 107) GSE16515 (n = 52) E-MEXP-950 (n = 50) 2. Validation: Mouse cell lines (n = 15) Human cell lines (n = 19) | Classical: (↑) Adhesion-associated genes (GATA6). More K-RAS-dependent | Good |
| Quasi-mesenchymal: (↑) Mesenchymal associated genes | Bad | |||
| Exocrine: (↑) Digestive exocrine enzyme genes | ||||
| Moffit et al. [33] | Transcriptional | 1. Microarray data Primary tumour (n = 145) Metastatic tumour (n = 61) Cell lines (n = 17) Pancreas normal samples (n = 46) Distant site adjacent samples (n = 88) 2. Validation Primary tumours (n = 15) PDXs (n = 37) Cell lines (n = 3) CAF lines (n = 6) | Classical: Classical Collison ((↑) adhesion-associated genes (GATA6)) and SMAD4 | Good |
| Basal: (↑) Genes also highly expressed in basal tumours in bladder and breast cancer | Bad | |||
| Normal stroma: (↑) Pancreatic stellate cells, smooth muscle actin, vimentin and desmin markers | Good | |||
| Activated stroma: (↑) Macrophages, tumour promotion and fibroblast activation-associated genes | Bad | |||
| Bailey et al. [10] | Mutational Transcriptional | Primary PDAC tumour samples and rare acinar cell carcinoma (n = 382) PDAC exomes (n = 74) | Squamous: Hypermethylation and (↓) pancreatic endodermal cell fate genes. TP53, KDM6A and TP63ΔN | Bad |
| Pancreatic progenitor: (↑) Pancreatic early development genes (PDX1) | Good | |||
| ADEX: (↑) K-RAS activation and pancreatic late development and differentiation genes | ||||
| Immunogenic: (↑) Immune suppression and strong immune infiltration | ||||
| Zhao et al. [34] | Transcriptional (metanalysis) | 1. Microarray datasets of PDAC primary tumour samples (n = 1268) TCGA (n = 172) GSE79670 (n = 51) TCGC PACA-AU (n = 71) MTAB-1791 (n = 195) ICGC array (n = 178) GSE71729 (n = 145) GSE62165 (n = 118) GSE62452 (n = 69) GSE57495 (n = 63) GSE60980 (n = 49) GSE77858 (n = 46) GSE55643 (n = 45) GSE15471 (n = 39) | L1: (↑) Metabolic genes | |
| L2: (↑) Metabolic, cell proliferation and epithelium genes (CDKN2A) | Bad | |||
| L3: (↑) Collagen and ECM associated genes | ||||
| L4: (↑) Immune profile | Good | |||
| L5: (↑) Neuroendocrine and insulin related pathways | Good | |||
| L6: (↑) Metabolic and digestive enzyme genes | ||||
| Lomberk et al. [35] | Epigenetic | 1. PDXs (n = 24) 2. Clinical samples microarray datasets GSE71729 (n = 145) ICGC (n = 178) TCGA (n = 172) | Classical: (↑) TFs involved in pancreatic development, metabolic regulators and Ras signalling | Good |
| Basal: (↑) TF proliferative and transcription nodes | Bad | |||
| Maurer et al. [36] | Transcriptional Computational modelling | 1. Primary PDAC tumour samples (n = 122) 2. Clinical samples microarray datasets GSE71729 (UNC) (n = 125) ICGC (n = 93) TCGA (n= 127) | Classical: Classical Moffit | Good |
| Basal: Basal Moffit | Bad | |||
| Immune-rich: (↑) immune and interleukin levels | Good | |||
| ECM-rich: (↑) matrix extracellular pathways | Bad | |||
| Dijk et al. [37] | Transcriptional | 1. Primary PDAC tumour samples (n = 90) 2. Pancreatic cancer PDXs cohort (n = 14) 3. PDAC Cell lines cohort (n = 51) | Epithelial: (↑) MYC, mitochondrial components and ribosome signature | Good |
| Mesenchymal: (↑) K-RAS, pathways related to EMT, stromal signalling and TGF-β | Bad | |||
| Compound pancreatic: Similar to the mesenchymal subtype and (↑) endocrine pathways | Good | |||
| Chan-Seng-Yue et al. [38] | Whole genome sequencing Transcriptional | Laser capture microdissected samples from late-stage PDAC 1. WGS (n = 314) 2. Bulk RNAseq (n = 248) 3. Single-cell RNAseq (n = 15) | Classical A/B: (↑) SMAD4 and GATA6 alterations | Good |
| Basal-like A/B: (↑) EMT and TGF-β pathways, loss of CDKN2A, TP53 mutations, K-RAS imbalance | Bad | |||
| Hybrid | Mid | |||
| Nicolle et al. [39] | Transcriptional | PDXs (n = 76) | Graded types between classical and basal based on tumour differentiation | Grade dependant |
| Authors | Common Subtypes | Others | ||
|---|---|---|---|---|
| Collisson et al. [31] | Classical | Quasi-mesenchymal | Exocrine-like | |
| Moffit et al. [33] | Classical | Basal-like | Normal and activated stroma | |
| Bailey et al. [39] | Progenitor | Squamous | ADEX | Immunogenic |
| Zhao et al. [34] | L1 | L2 | L6 | L3, L4 and L5 |
| Lomberk et al. [35] | Classical | Basal | ||
