Addition of a Viral Immunomodulatory Domain to Etanercept Generates a Bifunctional Chemokine and TNF Inhibitor


Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Cloning of hTNFR2 Fusion Constructs and Generation of Recombinant Baculoviruses
2.3. Expression and Purification of Recombinant Protein Constructs
2.4. TNF-Induced Cytotoxicity Assay
2.5. Cell Migration Assay
2.6. Surface Plasmon Resonance
2.7. Collagen-Induced Arthritis (CIA) Murine Model
3. Results
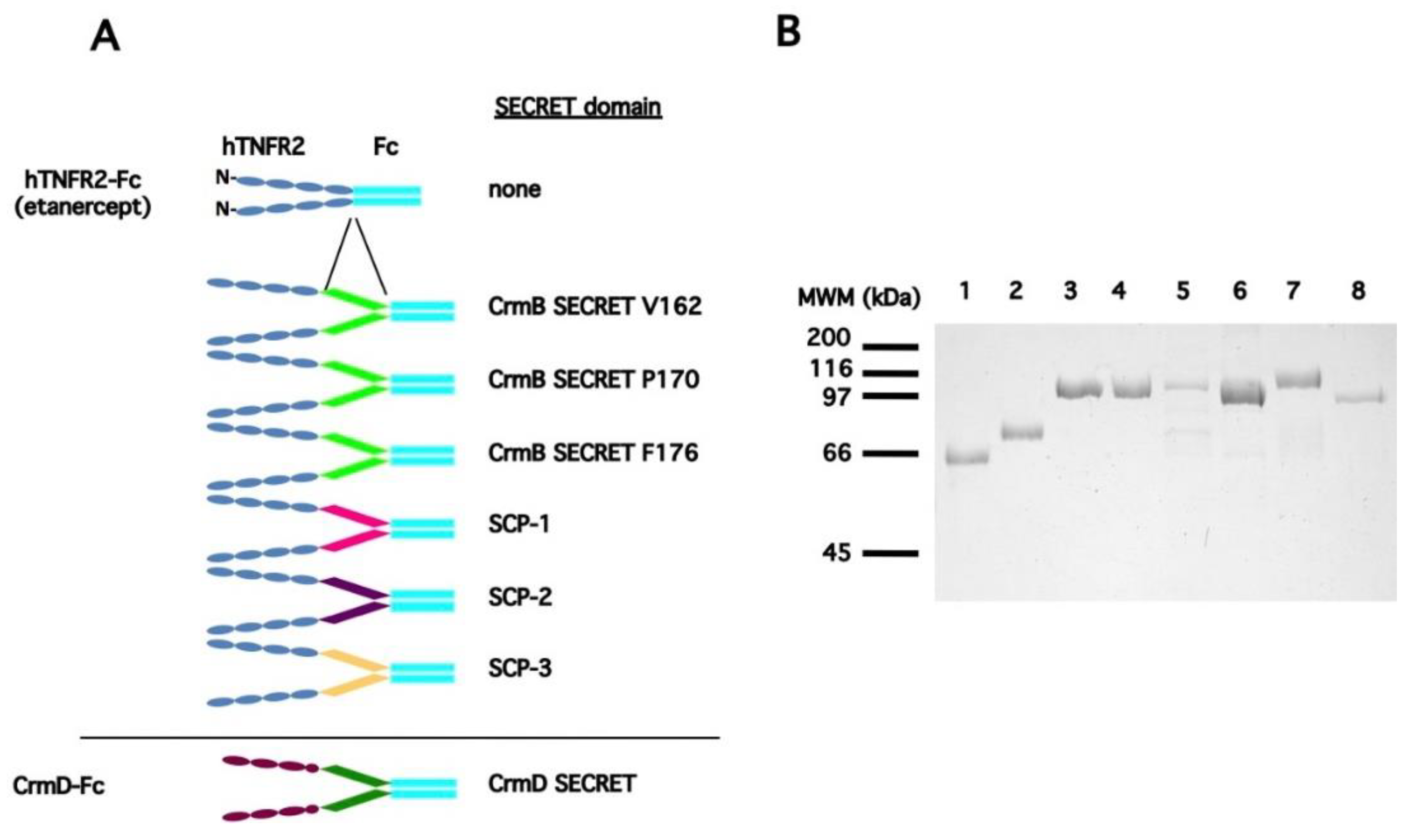
3.1. Generation of a Collection of hTNFR2-SECRET Fusion Proteins
3.2. Analysis of the TNF Inhibitory Activity of the hTNFR2–SECRET–Fc Constructs
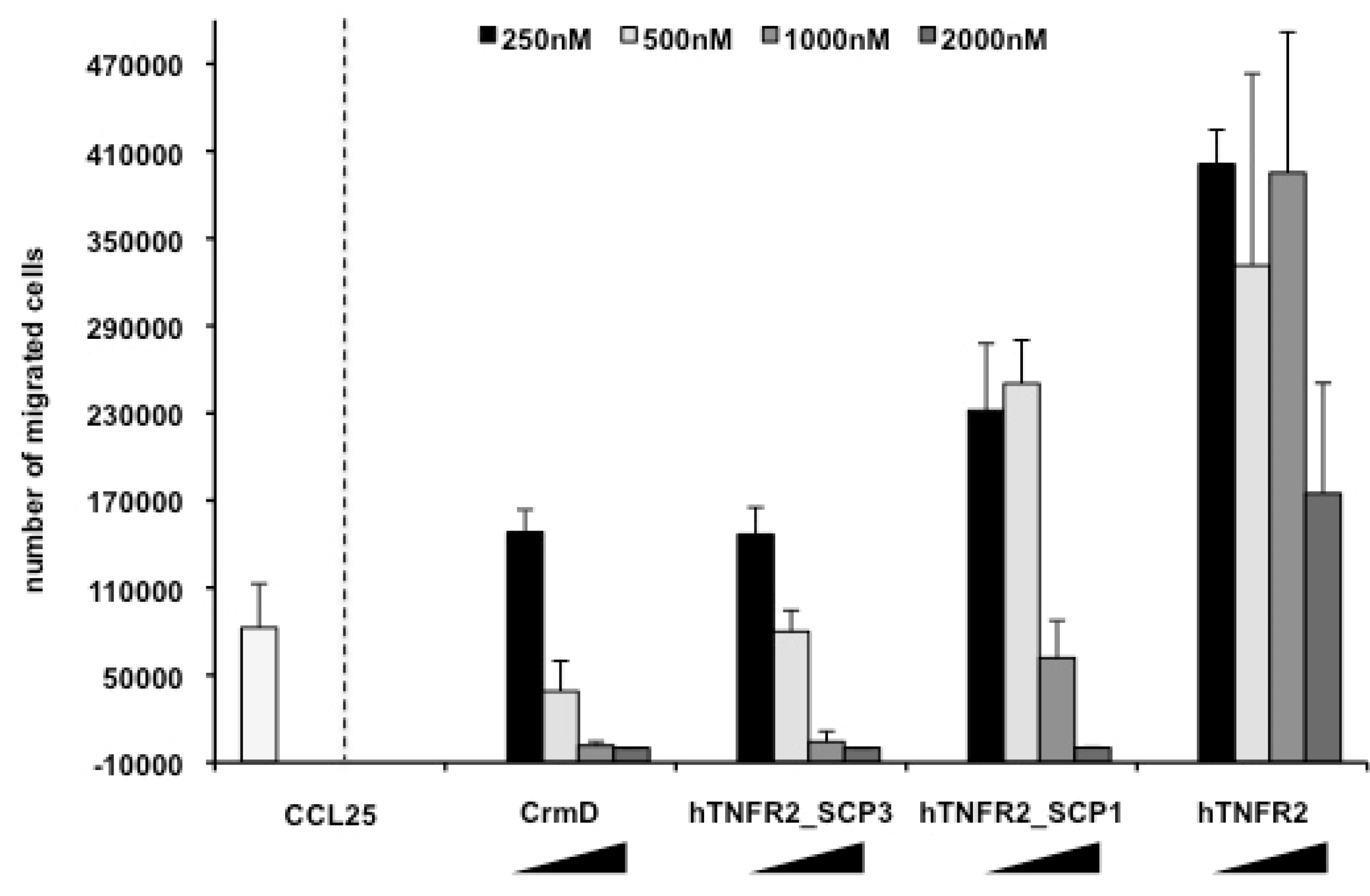
3.3. Chemokine Inhibitory Properties of Fusion Proteins hTNFR2-SCP1 and hTNFR2-SCP3
3.4. The Fusion Protein hTNFR2-SCP3 Binds TNF and Chemokines with High Affinity
3.5. The hTNFR2-SCP3 Fusion Protein Can Delay the Development Clinical Signs Associated with Arthritis in a Murine Model
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alcami, A. Viral mimicry of cytokines, chemokines and their receptors. Nat. Rev. Immunol. 2003, 3, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Benfield, C.T.O.; De Motes, C.M.; Mazzon, M.; Ember, S.W.J.; Ferguson, B.J.; Sumner, R.P. Vaccinia virus immune evasion: Mechanisms, virulence and immunogenicity. J. Gen. Virol. 2013, 94, 2367–2392. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Bowie, A.G. The interplay between viruses and innate immune signaling: Recent insights and therapeutic opportunities. Biochem. Pharmacol. 2008, 75, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Felix, J.; Savvides, S.N. Mechanisms of immunomodulation by mammalian and viral decoy receptors: Insights from structures. Nat. Rev. Immunol. 2017, 17, 112–129. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Simon, A.; Van Der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Dinarello, C.A. Anakinra Therapy for Non-cancer Inflammatory Diseases. Front. Pharmacol. 2018, 9, 1157. [Google Scholar] [CrossRef]
