Circulating Tumor DNA as a Prognostic Determinant in Small Cell Lung Cancer Patients Receiving Atezolizumab

 , ,
, ,  and
and
Abstract
1. Introduction
2. Patients and Methods
2.1. Summary of the IFCT-1603 Trial
2.2. ctDNA Analysis
2.3. Statistics
3. Results
3.1. Patients
3.2. ctDNA Analysis
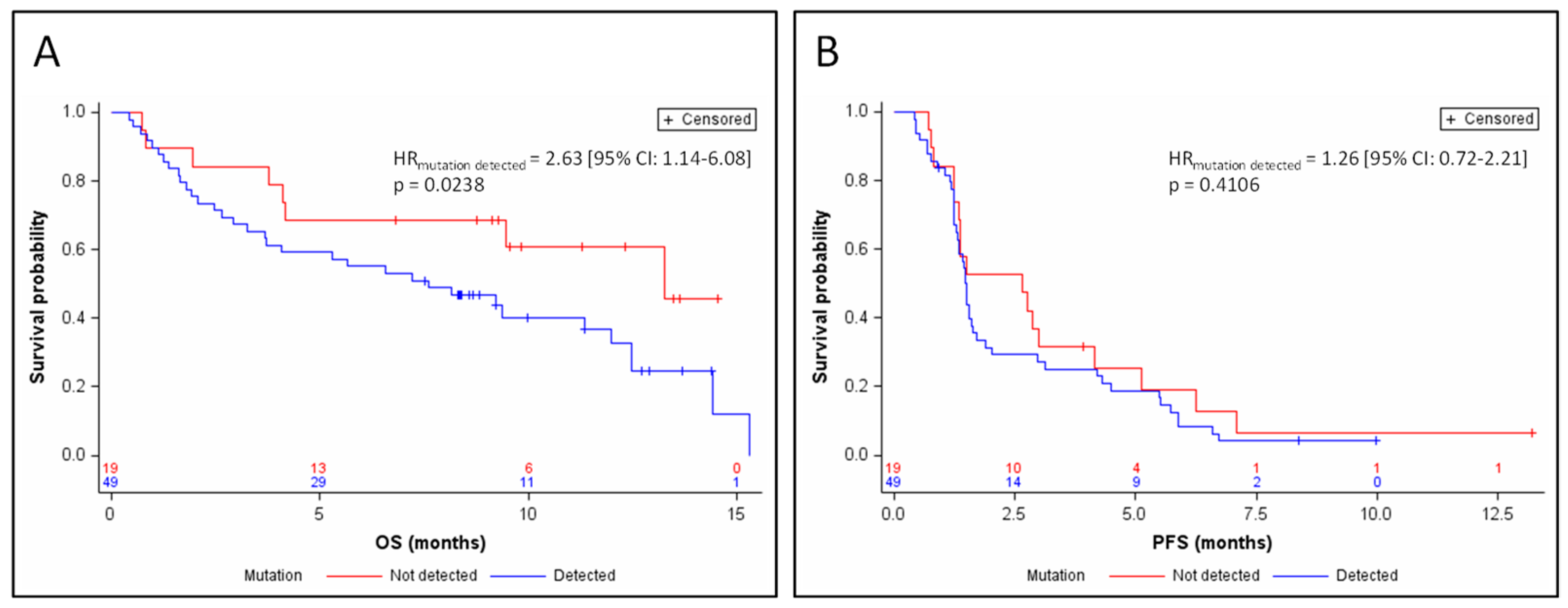
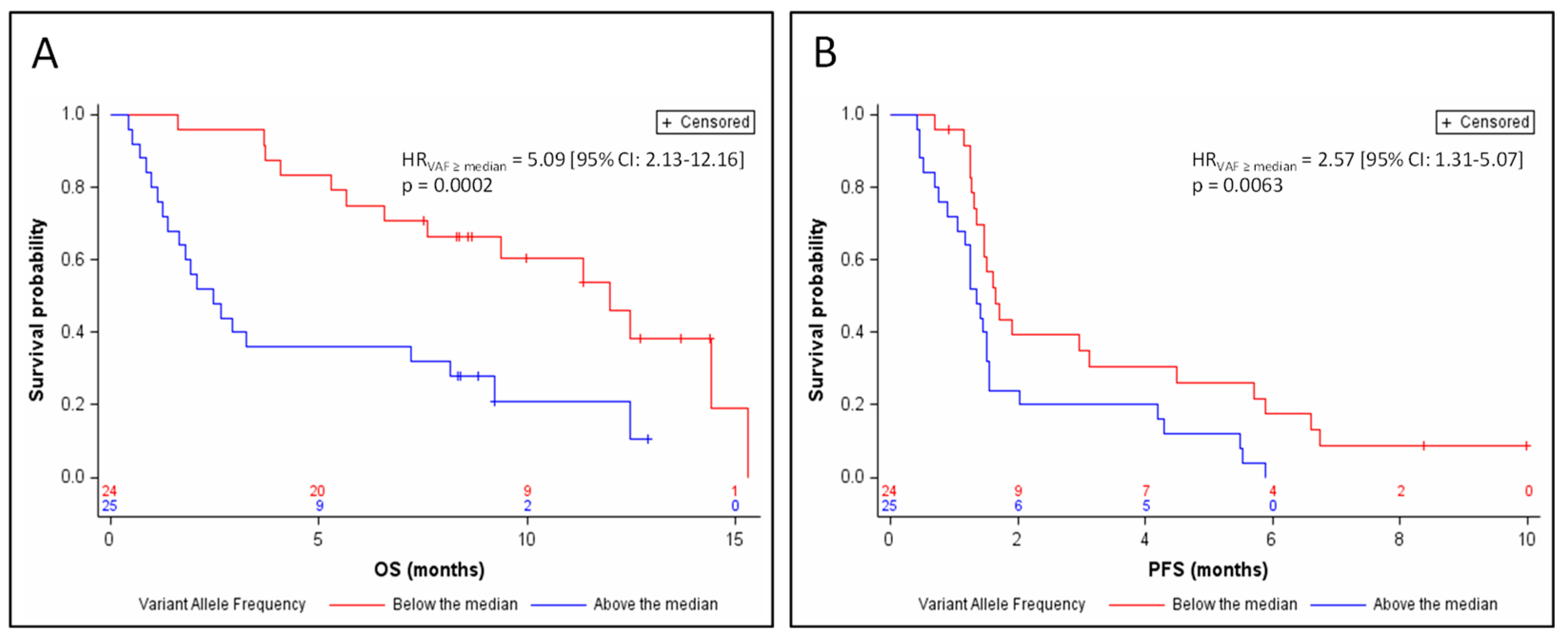
3.3. Response and Survival
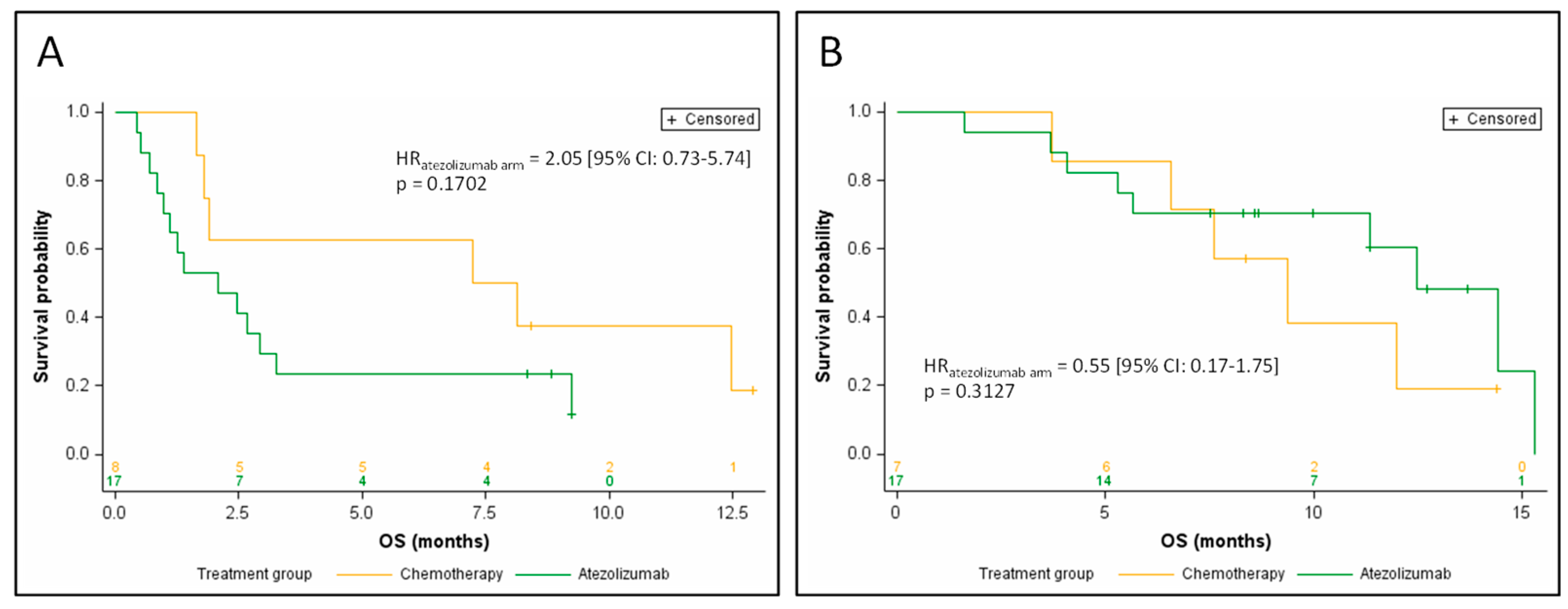
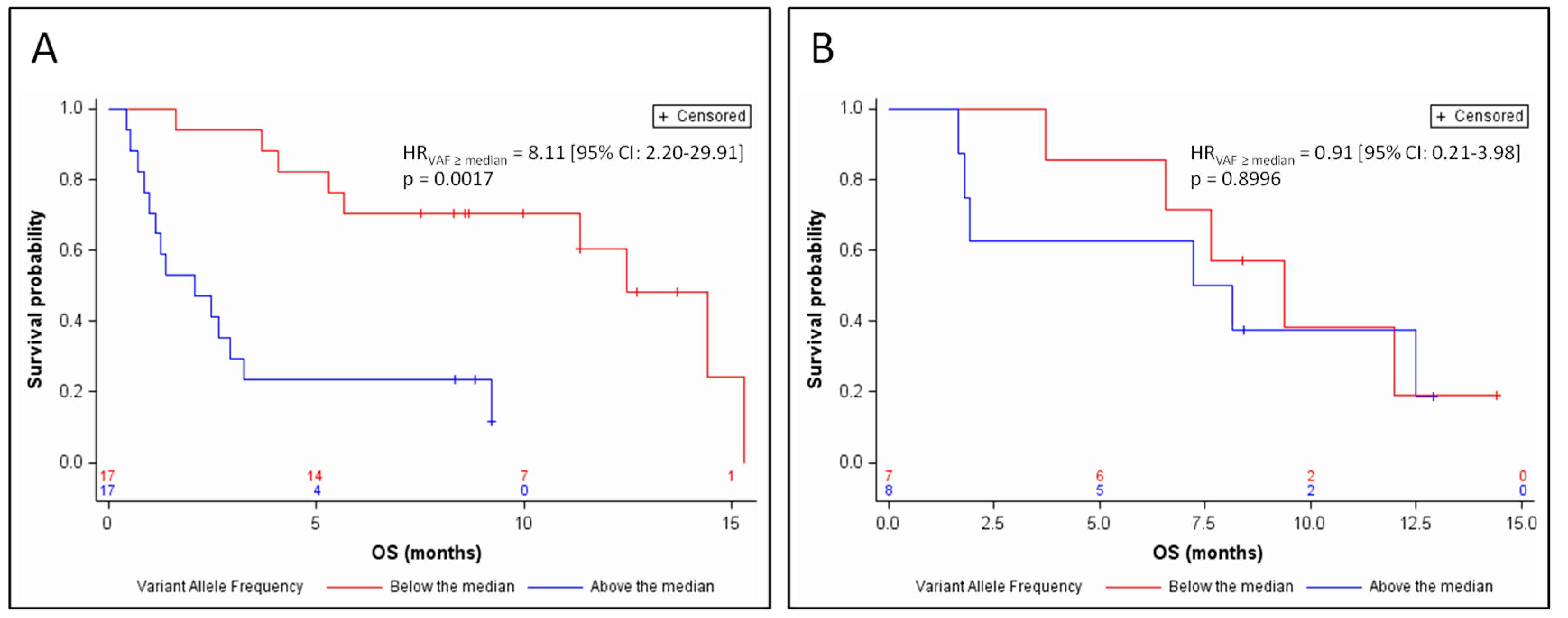
3.4. Efficacy of Atezolizumab, Based on ctDNA Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Spigel, D.R.; Socinski, M.A. Rationale for chemotherapy, immunotherapy, and checkpoint blockade in SCLC: Beyond traditional treatment approaches. J. Thorac. Oncol. 2013, 8, 587–598. [Google Scholar] [CrossRef]
- Rudin, C.M.; Ismaila, N.; Hann, C.L.; Malhotra, N.; Movsas, B.; Norris, K.; Pietanza, M.C.; Ramalingam, S.S.; Turrisi, A.T.; Giaccone, G. Treatment of Small-Cell Lung Cancer: American Society of Clinical Oncology Endorsement of the American College of Chest Physicians Guideline. J. Clin. Oncol. 2015, 33, 4106–4111. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, J.R.; von Pawel, J.; Pujol, J.-L.; Papai, Z.; Quoix, E.; Ardizzoni, A.; Poulin, R.; Preston, A.J.; Dane, G.; Ross, G. Phase III study of oral compared with intravenous topotecan as second-line therapy in small-cell lung cancer. J. Clin. Oncol. 2007, 25, 2086–2092. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomised, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef]
