Fat Embolism Syndrome in Sickle Cell Disease

Abstract
1. Introduction
2. The Literature
3. Our Experience
3.1. Clinical Features
3.2. Laboratory Investigations
3.3. Imaging
3.4. Other Investigations
3.5. Management and Outcomes
3.6. Reflection
4. Diagnosis and Management
4.1. Red Cell Exchange Transfusion
4.2. Therapeutic Plasma Exchange
4.3. Pre-Emptive Red Cell Exchange Transfusion
4.4. Long Term Management
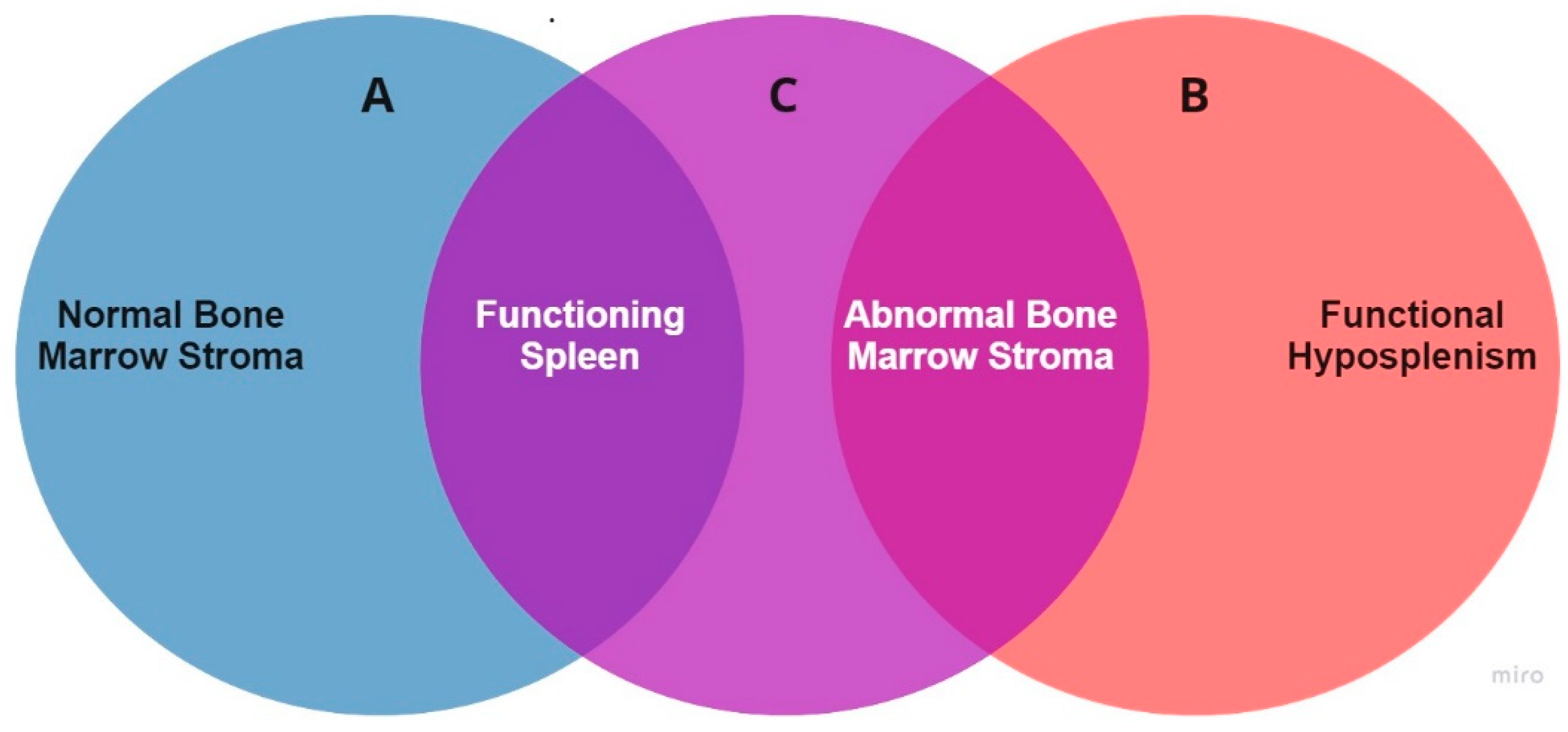
5. The Elusive Pathogenesis
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mellor, A.; Soni, N. Fat embolism. Anaesthesia 2001, 56, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.D.; Yaekoub, A.Y.; Matta, F.; Kleerekoper, M. Fat embolism syndrome. Am. J. Med. Sci. 2008, 336, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Wade, L.J.; Stevenson, L.D. Necrosis of the bone marrow with fat embolismin sickle cell anemia. Am. J. Pathol. 1941, 17, 47–54. [Google Scholar] [PubMed]
- Styles, L.A.; Schalkwijk, C.G.; Aarsman, A.J.; Vichinsky, E.P.; Lubin, B.H.; Kuypers, F.A. Phospholipase A2 levels in acute chest syndrome of sickle cell disease. Blood 1996, 87, 2573–2578. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E.; Williams, R.; Das, M.; Earles, A.N.; Lewis, N.; Adler, A.; McQuitty, J. Pulmonary fat embolism: A distinct cause of severe acute chest syndrome in sickle cell anemia. Blood 1994, 83, 3107–3112. [Google Scholar] [CrossRef]
- Ataga, K.I.; Orringer, E.P. Bone marrow necrosis in sickle cell disease: A description of three cases and a review of the literature. Am. J. Med. Sci. 2000, 320, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Godeau, B.; Schaeffer, A.; Bachir, D.; Fleury-Feith, J.; Galacteros, F.; Verra, F.; Escudier, E.; Vaillant, J.N.; Brun-Buisson, C.; Rahmouni, A.; et al. Bronchoalveolar lavage in adult sickle cell patients with acute chest syndrome: Value for diagnostic assessment of fat embolism. Am. J. Respir. Crit. Care Med. 1996, 153, 1691–1696. [Google Scholar] [CrossRef]
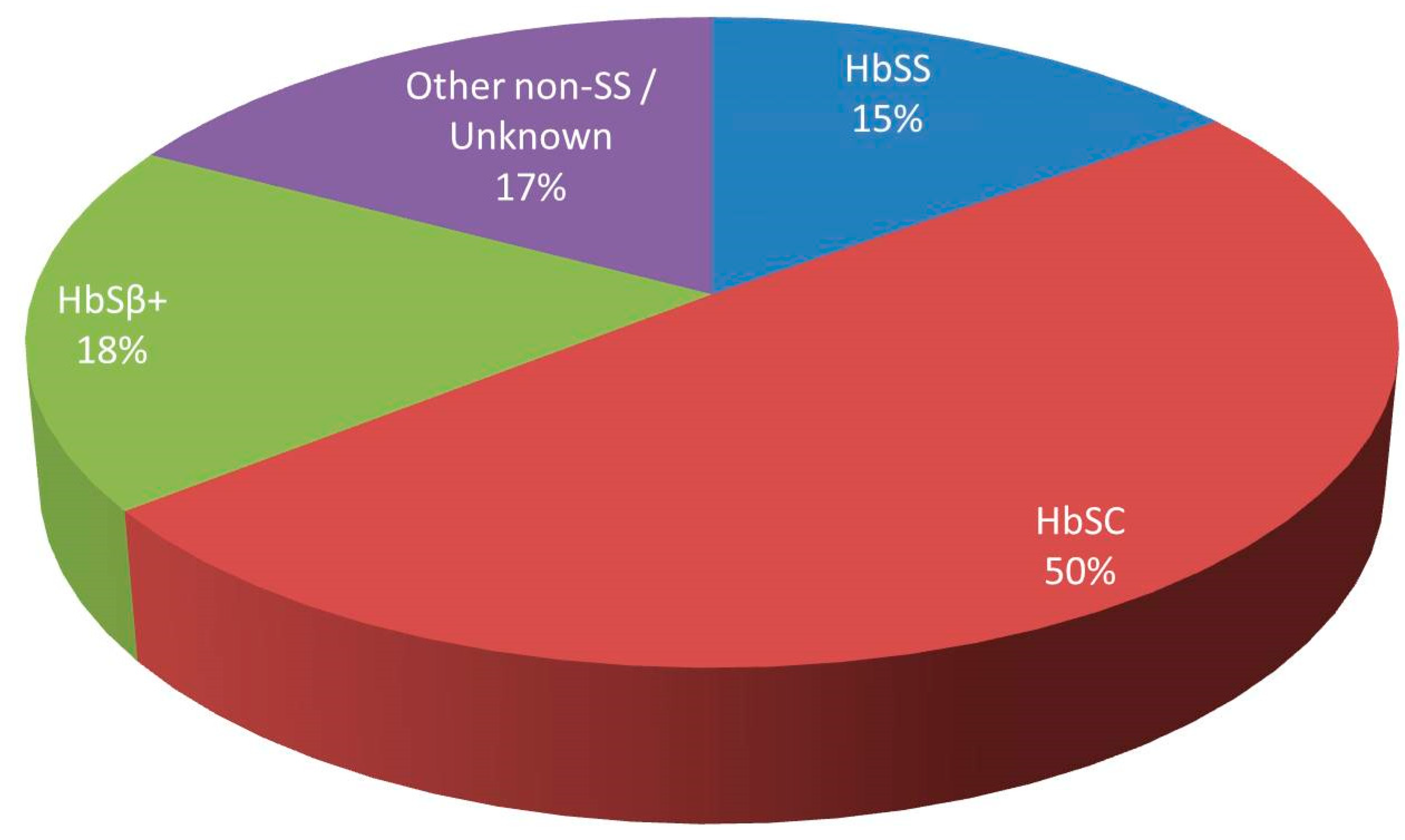
- Tsitsikas, D.A.; Gallinella, G.; Patel, S.; Seligman, H.; Greaves, P.; Amos, R.J. Bone marrow necrosis and fat embolism syndrome in sickle cell disease: Increased susceptibility of patients with non-SS genotypes and a possible association with human parvovirus B19 infection. Blood Rev. 2014, 28, 23–30. [Google Scholar] [CrossRef]