- Bagnasco, D.; Caminati, M.; Ferrando, M.; Aloè, T.; Testino, E.; Canonica, G.W.; Passalacqua, G. Anti-IL-5 and IL-5Ra: Efficacy and Safety of New Therapeutic Strategies in Severe Uncontrolled Asthma. BioMed Res. Int. 2018, 2018, 5698212. [Google Scholar] [CrossRef]
- Chiu, Y.G.; Ritchlin, C.T. Denosumab: Targeting the RANKL pathway to treat rheumatoid arthritis. Expert Opin. Biol. Ther. 2017, 17, 119–128. [Google Scholar] [CrossRef]
- Klonowska, J.; Gleń, J.; Nowicki, R.J.; Trzeciak, M. New Cytokines in the Pathogenesis of Atopic Dermatitis—New Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 3086. [Google Scholar] [CrossRef]
- Patterson, M.F.; Borish, L.; Kennedy, J.L. The past, present, and future of monoclonal antibodies to IL-5 and eosinophilic asthma: A review. J. Asthma Allergy 2015, 8, 125–134. [Google Scholar]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Interleukin (IL-6) Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10, a028456. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.C.; Ortigosa, L.C.; Benard, G. Anti-TNF-alpha agents in the treatment of immune-mediated inflammatory diseases: Mechanisms of action and pitfalls. Immunotherapy 2010, 2, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Udalova, I.; Monaco, C.; Nanchahal, J.; Feldmann, M. Anti-TNF Therapy. Microbiol. Spectr. 2016, 4, 1–11. [Google Scholar]
- Brunetti, C.R.; Paulose-Murphy, M.; Singh, R.; Qin, J.; Barrett, J.W.; Tardivel, A.; Schneider, P.; Essani, K.; McFadden, G. A secreted high-affinity inhibitor of human TNF from Tanapox virus. Proc. Natl. Acad. Sci. USA 2003, 100, 4831–4836. [Google Scholar] [CrossRef] [PubMed]
- Pontejo, S.M.; Alejo, A.; Alcami, A. Comparative Biochemical and Functional Analysis of Viral and Human Secreted Tumor Necrosis Factor (TNF) Decoy Receptors. J. Biol. Chem. 2015, 290, 15973–15984. [Google Scholar] [CrossRef] [PubMed]
- Pontejo, S.M.; Alejo, A.; Alcami, A. Poxvirus-encoded TNF decoy receptors inhibit the biological activity of transmembrane TNF. J. Gen. Virol. 2015, 96, 3118–3123. [Google Scholar] [CrossRef]
- Alejo, A.; Ruiz-Argüello, M.B.; Ho, Y.; Smith, V.P.; Saraiva, M.; Alcami, A. A chemokine-binding domain in the tumor necrosis factor receptor from variola (smallpox) virus. Proc. Natl. Acad. Sci. USA 2006, 103, 5995–6000. [Google Scholar] [CrossRef]