- Rudin, C.M.; Awad, M.M.; Navarro, A.; Gottfried, M.; Peters, S.; Csőszi, T.; Cheema, P.K.; Rodriguez-Abreu, D.; Wollner, M.; Yang, J.C.-H.; et al. Pembrolizumab or Placebo Plus Etoposide and Platinum as First-Line Therapy for Extensive-Stage Small-Cell Lung Cancer: Randomized, Double-Blind, Phase III KEYNOTE-604 Study. J. Clin. Oncol. 2020, 38, 2369–2379. [Google Scholar] [CrossRef]
- Pujol, J.-L.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Uwer, L.; Hureaux, J.; Guisier, F.; Carmier, D.; Madelaine, J.; Otto, J.; et al. A Randomized Non-Comparative Phase II Study of Anti-Programmed Cell Death-Ligand 1 Atezolizumab or Chemotherapy as Second-Line Therapy in Patients With Small Cell Lung Cancer: Results From the IFCT-1603 Trial. J. Thorac. Oncol. 2019, 14, 903–913. [Google Scholar] [CrossRef]
- Reck, M.; Vicente, D.; Ciuleanu, T.; Gettinger, S.; Peters, S.; Horn, L.; Audigier-Valette, C.; Pardo, N.; Juan-Vidal, O.; Cheng, Y.; et al. Efficacy and safety of nivolumab (nivo) monotherapy versus chemotherapy (chemo) in recurrent small cell lung cancer (SCLC): Results from CheckMate 331. Ann. Oncol. 2018, 29, x43. [Google Scholar] [CrossRef]
- Owonikoko, T.K.; Kim, H.R.; Govindan, R.; Ready, N.; Reck, M.; Peters, S.; Dakhil, S.R.; Navarro, A.; Rodriguez-Cid, J.; Schenker, M.; et al. Nivolumab (nivo) plus ipilimumab (ipi), nivo, or placebo (pbo) as maintenance therapy in patients (pts) with extensive disease small cell lung cancer (ED-SCLC) after first-line (1L) platinum-based chemotherapy (chemo): Results from the double-blind, randomized phase III CheckMate 451 study. Ann. Oncol. 2019, 30, ii77. [Google Scholar] [CrossRef]
