Loss of Fibroblast-Dependent Androgen Receptor Activation in Prostate Cancer Cells is Involved in the Mechanism of Acquired Resistance to Castration

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Indirect Coculture of Prostate Cancer Cell Lines (E9, F10, and AIDL Cells) with Fibroblasts
2.4. Stimulation of Cell Growth by Treatment with Growth Factors and Cytokines
2.5. Enzyme-Linked Immunosorbent Assay
2.6. Preparation of Cell Lysates
2.7. Western Blot Analyses
2.8. Animal Studies
2.9. In Vivo Xenograft Model
2.10. Histopathology and Immunohistochemistry
2.11. Statistical Analysis
3. Results
3.1. Effects of ADT on Tumor Growth and Serum PSA Kinetics of Xenograft Derived from Co-Inoculation of E9, F10, and AIDL Cells with pcPrF-M5 Cells In Vivo
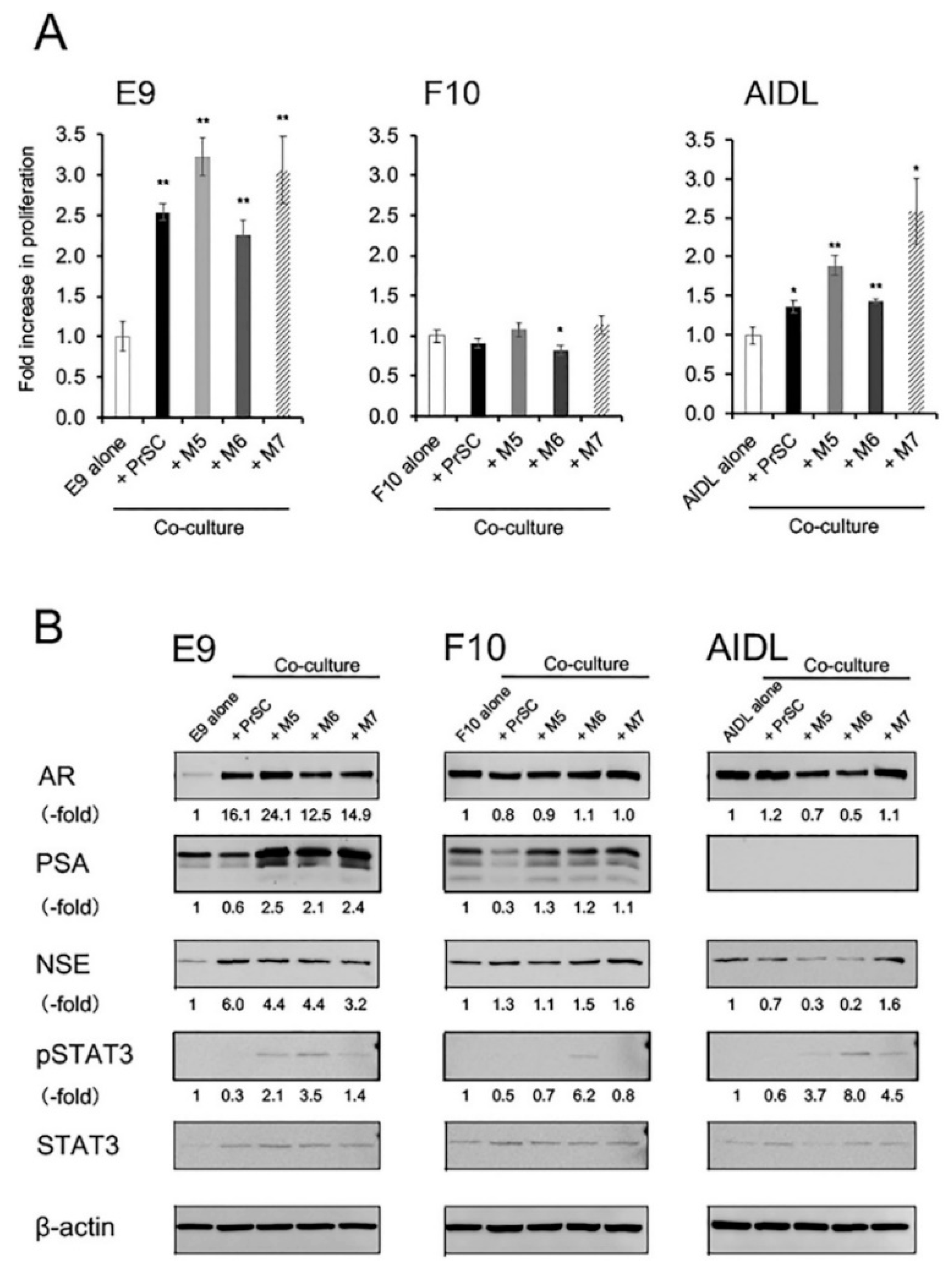
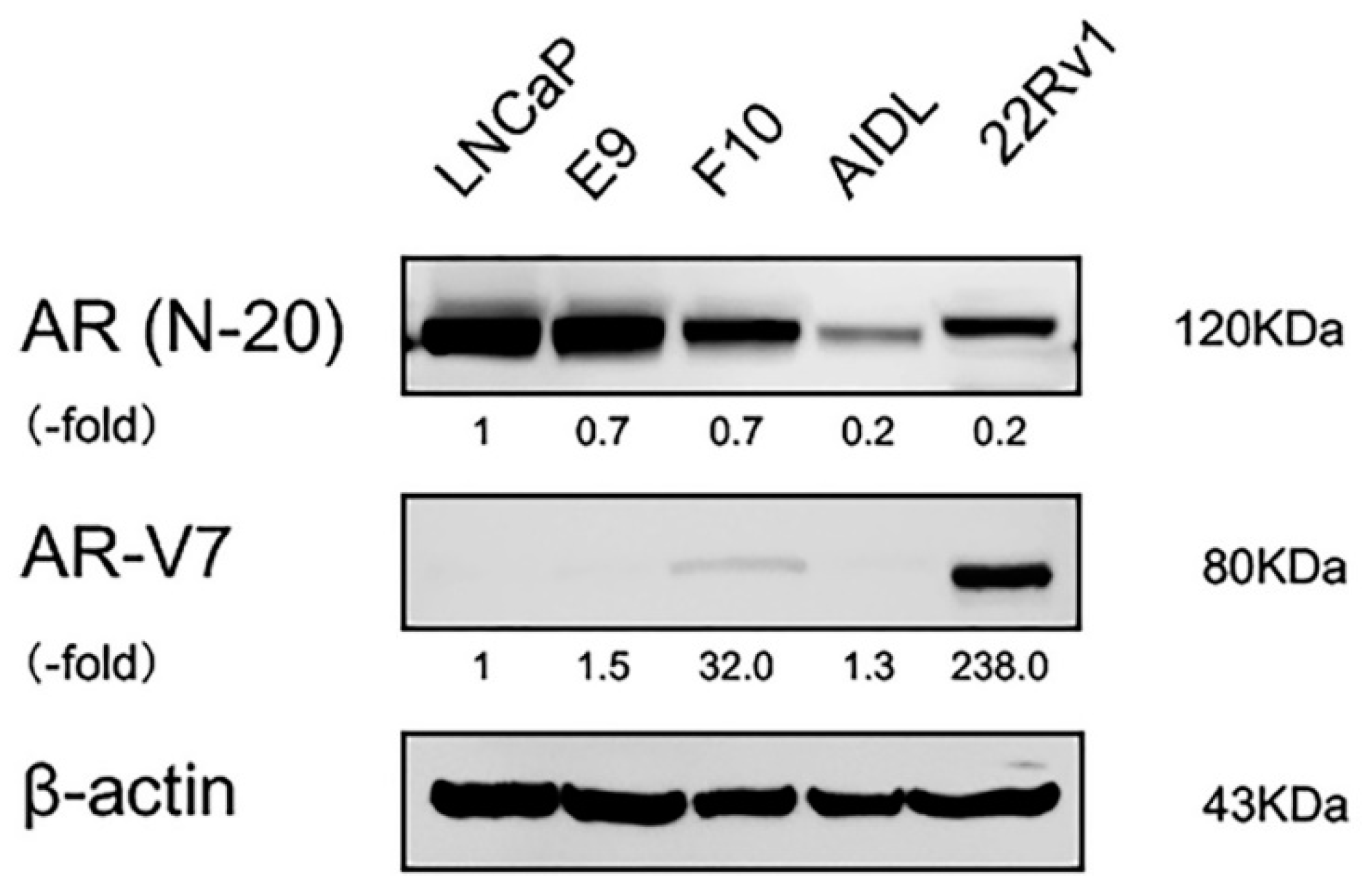
3.2. Effects of Indirect Coculture with Fibroblasts on E9, F10, and AIDL Cells In Vitro
3.3. Effects of Growth Factors and Cytokines on E9, F10, and AIDL Cells In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gronberg, H. Prostate cancer epidemiology. Lancet 2003, 361, 859–864. [Google Scholar] [CrossRef]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J. Urol. 2002, 168, 9–12. [Google Scholar] [CrossRef]
- Fizazi, K.; Higano, C.S.; Nelson, J.B.; Gleave, M.; Miller, K.; Morris, T.; Nathan, F.E.; McIntosh, S.; Pemberton, K.; Moul, J.W. Phase III, randomized, placebo-controlled study of docetaxel in combination with zibotentan in patients with metastatic castration-resistant prostate cancer. J. Clin. Oncol 2013, 31, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Taplin, M.E.; Ho, S.M. Clinical review 134: The endocrinology of prostate cancer. J. Clin. Endocrinol. Metab. 2001, 86, 3467–3477. [Google Scholar] [CrossRef] [PubMed][Green Version]
- So, A.; Gleave, M.; Hurtado-Col, A.; Nelson, C. Mechanisms of the development of androgen independence in prostate cancer. World J. Urol. 2005, 23, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z.; Klocker, H.; Bartsch, G.; Hobisch, A. Androgen receptors in prostate cancer. Endocr. Relat. Cancer 2002, 9, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Jennbacken, K.; Tesan, T.; Wang, W.; Gustavsson, H.; Damber, J.E.; Welen, K. N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocr. Relat. Cancer 2010, 17, 469–479. [Google Scholar] [CrossRef]
- Nelson, P.S. Molecular states underlying androgen receptor activation: A framework for therapeutics targeting androgen signaling in prostate cancer. J. Clin. Oncol. 2012, 30, 644–646. [Google Scholar] [CrossRef]
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Radmayr, C.; Trapman, J.; Hittmair, A.; Bartsch, G.; Klocker, H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994, 54, 5474–5478. [Google Scholar] [CrossRef]
- Ueda, T.; Mawji, N.R.; Bruchovsky, N.; Sadar, M.D. Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. J. Biol. Chem. 2002, 277, 38087–38094. [Google Scholar] [CrossRef]
- Kim, J.; Coetzee, G.A. Prostate specific antigen gene regulation by androgen receptor. J. Cell Biochem. 2004, 93, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.L.; Kyprianou, N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr. Relat. Cancer 2008, 15, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Mizokami, A.; Tsunoda, T.; Iguchi, K.; Kato, M.; Hori, Y.; Arima, K.; Namiki, M.; Sugimura, Y. Heterogenous induction of carcinoma-associated fibroblast-like differentiation in normal human prostatic fibroblasts by co-culturing with prostate cancer cells. J. Cell Biochem. 2011, 112, 3604–3611. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Sasaki, T.; Iguchi, K.; Kajiwara, S.; Kato, M.; Kanda, H.; Hirokawa, Y.; Arima, K.; Mizokami, A.; Sugimura, Y. Interleukin-6 induces VEGF secretion from prostate cancer cells in a manner independent of androgen receptor activation. Prostate 2018, 78, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Tanner, M.J.; Welliver, R.C., Jr.; Chen, M.; Shtutman, M.; Godoy, A.; Smith, G.; Mian, B.M.; Buttyan, R. Effects of androgen receptor and androgen on gene expression in prostate stromal fibroblasts and paracrine signaling to prostate cancer cells. PLoS ONE 2011, 6, e16027. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Mancini, A.; Ranieri, G.; Di Pasquale, B.; Marampon, F.; Di Clemente, L.; Ricevuto, E.; Festuccia, C. Phenotypic characterization of human prostatic stromal cells in primary cultures derived from human tissue samples. Int. J. Oncol. 2013, 42, 2116–2122. [Google Scholar] [CrossRef]
- Sasaki, T.; Ishii, K.; Iwamoto, Y.; Kato, M.; Miki, M.; Kanda, H.; Arima, K.; Shiraishi, T.; Sugimura, Y. Fibroblasts prolong serum prostate-specific antigen decline after androgen deprivation therapy in prostate cancer. Lab. Invest. 2016, 96, 338–349. [Google Scholar] [CrossRef]
- Halin, S.; Hammarsten, P.; Wikstrom, P.; Bergh, A. Androgen-insensitive prostate cancer cells transiently respond to castration treatment when growing in an androgen-dependent prostate environment. Prostate 2007, 67, 370–377. [Google Scholar] [CrossRef]
- Ishii, K.; Imamura, T.; Iguchi, K.; Arase, S.; Yoshio, Y.; Arima, K.; Hirano, K.; Sugimura, Y. Evidence that androgen-independent stromal growth factor signals promote androgen-insensitive prostate cancer cell growth in vivo. Endocr. Relat. Cancer 2009, 16, 415–428. [Google Scholar] [CrossRef]
- Horoszewicz, J.S.; Leong, S.S.; Chu, T.M.; Wajsman, Z.L.; Friedman, M.; Papsidero, L.; Kim, U.; Chai, L.S.; Kakati, S.; Arya, S.K.; et al. The LNCaP cell line--a new model for studies on human prostatic carcinoma. Prog. Clin. Biol. Res. 1980, 37, 115–132. [Google Scholar]
- Wan, X.S.; Zhou, Z.; Steele, V.; Kopelovich, L.; Kennedy, A.R. Establishment and characterization of sublines of LNCaP human prostate cancer cells. Oncol. Rep. 2003, 10, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, K.; Ishii, K.; Nakano, T.; Otsuka, T.; Usui, S.; Sugimura, Y.; Hirano, K. Isolation and characterization of LNCaP sublines differing in hormone sensitivity. J. Androl. 2007, 28, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, K.; Hayakawa, Y.; Ishii, K.; Matsumoto, K.; Usui, S.; Sugimura, Y.; Hirano, K. Characterization of the low pH/low nutrient-resistant LNCaP cell subline LNCaP-F10. Oncol. Rep. 2012, 28, 2009–2015. [Google Scholar] [CrossRef] [PubMed]
- Onishi, T.; Yamakawa, K.; Franco, O.E.; Kawamura, J.; Watanabe, M.; Shiraishi, T.; Kitazawa, S. Mitogen-activated protein kinase pathway is involved in alpha6 integrin gene expression in androgen-independent prostate cancer cells: Role of proximal Sp1 consensus sequence. Biochim. Biophys. Acta 2001, 1538, 218–227. [Google Scholar] [CrossRef]
- Titus, M.A.; Schell, M.J.; Lih, F.B.; Tomer, K.B.; Mohler, J.L. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin. Cancer Res. 2005, 11, 4653–4657. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Sugimura, Y. Identification of a new pharmacological activity of the phenylpiperazine derivative naftopidil: Tubulin-binding drug. J. Chem. Biol. 2015, 8, 5–9. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ishii, K.; Matsuoka, I.; Kajiwara, S.; Sasaki, T.; Miki, M.; Kato, M.; Kanda, H.; Arima, K.; Shiraishi, T.; Sugimura, Y. Additive naftopidil treatment synergizes docetaxel-induced apoptosis in human prostate cancer cells. J. Cancer Res. Clin. Oncol. 2018, 144, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Craft, N.; Chhor, C.; Tran, C.; Belldegrun, A.; DeKernion, J.; Witte, O.N.; Said, J.; Reiter, R.E.; Sawyers, C.L. Evidence for clonal outgrowth of androgen-independent prostate cancer cells from androgen-dependent tumors through a two-step process. Cancer Res. 1999, 59, 5030–5036. [Google Scholar]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef]
- Dutt, S.S.; Gao, A.C. Molecular mechanisms of castration-resistant prostate cancer progression. Future Oncol. 2009, 5, 1403–1413. [Google Scholar] [CrossRef]
- Tombal, B. What is the pathophysiology of a hormone-resistant prostate tumour? Eur. J. Cancer 2011, 47, S179–S188. [Google Scholar] [CrossRef]
- Yadav, S.S.; Stockert, J.A.; Hackert, V.; Yadav, K.K.; Tewari, A.K. Intratumor heterogeneity in prostate cancer. Urol. Oncol. 2018, 36, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, K.; Fukami, K.; Ishii, K.; Otsuka, T.; Usui, S.; Sugimura, Y.; Hirano, K. Low androgen sensitivity is associated with low levels of Akt phosphorylation in LNCaP-E9 cells. J. Androl. 2012, 33, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.K.; Hu, Y.C.; Yang, L.; Altuwaijri, S.; Chen, Y.T.; Kang, H.Y.; Chang, C. Suppression versus induction of androgen receptor functions by the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer LNCaP cells with different passage numbers. J. Biol. Chem. 2003, 278, 50902–50907. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, T. Degradation and beyond: Control of androgen receptor activity by the proteasome system. Cell Mol. Biol. Lett. 2006, 11, 109–131. [Google Scholar] [CrossRef] [PubMed]
- Kizaka-Kondoh, S.; Inoue, M.; Harada, H.; Hiraoka, M. Tumor hypoxia: A target for selective cancer therapy. Cancer Sci. 2003, 94, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.D.; