Targeting Endothelial Cell Metabolism by Inhibition of Pyruvate Dehydrogenase Kinase and Glutaminase-1

 , , ,
, , , 
 and
and 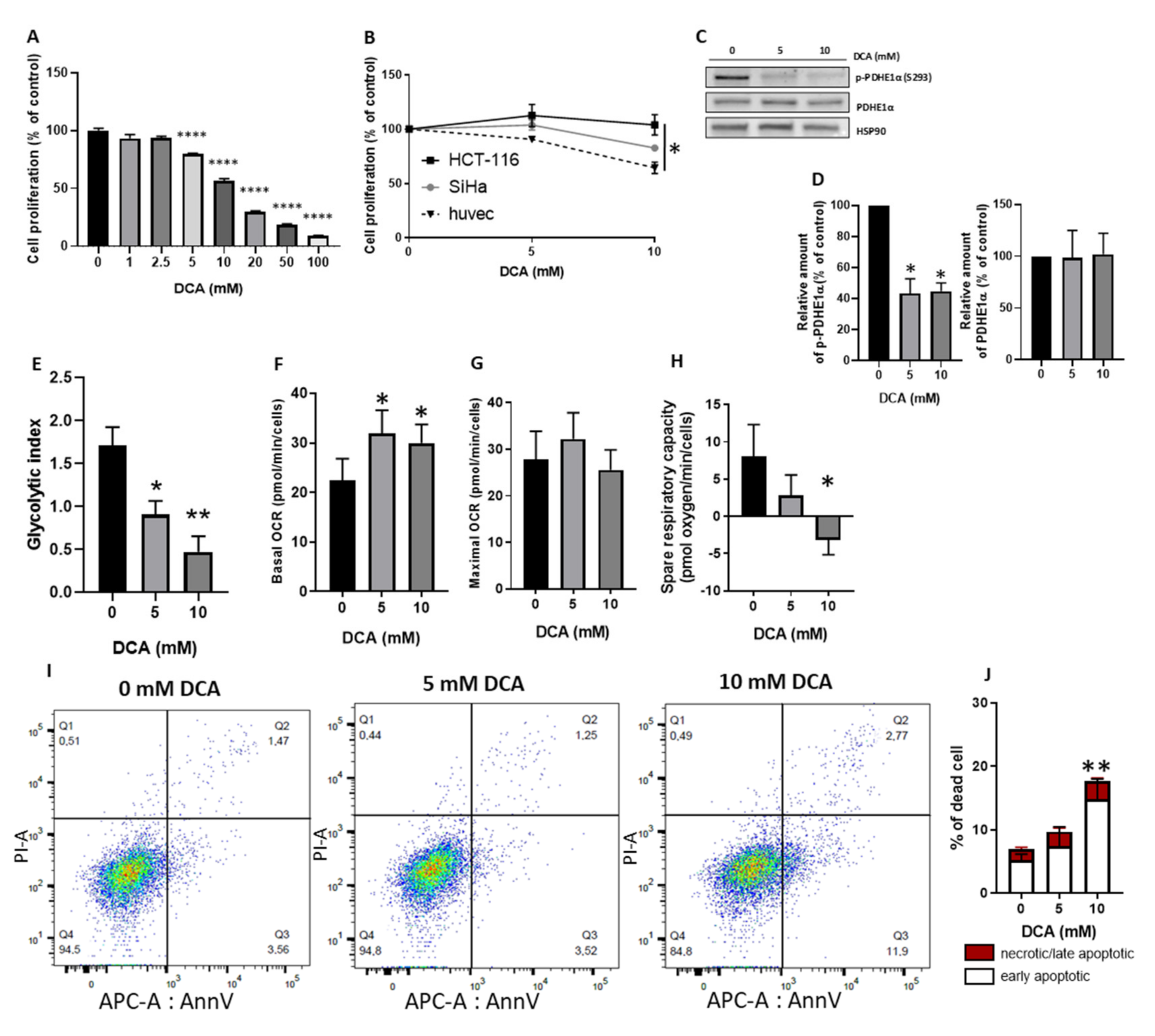{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Section
2.1. Cell Culture and Reagents
- rh VEGF: 5 ng/mL
- rh EGF: 5 ng/mL
- rh FGF basic: 5 ng/mL
- rh IGF-1: 15 ng/mL
- l-glutamine: 10 mM
- Heparin sulfate: 0.75 Units/mL
- Hydrocortisone: 1 µg/mL
- Ascorbic acid: 50 µg/mL
- Fetal bovine serum: 2%
2.2. Cell Proliferation
2.3. Cell Migration
2.4. Tube Formation
2.5. Western Blotting Analyses of PDH Phosphorylation
2.6. 13C NMR Spectroscopy Quantification of Metabolites
2.7. Oxygen Consumption Rate
2.8. Flow Cytometry Measurement of Apoptosis
2.9. Statistical Analysis
3. Results
3.1. DCA Reduces HUVECs Proliferation Similarly to Cancer Cells and Induces a Metabolic Shift from Glycolysis to Mitochondrial Respiration through PDH Activation