- Peters, S.; Pujol, J.-L.; Dafni, U.; Dómine, M.; Becker, A.; Andrade, J.; Curioni-Fontecedro, A.; Molinier, O.; Moro-Sibilot, D.; Nackaerts, K.; et al. Consolidation ipilimumab and nivolumab vs observation in limited stage SCLC after chemo-radiotherapy: Results from the ETOP/IFCT 4-12 STIMULI trial. Ann. Oncol. 2020, 31, S1211. [Google Scholar] [CrossRef]
- Herbreteau, G.; Vallée, A.; Charpentier, S.; Normanno, N.; Hofman, P.; Denis, M.G. Circulating free tumor DNA in non-small cell lung cancer (NSCLC): Clinical application and future perspectives. J. Thorac. Dis. 2019, 11, S113–S126. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Zhang, K.; Ding, J.; Li, J. Prognostic value of EGFR and KRAS in circulating tumor DNA in patients with advanced non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget 2017, 8, 33922–33932. [Google Scholar] [CrossRef] [PubMed]
- Herbreteau, G.; Vallée, A.; Knol, A.-C.; Théoleyre, S.; Quéreux, G.; Frénard, C.; Varey, E.; Hofman, P.; Khammari, A.; Dréno, B.; et al. Circulating Tumour DNA Is an Independent Prognostic Biomarker for Survival in Metastatic BRAF or NRAS-Mutated Melanoma Patients. Cancers 2020, 12, 1871. [Google Scholar] [CrossRef]
- Basnet, S.; Zhang, Z.-Y.; Liao, W.-Q.; Li, S.-H.; Li, P.-S.; Ge, H.-Y. The Prognostic Value of Circulating Cell-Free DNA in Colorectal Cancer: A Meta-Analysis. J. Cancer 2016, 7, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Almodovar, K.; Iams, W.T.; Meador, C.B.; Zhao, Z.; York, S.; Horn, L.; Yan, Y.; Hernandez, J.; Chen, H.; Shyr, Y.; et al. Longitudinal cell-free DNA analysis in patients with small cell lung cancer reveals dynamic insights into treatment efficacy and disease relapse. J. Thorac. Oncol. 2018, 13, 112–123. [Google Scholar] [CrossRef]