- Xue, X.; Lu, Q.; Wei, H.; Wang, N.; Chen, D.; He, G.; Huang, L.; Wang, H.; Wang, X. Structural Basis of Chemokine Sequestration by CrmD, a Poxvirus-Encoded Tumor Necrosis Factor Receptor. PLoS Pathog. 2011, 7, e1002162. [Google Scholar] [CrossRef]
- Alejo, A.; Ruiz-Argüello, M.B.; Pontejo, S.M.; de Marco, M.D.M.F.; Saraiva, M.; Hernáez, B.; Alcami, A. Chemokines cooperate with TNF to provide protective anti-viral immunity and to enhance inflammation. Nat. Commun. 2018, 9, 1790. [Google Scholar] [CrossRef]
- Hopkin, S.J.; Lewis, J.W.; Krautter, F.; Chimen, M.; McGettrick, H.M. Triggering the Resolution of Immune Mediated Inflammatory Diseases: Can Targeting Leukocyte Migration Be the Answer? Front. Pharmacol. 2019, 10, 184. [Google Scholar] [CrossRef]
- Saraiva, M.; Alcami, A. CrmE, a Novel Soluble Tumor Necrosis Factor Receptor Encoded by Poxviruses. J. Virol. 2001, 75, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Pontejo, S.M.; Sanchez, C.; Ruiz-Argüello, B.; Alcami, A. Insights into ligand binding by a viral tumor necrosis factor (TNF) decoy receptor yield a selective soluble human type 2 TNF receptor. J. Biol. Chem. 2019, 294, 5214–5227. [Google Scholar] [CrossRef] [PubMed]
- Caplazi, P.; Baca, M.; Barck, K.; Carano, R.A.D.; Devoss, J.; Lee, W.P.; Bolon, B.; Diehl, L. Mouse Models of Rheumatoid Arthritis. Vet. Pathol. 2015, 52, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, H.; Xu, P.; Yin, Q.; Wang, Y.; Opoku, Y.K.; Yang, J.; Song, L.; Sun, X.; Zhang, T.; et al. Ameliorative effects of a fusion protein dual targeting interleukin 17A and tumor necrosis factor alpha on imiquimod-induced psoriasis in mice. Biomed. Pharmacother. 2018, 108, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Song, L.; Wang, Y.; Xu, P.; Guo, X.; Yang, J.; Liu, H.; Wang, Y.; Wu, C.; Zhang, T.; et al. A novel fusion protein attenuates collagen-induced arthritis by targeting interleukin 17A and tumor necrosis factor alpha. Int. J. Pharm. 2018, 547, 72–82. [Google Scholar] [CrossRef]