- Tsitsikas, D.A.; May, J.E.; Gangaraju, R.; Abukar, J.; Amos, R.J.; Marques, M.B. Revisiting fat embolism in sickle syndromes: Diagnostic and emergency therapeutic measures. Br. J. Haematol. 2019, 186, e112–e115. [Google Scholar] [CrossRef] [PubMed]
- Saraf, S.L.; Molokie, R.E.; Nouraie, M.; Sable, C.A.; Luchtman-Jones, L.; Ensing, G.J.; Campbell, A.D.; Rana, S.R.; Niu, X.M.; Machado, R.F.; et al. Differences in the clinical and genotypic presentation of sickle cell disease around the world. Paediatr. Respir. Rev. 2014, 15, 4–12. [Google Scholar] [CrossRef]
- Piel, F.B.; Steinberg, M.H.; Rees, D.C. Sickle Cell Disease. N. Engl. J. Med. 2017, 377, 305. [Google Scholar] [CrossRef] [PubMed]
- Maroni, A.; Dauger, S.; Chomton, M. Fat Embolism Syndrome in a Child with Sickle Cell Disease. J. Pediatr. 2019, 214, 236. [Google Scholar] [CrossRef]
- Debus, J.; Dumont, B.; Le Breton, C.; Emery, M.; Peynaud-Debayle, E.; Affo, L. A case of bone marrow necrosis and fat embolism in a sickle-cell disease patient. Ann. Biol. Clin. 2019, 77, 318–322. [Google Scholar] [CrossRef]
- Lundeen, K.M.; Bhoopal, J.R.; Simegn, M.A.; Leatherman, J.W. Acute Hypoxemia and Coma in a Patient with Hemoglobin SC Disease. Chest 2019, 155, e21–e23. [Google Scholar] [CrossRef]
- Gendreau, S.; Scholer, M.; Cecchini, J.; Habibi, A.; Razazi, K.; De Prost, N.; Carteaux, G.; Bartolucci, P.; Gendre, T.; Brugieres, P.; et al. Cerebral fat embolism in sickle cell disease. Am. J. Hematol. 2020, 95, E41–E45. [Google Scholar] [CrossRef]
- Greaves, P.; Mathew, V.; Peters, C.; Rowe, S.; Amos, R.J.; Tsitsikas, D.A. Successful outcome of three patients with sickle-cell disease and fat embolism syndrome treated with intensive exchange transfusion. Clin. Case Rep. 2016, 5, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Gurd, A.R. Fat embolism: An aid to diagnosis. J. Bone Jt. Surg. Br. 1970, 52, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, S.A.; Ploysongsang, Y.; DiLisio, R.; Crissman, J.D.; Miller, E.; Hammerschmidt, D.E.; Jacob, H.S. Fat embolism prophylaxis with corticosteroids. A prospective study in high-risk patients. Ann. Intern. Med. 1983, 99, 438–443. [Google Scholar] [CrossRef]
- Lindeque, B.G.; Schoeman, H.S.; Dommisse, G.F.; Boeyens, M.C.; Vlok, A.L. Fat embolism and the fat embolism syndrome. A double-blind therapeutic study. J. Bone Jt. Surg. Br. 1987, 69, 128–131. [Google Scholar] [CrossRef]
- Dang, N.C.; Johnson, C.; Eslami-Farsani, M.; Haywood, L.J. Bone marrow embolism in sickle cell disease: A review. Am. J. Hematol. 2005, 79, 61–67. [Google Scholar] [CrossRef]
- Milner, P.F.; Brown, M. Bone marrow infarction in sickle cell anemia: Correlation with hematologic profiles. Blood 1982, 60, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Connelly-Smith, L.; Aqui, N.; Balogun, R.A.; Klingel, R.; Meyer, E.; Pham, H.P.; Schneiderman, J.; Witt, V.; Wu, Y.; et al. Guidelines on the Use of Therapeutic Apheresis in Clinical Practice—Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Eighth Special Issue. J. Clin. Apher. 2019, 34, 171–354. [Google Scholar] [CrossRef]
- Boga, C.; Kozanoglu, I.; Ozdogu, H.; Ozyurek, E. Plasma exchange in critically ill patients with sickle cell disease. Transfus. Apher. Sci. 2007, 37, 17–22. [Google Scholar] [CrossRef]
- Venkata Sasidhar Majjiga Tripathy, A.K.; Viswanathan, K.; Shukla, M. Thrombotic thrombocytopenic purpura and multiorgan system failure in a child with sickle cell-hemoglobin C disease. Clin. Pediatr. 2010, 49, 992–996. [Google Scholar] [CrossRef]
- Shelat, S.G. Thrombotic thrombocytopenic purpura and sickle cell crisis. Clin. Appl. Thromb. Hemost. 2010, 16, 224–227. [Google Scholar] [CrossRef]
- Shome, D.K.; Ramadorai, P.; Al-Ajmi, A.; Ali, F.; Malik, N. Thrombotic microangiopathy in sickle cell disease crisis. Ann. Hematol. 2013, 92, 509–515. [Google Scholar] [CrossRef]
- Bolaños-Meade, J.; Keung, Y.K.; López-Arvizu, C.; Florendo, R.; Cobos, E. Thrombotic thrombocytopenic purpura in a patient with sickle cell crisis. Ann. Hematol. 1999, 78, 558–559. [Google Scholar] [CrossRef]
- Lee, H.E.; Marder, V.J.; Logan, L.J.; Friedman, S.; Miller, B.J. Life-threatening thrombotic thrombocytopenic purpura (TTP) in a patient with sickle cell-hemoglobin C disease. Ann. Hematol. 2003, 82, 702–704. [Google Scholar] [CrossRef] [PubMed]
- Myers, C.F.; Ipe, T.S. Bone Marrow Necrosis in Sickle Cell-Beta Thalassemia Patient Mimicking Thrombotic Thrombocytopenic Purpura. Ann. Clin. Lab. Sci. 2018, 48, 670–673. [Google Scholar]