- Nong, J.; Gong, Y.; Guan, Y.; Yi, X.; Yi, Y.; Chang, L.; Yang, L.; Lv, J.; Guo, Z.; Jia, H.; et al. Circulating tumor DNA analysis depicts subclonal architecture and genomic evolution of small cell lung cancer. Nat. Commun. 2018, 9, 3114. [Google Scholar] [CrossRef]
- Mohan, S.; Foy, V.; Ayub, M.; Leong, H.S.; Schofield, P.; Sahoo, S.; Descamps, T.; Kilerci, B.; Smith, N.K.; Carter, M.; et al. Profiling of Circulating Free DNA Using Targeted and Genome-wide Sequencing in Patients with SCLC. J. Thorac. Oncol. 2020, 15, 216–230. [Google Scholar] [CrossRef]
- Pécuchet, N.; Rozenholc, Y.; Zonta, E.; Pietrasz, D.; Didelot, A.; Combe, P.; Gibault, L.; Bachet, J.-B.; Taly, V.; Fabre, E.; et al. Analysis of Base-Position Error Rate of Next-Generation Sequencing to Detect Tumor Mutations in Circulating DNA. Clin. Chem. 2016, 62, 1492–1503. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Minor, D.; Ribas, A.; Lebbe, C.; O’Hagan, A.; Arya, N.; Guckert, M.; Schadendorf, D.; Kefford, R.F.; Grob, J.-J.; et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 3205–3211. [Google Scholar] [CrossRef]
- McEvoy, A.C.; Warburton, L.; Al-Ogaili, Z.; Celliers, L.; Calapre, L.; Pereira, M.R.; Khattak, M.A.; Meniawy, T.M.; Millward, M.; Ziman, M.; et al. Correlation between circulating tumour DNA and metabolic tumour burden in metastatic melanoma patients. BMC Cancer 2018, 18, 726. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, F.; Fortarezza, F.; Lunardi, F.; Calabrese, F. Are there any theranostic biomarkers in small cell lung carcinoma? J. Thorac. Dis. 2019, 11, S102–S112. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.C.; Jacobson, G.M.; Walker, L.E.; Blake, N.W.; Marshall, E.; Coulson, J.M. SCG3 Transcript in Peripheral Blood Is a Prognostic Biomarker for REST-Deficient Small Cell Lung Cancer. Clin. Cancer Res. 2009, 15, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Messaritakis, I.; Politaki, E.; Plataki, M.; Karavassilis, V.; Kentepozidis, N.; Koinis, F.; Samantas, E.; Georgoulias, V.; Kotsakis, A. Heterogeneity of circulating tumor cells (CTCs) in patients with recurrent small cell lung cancer (SCLC) treated with pazopanib. Lung Cancer 2017, 104, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Wang, X.; Ranganathan, A.; Torigian, D.; Troxel, A.; Evans, T.; Cohen, R.B.; Vaidya, B.; Rao, C.; Connelly, M.; et al. Circulating tumor cells as a predictive biomarker in patients with small cell lung cancer undergoing chemotherapy. Lung Cancer 2017, 112, 118–125. [Google Scholar] [CrossRef]
- Hiltermann, T.J.N.; Pore, M.M.; van den Berg, A.; Timens, W.; Boezen, H.M.; Liesker, J.J.W.; Schouwink, J.H.; Wijnands, W.J.A.; Kerner, G.S.M.A.; Kruyt, F.A.E.; et al. Circulating tumor cells in small-cell lung cancer: A predictive and prognostic factor. Ann. Oncol. 2012, 23, 2937–2942. [Google Scholar] [CrossRef]
- Naito, T.; Tanaka, F.; Ono, A.; Yoneda, K.; Takahashi, T.; Murakami, H.; Nakamura, Y.; Tsuya, A.; Kenmotsu, H.; Shukuya, T.; et al. Prognostic Impact of Circulating Tumor Cells in Patients with Small Cell Lung Cancer. J. Thorac. Oncol. 2012, 7, 512–519. [Google Scholar] [CrossRef]
- Hou, J.-M.; Krebs, M.G.; Lancashire, L.; Sloane, R.; Backen, A.; Swain, R.K.; Priest, L.J.C.; Greystoke, A.; Zhou, C.; Morris, K.; et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J. Clin. Oncol. 2012, 30, 525–532. [Google Scholar] [CrossRef]