- Grell, M.; Wajant, H.; Zimmermann, G.; Scheurich, P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc. Natl. Acad. Sci. USA 1998, 95, 570–575. [Google Scholar] [CrossRef]
- Lee, A.W.; Deruaz, M.; Lynch, C.; Davies, G.; Singh, K.; Alenazi, Y.; Eaton, J.R.O.; Kawamura, A.; Shaw, J.; Proudfoot, A.E.I.; et al. A knottin scaffold directs the CXC-chemokine–binding specificity of tick evasins. J. Biol. Chem. 2019, 294, 11199–11212. [Google Scholar] [CrossRef]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Szekanecz, Z.; Koch, A.E. Successes and failures of chemokine-pathway targeting in rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 5–13. [Google Scholar] [CrossRef]
- Alcami, A.; Lira, S.A. Modulation of chemokine activity by viruses. Curr. Opin. Immunol. 2010, 22, 482–487. [Google Scholar] [CrossRef]
- Parry, C.M.; Simas, J.P.; Smith, V.P.; Stewart, C.A.; Minson, A.C.; Efstathiou, S.; Alcami, A. A Broad Spectrum Secreted Chemokine Binding Protein Encoded by a Herpesvirus. J. Exp. Med. 2000, 191, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Van Berkel, V.; Barrett, J.; Tiffany, H.L.; Fremont, D.H.; Murphy, P.M.; McFadden, G.; Speck, S.H.; Virgin, H.W. Identification of a Gammaherpesvirus Selective Chemokine Binding Protein That Inhibits Chemokine Action. J. Virol. 2000, 74, 6741–6747. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, A.; Stevenson, P.G.; Simas, J.P.; Efstathiou, S. A Secreted Chemokine Binding Protein Encoded by Murine Gammaherpesvirus-68 Is Necessary for the Establishment of a Normal Latent Load. J. Exp. Med. 2001, 194, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Bursill, C.A.; Choudhury, R.P.; Ali, Z.; Greaves, D.R.; Channon, K.M. Broad-Spectrum CC-Chemokine Blockade by Gene Transfer Inhibits Macrophage Recruitment and Atherosclerotic Plaque Formation in Apolipoprotein E–Knockout Mice. Circulation 2004, 110, 2460–2466. [Google Scholar] [CrossRef] [PubMed]