| Maurer et al. [36] | Classical | Basal | Immune-rich and ECM-rich | |
| Dijk et al. [37] | Epithelial | Mesenchymal | Secretory | Compound pancreatic |
| Chan-Seng-Yue et al. [38] | Classical (A, B) | Basal-like (A, B) | Hybrid | |
| Nicolle et al. [39] | From Classical to Basal | |||
| Author | Type of Study | Type and Number of PDAC Samples | Dysregulated Pathways, Metabolites and Mutations | Prognosis |
|---|---|---|---|---|
| Daemen et al. [92] | Metabolic Transcriptional | 1. Metabolomic analysis Cell lines (n = 38) 2. Transcriptional analysis Cell lines (n = 38) | Slow-proliferating: (↓) amino acids and carbohydrates levels | |
| Glycolytic: (↑) Metabolites and genes in glycolytic, pentose phosphate and serine pathways | Bad | |||
| Lipogenic: (↑) Metabolites and genes in cholesterol and de novo lipid synthesis | Good | |||
| Zhao et al. [34] | Transcriptional | 1. Microarray datasets of primary tumour samples (n = 1268) TCGA (n = 172) GSE79670 (n = 51) TCGC PACA-AU (n = 71) MTAB-1791 (n = 195) ICGCarray (n = 178) GSE71729 (n = 145) GSE62165 (n = 118) GSE62452 (n = 69) GSE57495 (n = 63) GSE60980 (n = 49) GSE77858 (n = 46) GSE55643 (n = 45) GSE15471 (n = 39) | L1: (↑) Glycolytic and lipogenic genes | |
| L2: (↑) Glycolytic genes | Bad | |||
| L3: (↑) Protein metabolism and digestive enzyme activity genes | ||||
| Lomberk et al. [35] | Epigenetic Transcriptional | 1. PDXs (n = 24) 2. Clinical samples microarray datasets GSE71729 (n = 145) ICGC (n = 178) TCGA (n= 172) | Basal: (↑) MYC, glucose metabolism genes Classical: (↑) PPARs, lipid metabolism genes | Good |
| Maurer et al. [36] | Transcriptional Computational modelling | 1. Primary PDAC tumour samples (n = 122) 2. Clinical samples microarray datasets GSE71729 (UNC) (n = 125) ICGC (n = 93) TCGA (n = 127) | Classical: (↑) lipogenic pathways (cholesterol, retinol and steroid hormone biosynthesis) | Good |
| Karasinska et al. [93] | Transcriptional Mutational | 1. Transcriptional datasets TCGA (PAAD-US) (n = 61) ICGC (PACA-CA) (n = 148) COMPASS (n = 90) PanGen/POG (n = 26) 2. Mutational datasets TCGA (PAAD-US) (n = 60) ICGC (PACA-CA) (n = 86) | Quiescent: (↓) metabolic activity | |
| Glycolytic: Glycolytic genes. K-RAS and MYC oncogenes amplification (↓) expression MPC1 and MPC2 | Bad | |||
| Cholesterogenic: (↑) Cholesterol biosynthesis genes | Good | |||
| Mixed: (↑) Glycolytic and cholesterol biosynthesis genes | ||||
| Dijk et al. [37] | Transcriptional | 1. Primary PDAC tumour samples (n = 90) 2. Pancreatic cancer PDXs cohort (n = 14) 3. PDAC Cell lines cohort (n = 51) | Epithelial: (↑) lipogenic pathways | Good |
| Authors | Subtypes | |
|---|---|---|
| Collisson et al. [31] | Classical | Quasi-mesenchymal |
| Moffit et al. [33] | Classical | Basal-like |
| Bailey et al. [39] | Progenitor | Squamous |
| Daemen et al. [91] | Lipogenic | Glycolytic |
| Zhao et al. [92] | L1 (Glycolytic/lipogenic) | L2 (Glycolytic) |
| Lomberk et al. [35] | Classical (PPAR-dep) | Basal (MYC/K-RAS dep) |
| Maurer et al. [36] | Classical (lipid metabolism) | Basal |
| Karasinska et al. [34] | Cholesterogenic | Glycolytic |
| Dijk et al. [37] | Epithelial (lipid metabolism) | Mesenchymal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espiau-Romera, P.; Courtois, S.; Parejo-Alonso, B.; Sancho, P. Molecular and Metabolic Subtypes Correspondence for Pancreatic Ductal Adenocarcinoma Classification. J. Clin. Med. 2020, 9, 4128. https://doi.org/10.3390/jcm9124128
Espiau-Romera P, Courtois S, Parejo-Alonso B, Sancho P. Molecular and Metabolic Subtypes Correspondence for Pancreatic Ductal Adenocarcinoma Classification. Journal of Clinical Medicine. 2020; 9(12):4128. https://doi.org/10.3390/jcm9124128
Chicago/Turabian StyleEspiau-Romera, Pilar, Sarah Courtois, Beatriz Parejo-Alonso, and Patricia Sancho. 2020. "Molecular and Metabolic Subtypes Correspondence for Pancreatic Ductal Adenocarcinoma Classification" Journal of Clinical Medicine 9, no. 12: 4128. https://doi.org/10.3390/jcm9124128
APA StyleEspiau-Romera, P., Courtois, S., Parejo-Alonso, B., & Sancho, P. (2020). Molecular and Metabolic Subtypes Correspondence for Pancreatic Ductal Adenocarcinoma Classification. Journal of Clinical Medicine, 9(12), 4128. https://doi.org/10.3390/jcm9124128