- Kammeyer, R.; Devnani, R.; Mehta, R. Cerebral fat embolism syndrome mimicking thrombotic thrombocytopenic purpura in a patient with hemoglobin SC disease. Am. J. Hematol. 2016, 91, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Gangaraju, R.; May, J.E.; Williams LA 3rd Reddy, V.B.; MacLennan, P.; Marques, M.B. Fat embolism syndrome due to bone marrow necrosis in patients with hemoglobinopathies: A life-threatening complication mimicking thrombotic thrombocytopenic purpura. Am. J. Hematol. 2019, 94, E64–E66. [Google Scholar] [CrossRef]
- Nader, E.; Connes, P.; Lamarre, Y.; Renoux, C.; Joly, P.; Hardy-Dessources, M.D.; Cannas, G.; Lemonne, N.; Ballas, S.K. Plasmapheresis may improve clinical condition in sickle cell disease through its effects on red blood cell rheology. Am. J. Hematol. 2017, 92, E629–E630. [Google Scholar] [CrossRef]
- Harel, L.; Straussberg, R.; Rudich, H.; Cohen, A.H.; Amir, J. Raynaud’s phenomenon as a manifestation of parvovirus B19 infection: Case reports and review of parvovirus B19 rheumatic and vasculitic syndromes. Clin. Infect. Dis. 2000, 30, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.M.; Iwenofu, O.H.; Kerns, M.J.; Nuovo, G.J.; Dyrsen, M.E.; Segal, J.P. Fulminant and accelerated presentation of dermatomyositis in two previously healthy young adult males: A potential role for endotheliotropic viral infection. J. Cutan. Pathol. 2009, 36, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.M.; Nuovo, G.; Ferri, C.; Crowson, A.N.; Giuggioli, D.; Sebastiani, M. Parvoviral infection of endothelial cells and stromal fibroblasts: A possible pathogenetic role in scleroderma. J. Cutan. Pathol. 2004, 31, 43–50. [Google Scholar] [CrossRef]
- Crowson, A.N.; Magro, C.M.; Dawood, M.R. A causal role for parvovirus B19 infection in adult dermatomyositis and other autoimmune syndromes. J. Cutan. Pathol. 2000, 27, 505–515. [Google Scholar] [CrossRef]
- Takahashi, Y.; Murai, C.; Shibata, S.; Munakata, Y.; Ishii, T.; Ishii, K.; Saitoh, T.; Sawai, T.; Sugamura, K.; Sasaki, T. Human parvovirus B19 as a causative agent for rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 1998, 95, 8227–8232. [Google Scholar] [CrossRef]
- Chou, T.N.; Hsu, T.C.; Chen, R.M.; Lin, L.I.; Tsay, G.J. Parvovirus B19 infection associated with the production of anti-neutrophil cytoplasmic antibody (ANCA) and anticardiolipin antibody (aCL). Lupus 2000, 9, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Pironi, L.; Bonvicini, F.; Gionchetti, P.; D’Errico, A.; Rizzello, F.; Corsini, C.; Foroni, L.; Gallinella, G. Parvovirus b19 infection localized in the intestinal mucosa and associated with severe inflammatory bowel disease. J. Clin. Microbiol. 2009, 47, 1591–1595. [Google Scholar] [CrossRef]