- Ridiandries, A.; Ravindran, D.; Lindsay, L.; Hawkins, C.; Tan, J.T.M.; Williams, H.; Medbury, H.J.; Prosser, H.C.G.; Bursill, C.A. CC-chemokine class inhibition attenuates pathological angiogenesis while preserving physiological angiogenesis. FASEB J. 2017, 31, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- Buatois, V.; Fagète, S.; Magistrelli, G.; Chatel, L.; Fischer, N.; Kosco-Vilbois, M.H.; Ferlin, W.G. Pan–CC Chemokine Neutralization Restricts Splenocyte Egress and Reduces Inflammation in a Model of Arthritis. J. Immunol. 2010, 185, 2544–2554. [Google Scholar] [CrossRef] [PubMed]
- Lucas, A.; Yaron, J.R.; Zhang, L.; Macaulay, C.; McFadden, G. Serpins: Development for Therapeutic Applications. Methods Mol. Biol. 2018, 1826, 255–265. [Google Scholar]
- Kwiecien, J.M.; Dabrowski, W.; Marzec-Kotarska, B.; Kwiecien-Delaney, C.J.; Yaron, J.; Zhang, L.; Schutz, L.; Lucas, A.R. Myxoma virus derived immune modulating proteins, M-T7 and Serp-1, reduce early inflammation after spinal cord injury in the rat model. Folia Neuropathol. 2019, 57, 41–50. [Google Scholar] [CrossRef]
- Mitoma, H.; Horiuchi, T.; Tsukamoto, H.; Ueda, N. Molecular mechanisms of action of anti-TNF-alpha agents—Comparison among therapeutic TNF-alpha antagonists. Cytokine 2018, 101, 56–63. [Google Scholar] [CrossRef]
- Viejo-Borbolla, A.; Martin, A.P.; Muniz, L.R.; Shang, L.; Marchesi, F.; Thirunarayanan, N.; Harpaz, N.; Garcia, R.A.; Apostolaki, M.; Furtado, G.C.; et al. Attenuation of TNF-driven murine ileitis by intestinal expression of the viral immunomodulator CrmD. Mucosal Immunol. 2010, 3, 633–644. [Google Scholar] [CrossRef]
- López-Cotarelo, P.; Gómez-Moreira, C.; Criado-García, O.; Sánchez, L.; Rodríguez-Fernández, J.L. Beyond Chemoattraction: Multifunctionality of Chemokine Receptors in Leukocytes. Trends Immunol. 2017, 38, 927–941. [Google Scholar] [CrossRef] [PubMed]
- Nanki, T.; Hayashida, K.; El-Gabalawy, H.S.; Suson, S.; Shi, K.; Girschick, H.J.; Yavuz, S.; Lipsky, P.E. Stromal cell-derived factor-1-CXC chemokine receptor 4 interactions play a central role in CD4+ T cell accumulation in rheumatoid arthritis synovium. J. Immunol. 2000, 165, 6590–6598. [Google Scholar] [CrossRef] [PubMed]
- Nanki, T.; Nagasaka, K.; Hayashida, K.; Saita, Y.; Miyasaka, N. Chemokines regulate IL-6 and IL-8 production by fibroblast-like synoviocytes from patients with rheumatoid arthritis. J. Immunol. 2001, 167, 5381–5385. [Google Scholar] [CrossRef] [PubMed]