- Wang, P.-P.; Liu, S.-H.; Chen, C.-T.; Lv, L.; Li, D.; Liu, Q.-Y.; Liu, G.-L.; Wu, Y. Circulating tumor cells as a new predictive and prognostic factor in patients with small cell lung cancer. J. Cancer 2020, 11, 2113–2122. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total (n = 68) | Mutation Detected (n = 49) | No Mutation Detected (n = 19) | p | |||
|---|---|---|---|---|---|---|
| Age (years) | Mean ± SD | 65.14 +/− 7.28 | 65.34 +/− 7.56 | 64.62 +/− 6.68 | 0.6718 | |
| Median | 64.54 | 65.44 | 63.03 | |||
| Range | (51.11–85.47) | (51.11–85.47) | (57.2–85.05) | |||
| Gender | Female | n (%) | 29 (42.6) | 19 (38.8) | 10 (52.6) | 0.2999 |
| Male | n (%) | 39 (57.4) | 30 (61.2) | 9 (47.4) | ||
| Performance status | 0 | n (%) | 26 (38.2) | 18 (36.7) | 8 (42.1) | 0.3656 |
| 1 | n (%) | 33 (48.5) | 26 (53.1) | 7 (36.8) | ||
| 2 | n (%) | 9 (13.2) | 5 (10.2) | 4 (21.1) | ||
| Smoker (current or former) | Yes | n (%) | 66 (97.1) | 48 (98) | 18 (94.7) | 0.4838 |
| No | n (%) | 2 (2.9) | 1 (2) | 1 (5.3) | ||
| Stage at time of random allocation | Limited disease | n (%) | 16 (23.5) | 10 (20.4) | 6 (31.6) | 0.3530 |
| Extensive disease | n (%) | 52 (76.5) | 39 (79.6) | 13 (68.4) | ||
| Sensitive relapse (progression ≥90 days after last first-line chemotherapy dose) | Yes | n (%) | 44 (64.7) | 32 (65.3) | 12 (63.2) | 0.8679 |
| No | n (%) | 24 (35.3) | 17 (34.7) | 7 (36.8) | ||
| Mutated gene | TP53 | n (%) | - | 32 (65.3) | - | - |
| RB1 | n (%) | - | 25 (51) | - | ||
| NOTCH1 | n (%) | - | 3 (6.1) | - | ||
| NOTCH2 | n (%) | - | 6 (12.2) | - | ||
| NOTCH3 | n (%) | - | 4 (8.2) | - | ||
| Treatment arm | Chemotherapy | n (%) | 22 (32.4) | 15 (30.6) | 7 (36.8) | 0.6222 |