- Wierenga, K.J.; Pattison, J.R.; Brink, N.; Griffiths, M.; Miller, M.; Shah, D.J.; Williams, W.; Serjeant, B.E.; Serjeant, G.R. Glomerulonephritis after human parvovirus infection in homozygous sickle-cell disease. Lancet 1995, 346, 475–476. [Google Scholar] [CrossRef]
- Tolaymat, A.; Al Mousily, F.; MacWilliam, K.; Lammert, N.; Freeman, B. Parvovirus glomerulonephritis in a patient with sickle cell disease. Pediatr. Nephrol. 1999, 13, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Mallouh, A.A.; Qudah, A. Acute splenic sequestration together with aplastic crisis caused by human parvovirus B19 in patients with sickle cell disease. J Pediatr. 1993, 122, 593–595. [Google Scholar] [CrossRef]
- Saad, A.A.; Beshlawi, I.; Al-Rawas, A.H.; Zachariah, M.; Nazir, H.F.; Wali, Y. Human Parvovirus B19 in Children with Sickle Cell Disease; Poking the Spleen. Oman Med. J. 2017, 32, 425–428. [Google Scholar] [CrossRef]
- Wierenga, K.J.; Serjeant, B.E.; Serjeant, G.R. Cerebrovascular complications and parvovirus infection in homozygous sickle cell disease. J. Pediatr. 2001, 139, 438–442. [Google Scholar] [CrossRef]
- Mallouh, A.A.; Qudah, A. An epidemic of aplastic crisis caused by human parvovirus B19. Pediatr. Infect. Dis. J. 1995, 14, 31–34. [Google Scholar] [CrossRef]
- Lowenthal, E.A.; Wells, A.; Emanuel, P.D.; Player, R.; Prchal, J.T. Sickle cell acute chest syndrome associated with parvovirus B19 infection: Case series and review. Am. J. Hematol. 1996, 51, 207–213. [Google Scholar] [CrossRef]
- Park, S.Y.; Matte, A.; Jung, Y.; Ryu, J.; Anand, W.B.; Han, E.Y.; Liu, M.; Carbone, C.; Melisi, D.; Nagasawa, T.; et al. Pathologic angiogenesis in the bone marrow of humanized sickle cell mice is reversed by blood transfusion. Blood 2020, 135, 2071–2084. [Google Scholar] [CrossRef]
- Chmel, H.; Bertles, J.F. Hemoglobin S/C disease in a pregnant woman with crisis and fat embolization syndrome. Am. J. Med. 1975, 58, 563–566. [Google Scholar] [CrossRef]
- Godeau, B.; Dhainaut, J.F.; Bachir, D.; Galacteros, F. Pulmonary fat embolism after prostaglandin infusion in sickle cell disease with fatal outcome despite exchange blood transfusion. Am. J. Hematol. 1993, 43, 330–331. [Google Scholar] [CrossRef]