- Pablos, J.L.; Santiago, B.; Galindo, M.; Torres, C.; Brehmer, M.T.; Blanco, F.J.; García-Lázaro, F.J. Synoviocyte-derived CXCL12 is displayed on endothelium and induces angiogenesis in rheumatoid arthritis. J. Immunol. 2003, 170, 2147–2152. [Google Scholar] [CrossRef]
- Chen, Z.; Kim, S.J.; Essani, A.B.; Volin, M.V.; Vila, O.M.; Swedler, W.; Arami, S.; Volkov, S.; Sardin, L.V.; Sweiss, N.; et al. Characterising the expression and function of CCL28 and its corresponding receptor, CCR10, in RA pathogenesis. Ann. Rheum. Dis. 2015, 74, 1898–1906. [Google Scholar] [CrossRef]
- Finch, N.K.; Ettinger, R.; Karnell, J.L.; Herbst, R.; Sleeman, M.A. Effects of CXCL13 inhibition on lymphoid follicles in models of autoimmune disease. Eur. J. Clin. Investig. 2013, 43, 501–509. [Google Scholar] [CrossRef]
- Zhong, C.; Wang, J.; Li, B.; Xiang, H.; Ultsch, M.; Coons, M.; Wong, T.; Chiang, N.Y.; Clark, S.; Clark, R.; et al. Development and Preclinical Characterization of a Humanized Antibody Targeting CXCL. Clin. Cancer Res. 2013, 19, 4433–4445. [Google Scholar] [CrossRef]
- Nanki, T.; Urasaki, Y.; Imai, T.; Nishimura, M.; Muramoto, K.; Kubota, T.; Miyasaka, N. Inhibition of fractalkine ameliorates murine collagen-induced arthritis. J. Immunol. 2004, 173, 7010–7016. [Google Scholar] [CrossRef]
- Lee, A.Y.; Körner, H. CCR6 and CCL20: Emerging players in the pathogenesis of rheumatoid arthritis. Immunol. Cell Biol. 2014, 92, 354–358. [Google Scholar] [CrossRef]
- Hirota, K.; Yoshitomi, H.; Hashimoto, M.; Maeda, S.; Teradaira, S.; Sugimoto, N.; Yamaguchi, T.; Nomura, T.; Ito, H.; Nakamura, T.; et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J. Exp. Med. 2007, 204, 2803–2812. [Google Scholar] [CrossRef]
- Yokoyama, W.; Kohsaka, H.; Kaneko, K.; Walters, M.; Takayasu, A.; Fukuda, S.; Miyabe, C.; Miyabe, Y.; Love, P.E.; Nakamoto, N.; et al. Abrogation of CC chemokine receptor 9 ameliorates collagen-induced arthritis of mice. Arthritis Res. Ther. 2014, 16, 445. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a Protein Expressed | b SECRET Domain | Predicted pI/MWof the Secreted Protein |