| Atezolizumab | n (%) | 46 (67.6) | 34 (69.4) | 12 (63.2) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herbreteau, G.; Langlais, A.; Greillier, L.; Audigier-Valette, C.; Uwer, L.; Hureaux, J.; Moro-Sibilot, D.; Guisier, F.; Carmier, D.; Madelaine, J.; et al. Circulating Tumor DNA as a Prognostic Determinant in Small Cell Lung Cancer Patients Receiving Atezolizumab. J. Clin. Med. 2020, 9, 3861. https://doi.org/10.3390/jcm9123861
Herbreteau G, Langlais A, Greillier L, Audigier-Valette C, Uwer L, Hureaux J, Moro-Sibilot D, Guisier F, Carmier D, Madelaine J, et al. Circulating Tumor DNA as a Prognostic Determinant in Small Cell Lung Cancer Patients Receiving Atezolizumab. Journal of Clinical Medicine. 2020; 9(12):3861. https://doi.org/10.3390/jcm9123861
Chicago/Turabian StyleHerbreteau, Guillaume, Alexandra Langlais, Laurent Greillier, Clarisse Audigier-Valette, Lionel Uwer, José Hureaux, Denis Moro-Sibilot, Florian Guisier, Delphine Carmier, Jeannick Madelaine, and et al. 2020. "Circulating Tumor DNA as a Prognostic Determinant in Small Cell Lung Cancer Patients Receiving Atezolizumab" Journal of Clinical Medicine 9, no. 12: 3861. https://doi.org/10.3390/jcm9123861
APA StyleHerbreteau, G., Langlais, A., Greillier, L., Audigier-Valette, C., Uwer, L., Hureaux, J., Moro-Sibilot, D., Guisier, F., Carmier, D., Madelaine, J., Otto, J., Souquet, P.-J., Gounant, V., Merle, P., Molinier, O., Renault, A., Rabeau, A., Morin, F., Denis, M. G., & Pujol, J.-L. (2020). Circulating Tumor DNA as a Prognostic Determinant in Small Cell Lung Cancer Patients Receiving Atezolizumab. Journal of Clinical Medicine, 9(12), 3861. https://doi.org/10.3390/jcm9123861