- Huang, J.C.; Gay, R.; Khella, S.L. Sickling crisis, fat embolism, and coma after steroids. Lancet 1994, 344, 951–952. [Google Scholar] [CrossRef]
- Garza, J.A. Massive fat and necrotic bone marrow embolization in a previously undiagnosed patient with sickle cell disease. Am. J. Forensic Med. Pathol. 1990, 11, 83–88. [Google Scholar] [CrossRef]
- Elenga, N.; Celicourt, D.; Muanza, B.; Elana, G.; Hocquelet, S.; Tarer, V.; Maillard, F.; Sibille, G.; Divialle Doumdo, L.; Petras, M.; et al. Dengue in hospitalized children with sickle cell disease: A retrospective cohort study in the French departments of America. J. Infect. Public Health 2020, 13, 186–192. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. HLH Across Speciality Collaboration, UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Arlet, J.B.; de Luna, G.; Khimoud, D.; Odièvre, M.H.; de Montalembert, M.; Joseph, L.; Chantalat-Auger, C.; Flamarion, E.; Bartolucci, P.; Lionnet, F.; et al. Prognosis of patients with sickle cell disease and COVID-19: A French experience. Lancet Haematol. 2020, 7, e632–e634. [Google Scholar] [CrossRef]
- Panepinto, J.A.; Brandow, A.; Mucalo, L.; Yusuf, F.; Singh, A.; Taylor, B.; Woods, K.; Payne, A.B.; Peacock, G.; Schieve, L.A. Coronavirus Disease among Persons with Sickle Cell Disease, United States, March 20-May 21, 2020. Emerg. Infect. Dis. 2020, 26, 2473–2476. [Google Scholar] [CrossRef]
- Telfer, P.; de la Fuente, J.; Sohal, M.; Brown, R.; Eleftheriou, P.; Roy, N.; Piel, F.B.; Chakravorty, S.; Gardner, K.; Velangi, M.; et al. Real-time national survey of COVID-19 in hemoglobinopathy and rare inherited anemia patients. Haematologica 2020. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Pt | GT | BMN D | FES D | HPV B19 | PD | Mx | OC On DC/On FU |
|---|---|---|---|---|---|---|---|
| 1 | SC | CL | CL/MRI | Y | MD/AVN | RCE | SNI/nCR |
| 2 | SS | BMBx | CL | NT | MD/AVN | RCE/TPE | CR |
| 3 | SC | CL | CL | Y | MD | RCE | CR |
| 4 | SC | CL | CL | NT | MD | RCE | CR |
| 5 | SC | BMBx | CL/MRI | N | MD | RCE | MNI/CR |
| 6 | SC | A | A | N | MD | ST | D |
| 7 | SC | CL | CL/MRI | N | MD | RCE | SNI/SNI |
| 8 | Sβ+ | BMBx | CL | N | MD | RCE/TPE | D |
| Pt | Respiratory Failure (Hours) | Fever (Hours) | Neurology (Hours) | >50% PLT (Hours) | >50% Cr (Hours) | Liver imp. (Hours) | Other (Hours) | LOS (Days) |
|---|---|---|---|---|---|---|---|---|
| 1 | 25 | 25 | Coma 32 | 37 | No | No | No | 407 (334) |
| 2 | 48 | 0 | Altered level of consciousness 32 | 31 | 47 | Severe hyperbilirubinaemia 84 | E. Coli sepsis 0 | 65 |
| 3 | 23 | No | Altered level of consciousness 23 | 0 | 0 | Severe transaminitis 23 | Skin lesions 48 | 34 |
| 4 | 0 | 0 | 3rd branch V nerve sensory loss 0 | 24 | 30 | Mild transaminitis 0 | No | 14 |
| 5 | 0 | 12 | Left arm Hemiparesis 28 | 0 | 28 | Mild transaminitis 20 | Hypertension 0 | 126 |
| 6 | 22 | 22 | Altered level of consciousness 22 | 27 | 34 | Severe transaminitis 22 | No | 2 RIP |
| 7 | 60 | 56 | Coma 60 | No | 60 | No | No | 211 (160) |
| 8 | 71 | No | Altered level of consciousness 71 | 71 | 71 | Severe transaminitis 71 | No | 7 RIP |
| Baseline Mean (Range) | Presentation Mean (Range) | Max/Nadir Value Mean (Range) | Max/Nadir Hours Mean (Range) | |
|---|---|---|---|---|
| Ferritin mcg/L | 109 (10–214) | 10,119 (2274–40,000) | 18,172 (2274–50,014) | 42 (0–76) |
| LDH U/L | 298 (126–630) | 2167 (678–5287) | 3516 (1105–6852) | 15 (0–37) |
| CRP mg/L | <5 | 52 (<5–235) | 317 (196–493) | 102 (38–198) |
| Haemoglobin g/L | 113 (75–130) | 108 (69–136) | 86 (45–116) | 31 (7–75) |
| Reticulocytes × 109/L | 121 (75–165) | 127 (78–196) | 43 (12–81) | 105 (7–267) |
| PLT × 109/L | 246 (150–330) | 236 (85–338) | 110 (31–231) | 46 (0–75) |
| Creatinine umol/L | 70 (48–90) | 82 (74–146) | 284 (82–936) | 84 (37–140) |
| ALP U/L | 77 (48–100) | 138 (43–410) | 475 (283–774) | 115 (16–246) |
| ALT U/L | 21 (11–35) | 88 (18–309) | 2475 (18–11,262) | 131 (0–397) |
| Bilirubin umol/L | 23 (8–65) | 40 (10–119) | 156 (18–766) | 66 (0–153) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsitsikas, D.A.; Bristowe, J.; Abukar, J. Fat Embolism Syndrome in Sickle Cell Disease. J. Clin. Med. 2020, 9, 3601. https://doi.org/10.3390/jcm9113601
Tsitsikas DA, Bristowe J, Abukar J. Fat Embolism Syndrome in Sickle Cell Disease. Journal of Clinical Medicine. 2020; 9(11):3601. https://doi.org/10.3390/jcm9113601
Chicago/Turabian StyleTsitsikas, Dimitris A., Jessica Bristowe, and Jibril Abukar. 2020. "Fat Embolism Syndrome in Sickle Cell Disease" Journal of Clinical Medicine 9, no. 11: 3601. https://doi.org/10.3390/jcm9113601
APA StyleTsitsikas, D. A., Bristowe, J., & Abukar, J. (2020). Fat Embolism Syndrome in Sickle Cell Disease. Journal of Clinical Medicine, 9(11), 3601. https://doi.org/10.3390/jcm9113601