|---|---|---|
| CrmD | na | 5.73/60053.42 |
| hTNFR2 | none | 6.89/52797.53 |
| hTNFR2-CrmD SECRET N181 * | ECTV_CrmD N181-D320 | 5.74/68594.08 |
| hTNFR2-CrmD SECRET P153 * | ECTV_CrmD P153-D320 | 5.62/71693.57 |
| hTNFR2-CrmD SECRET F163 * | ECTV_CrmD F163-D320 | 5.68/70650.41 |
| hTNFR2-CrmB SECRET V162 | CPXV_CrmB V162-L355 | 5.80/74763.68 |
| hTNFR2-CrmB SECRET P170 | CPXV_CrmB P170-L355 | 5.86/73915.75 |
| hTNFR2-CrmB SECRET F176 | CPXV_CrmB F176-L355 | 5.86/73320.09 |
| hTNFR2-SCP1 | CPXV_V218 S19-G193 | 6.16/73188.66 |
| hTNFR-SCP2 | ECTV_E12 N22-N202 | 5.54/73124.16 |
| hTNFR-SCP3 | ECTV_E184 Y18-F181 | 6.22/72356.33 |
| hTNFR2_SCP3 | a CrmD | a hTNFR2 | |||
|---|---|---|---|---|---|
| Ligand | ka b ± SE × 105 (1/Ms) | kd c ± SE × 10−3 (1/s) | KD d (nM) | KD (nM) | KD (nM) |
| hTNF | 31.78 ± 0.25 | 0.751 ± 0.02 | 0.23 | 0.41 | 0.28 |
| hCCL25 | 5.54 ± 0.35 | 3.74 ± 0.1.8 | 6.75 | 4.86 | - |
| hCXCL12 | 1.66 ± 0.1 | 8.25 ± 0.2 | 49.76 | 16.6 | - |
| hCXCL13 | 0.72 ± 0.04 | 1.55 ± 0.08 | 21.40 | 13.2 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alejo, A.; Sánchez, C.; Amu, S.; Fallon, P.G.; Alcamí, A. Addition of a Viral Immunomodulatory Domain to Etanercept Generates a Bifunctional Chemokine and TNF Inhibitor. J. Clin. Med. 2020, 9, 25. https://doi.org/10.3390/jcm9010025
Alejo A, Sánchez C, Amu S, Fallon PG, Alcamí A. Addition of a Viral Immunomodulatory Domain to Etanercept Generates a Bifunctional Chemokine and TNF Inhibitor. Journal of Clinical Medicine. 2020; 9(1):25. https://doi.org/10.3390/jcm9010025
Chicago/Turabian StyleAlejo, Alí, Carolina Sánchez, Sylvie Amu, Padraic G. Fallon, and Antonio Alcamí. 2020. "Addition of a Viral Immunomodulatory Domain to Etanercept Generates a Bifunctional Chemokine and TNF Inhibitor" Journal of Clinical Medicine 9, no. 1: 25. https://doi.org/10.3390/jcm9010025
APA StyleAlejo, A., Sánchez, C., Amu, S., Fallon, P. G., & Alcamí, A. (2020). Addition of a Viral Immunomodulatory Domain to Etanercept Generates a Bifunctional Chemokine and TNF Inhibitor. Journal of Clinical Medicine, 9(1), 25. https://doi.org/10.3390/jcm9010025