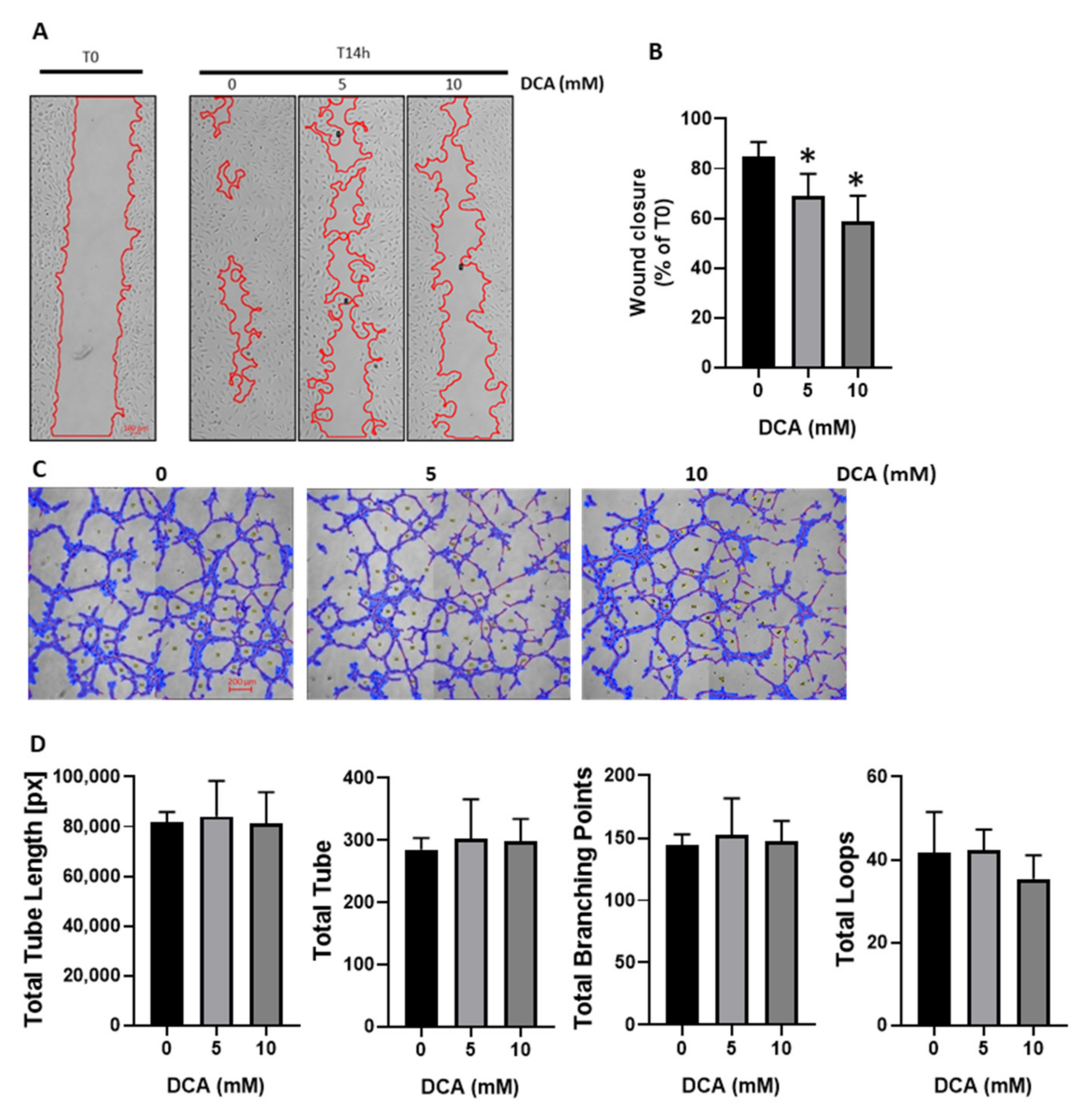
3.2. DCA Reduces HUVECs Migration without Impacting Tube Formation
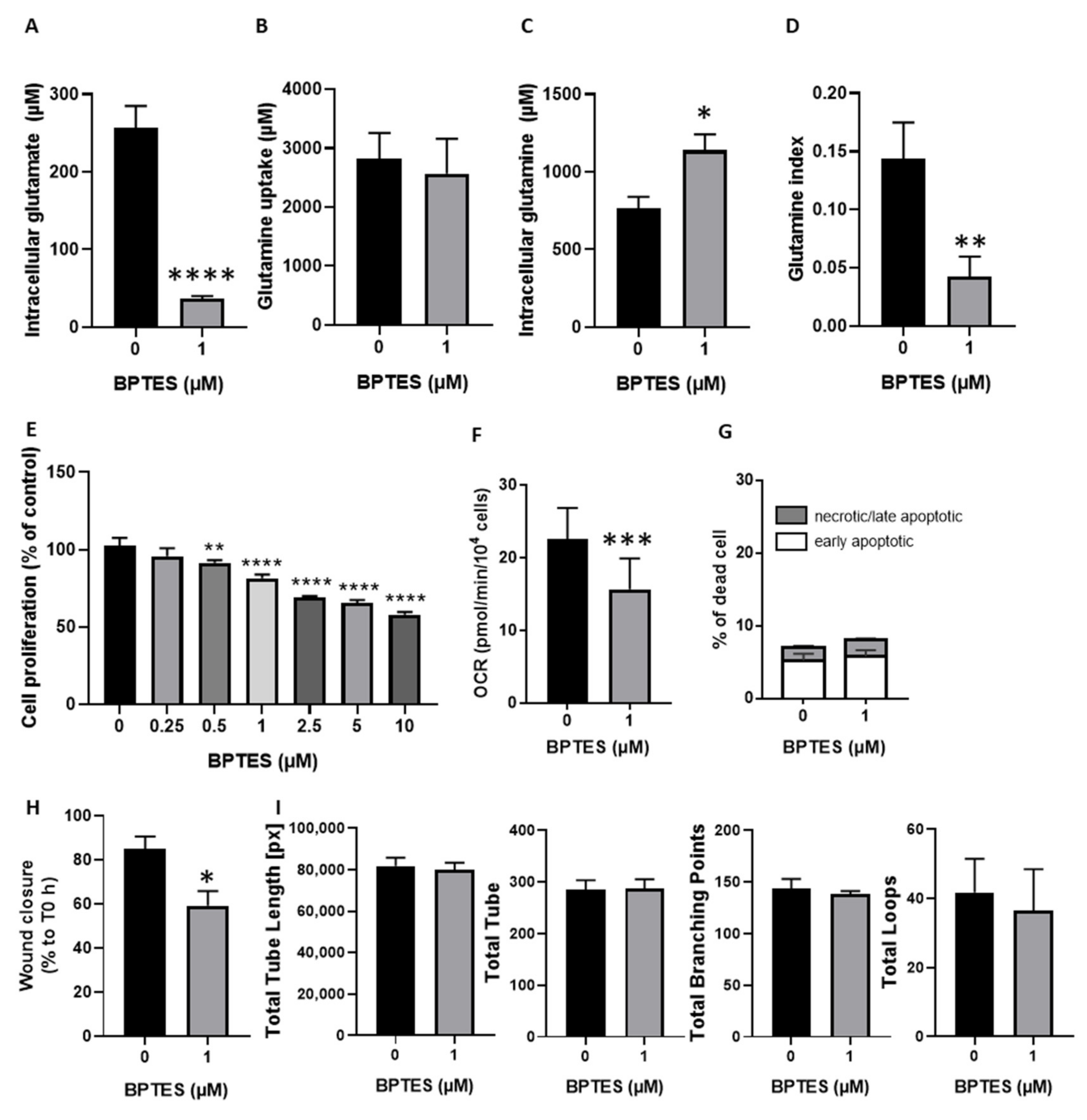
3.3. Inhibition of Glutamine Conversion to Glutamate Reduces Huvecs Proliferation, Migration and Respiration without Change in Apoptosis Induction and Tube Formation
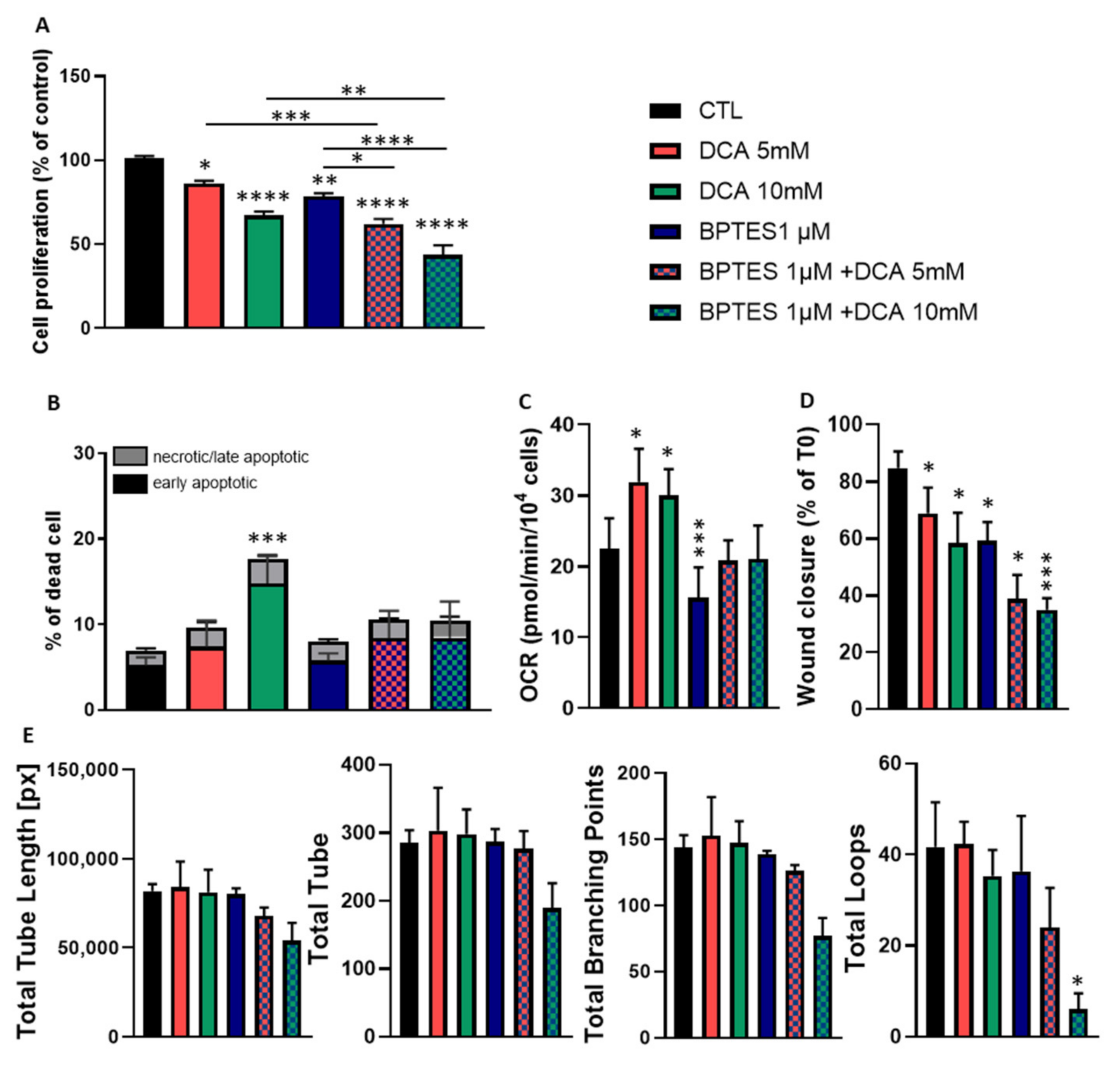
3.4. Combined Exposure to BPTES and DCA Increases the Impact on HUVECs Proliferation and Migration, but Normalize OCR and Apoptosis
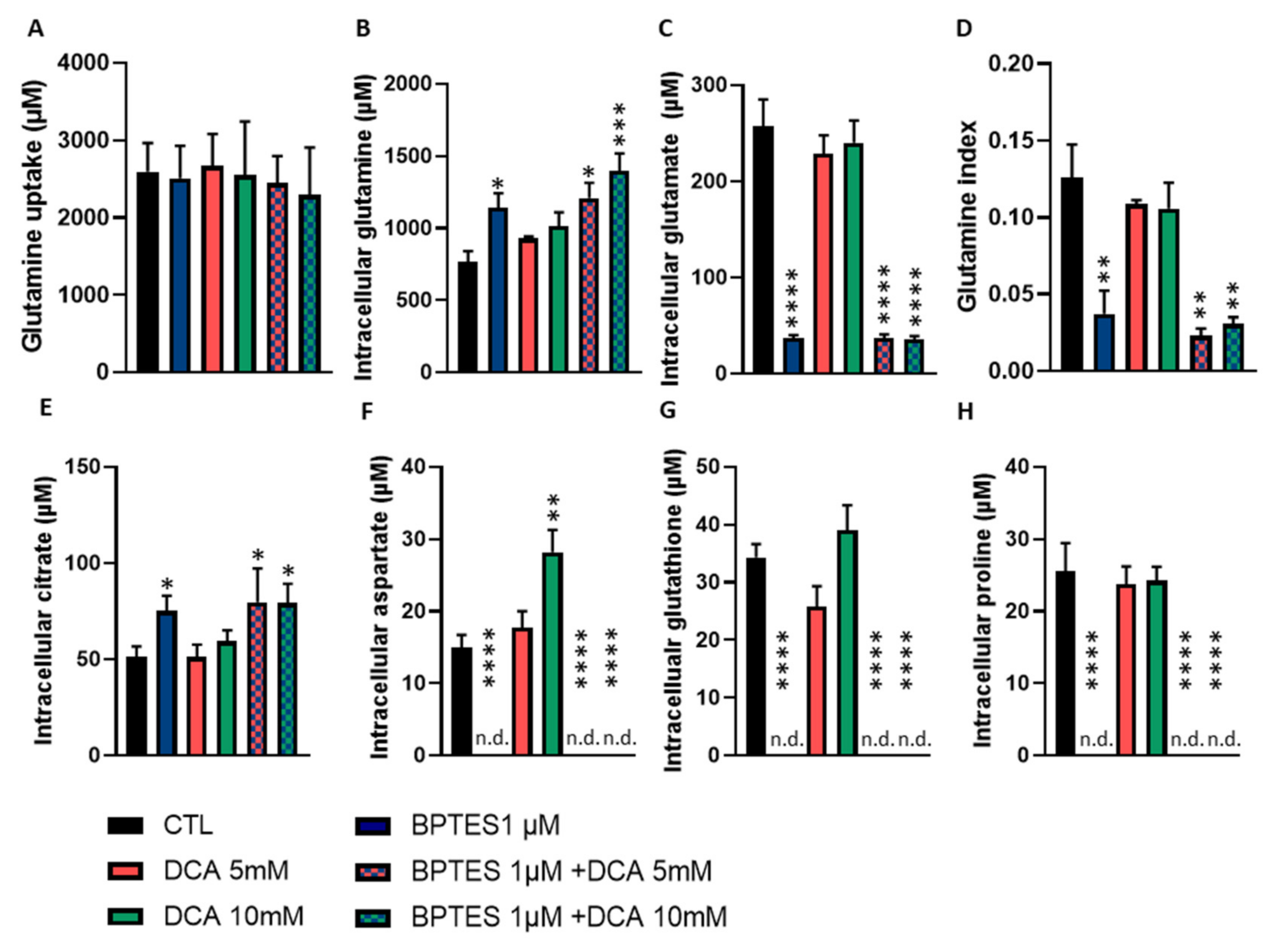
3.5. BPTES and DCA Modulate Glutamine Metabolism and Reveal the Importance of Citrate on HUVECs Homeostasis
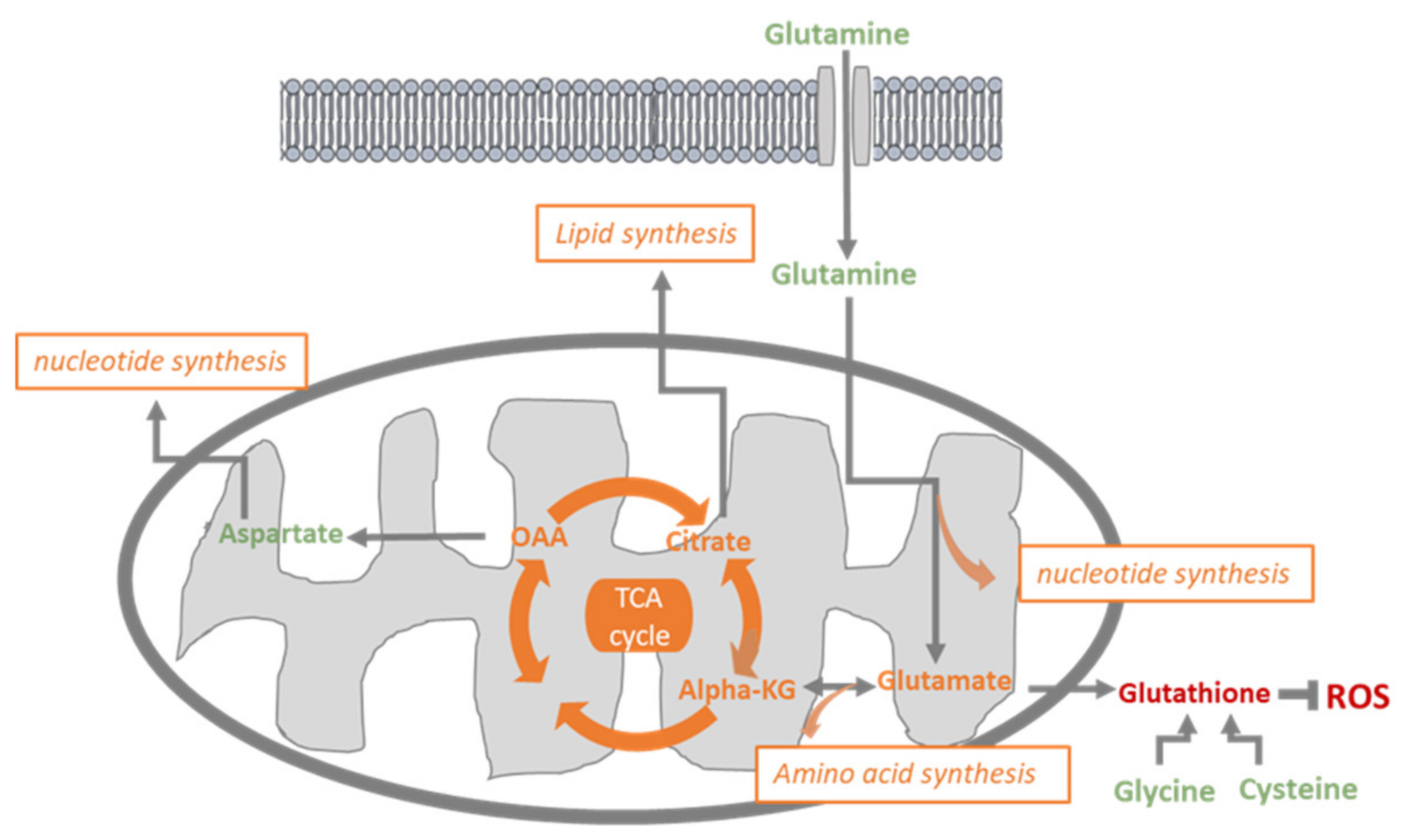
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, G.; Soro-Arnaiz, I.; De Bock, K. The Warburg Effect in Endothelial Cells and its Potential as an Anti-angiogenic Target in Cancer. Front Cell Dev. Biol. 2018, 6, 1002018. [Google Scholar] [CrossRef]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Bruning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef]
- Peyton, K.J.; Liu, X.M.; Yu, Y.; Yates, B.; Behnammanesh, G.; Durante, W. Glutaminase-1 stimulates the proliferation, migration, and survival of human endothelial cells. Biochem. Pharmacol. 2018, 156, 204–214. [Google Scholar] [CrossRef]
- Eelen, G.; Dubois, C.; Cantelmo, A.R.; Goveia, J.; Bruning, U.; DeRan, M.; Jarugumilli, G.; Van Rijssel, J.; Saladino, G.; Comitani, F.; et al. Role of glutamine synthetase in angiogenesis beyond glutamine synthesis. Nature 2018, 561, 63–69. [Google Scholar] [CrossRef]
- Wu, D.; Huang, R.T.; Hamanaka, R.B.; Krause, M.; Oh, M.J.; Kuo, C.H.; Nigdelioglu, R.; Meliton, A.Y.; Witt, L.; Dai, G.; et al. HIF-1alpha is required for disturbed flow-induced metabolic reprogramming in human and porcine vascular endothelium. Elife 2017, 6. [Google Scholar] [CrossRef]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Vegran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frerart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef]
- Sutendra, G.; Michelakis, E.D. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front. Oncol. 2013, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Dromparis, P.; Kinnaird, A.; Stenson, T.H.; Haromy, A.; Parker, J.M.; McMurtry, M.S.; Michelakis, E.D. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene 2013, 32, 1638–1650. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Zhao, X.; Ren, W.; Wang, X.; Yu, K.F.; Li, D.; Zhang, X.; Zhang, Q. Antitumor activity of dichloroacetate on C6 glioma cell: In vitro and in vivo evaluation. Onco. Targets Ther. 2013, 6, 189–198. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Kinnaird, A.; Dromparis, P.; Saleme, B.; Gurtu, V.; Watson, K.; Paulin, R.; Zervopoulos, S.; Stenson, T.; Sutendra, G.; Pink, D.B.; et al. Metabolic Modulation of Clear-cell Renal Cell Carcinoma with Dichloroacetate, an Inhibitor of Pyruvate Dehydrogenase Kinase. Eur. Urol. 2016, 69, 734–744. [Google Scholar] [CrossRef]
- Kankotia, S.; Stacpoole, P.W. Dichloroacetate and cancer: New home for an orphan drug. Biochim. Biophys Acta 2014, 1846, 617–629. [Google Scholar] [CrossRef]
- Dunbar, E.M.; Coats, B.S.; Shroads, A.L.; Langaee, T.; Lew, A.; Forder, J.R.; Shuster, J.J.; Wagner, D.A.; Stacpoole, P.W. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Investig. New Drugs 2014, 32, 452–464. [Google Scholar] [CrossRef]
- Chu, Q.S.; Sangha, R.; Spratlin, J.; Vos, L.J.; Mackey, J.R.; McEwan, A.J.; Venner, P.; Michelakis, E.D. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Investigig. New Drugs 2015, 33, 603–610. [Google Scholar] [CrossRef]
- Schoonjans, C.A.; Joudiou, N.; Brusa, D.; Corbet, C.; Feron, O.; Gallez, B. Acidosis-induced metabolic reprogramming in tumor cells enhances the anti-proliferative activity of the PDK inhibitor dichloroacetate. Cancer Lett. 2020, 470, 18–28. [Google Scholar] [CrossRef]
- De Preter, G.; Neveu, M.A.; Danhier, P.; Brisson, L.; Payen, V.L.; Porporato, P.E.; Jordan, B.F.; Sonveaux, P.; Gallez, B. Inhibition of the pentose phosphate pathway by dichloroacetate unravels a missing link between aerobic glycolysis and cancer cell proliferation. Oncotarget 2016, 7, 2910–2920. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Feron, O.; Belhassen, L.; Kobzik, L.; Smith, T.W.; Kelly, R.A.; Michel, T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J. Biol. Chem. 1996, 271, 22810–22814. [Google Scholar]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Bruning, U.; Morales-Rodriguez, F.; Kalucka, J.; Goveia, J.; Taverna, F.; Queiroz, K.C.S.; Dubois, C.; Cantelmo, A.R.; Chen, R.; Loroch, S.; et al. Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation. Cell Metab. 2018, 28, 866–880. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoonjans, C.A.; Mathieu, B.; Joudiou, N.; Zampieri, L.X.; Brusa, D.; Sonveaux, P.; Feron, O.; Gallez, B. Targeting Endothelial Cell Metabolism by Inhibition of Pyruvate Dehydrogenase Kinase and Glutaminase-1. J. Clin. Med. 2020, 9, 3308. https://doi.org/10.3390/jcm9103308
Schoonjans CA, Mathieu B, Joudiou N, Zampieri LX, Brusa D, Sonveaux P, Feron O, Gallez B. Targeting Endothelial Cell Metabolism by Inhibition of Pyruvate Dehydrogenase Kinase and Glutaminase-1. Journal of Clinical Medicine. 2020; 9(10):3308. https://doi.org/10.3390/jcm9103308
Chicago/Turabian StyleSchoonjans, Céline A., Barbara Mathieu, Nicolas Joudiou, Luca X. Zampieri, Davide Brusa, Pierre Sonveaux, Olivier Feron, and Bernard Gallez. 2020. "Targeting Endothelial Cell Metabolism by Inhibition of Pyruvate Dehydrogenase Kinase and Glutaminase-1" Journal of Clinical Medicine 9, no. 10: 3308. https://doi.org/10.3390/jcm9103308
APA StyleSchoonjans, C. A., Mathieu, B., Joudiou, N., Zampieri, L. X., Brusa, D., Sonveaux, P., Feron, O., & Gallez, B. (2020). Targeting Endothelial Cell Metabolism by Inhibition of Pyruvate Dehydrogenase Kinase and Glutaminase-1. Journal of Clinical Medicine, 9(10), 3308. https://doi.org/10.3390/jcm9103308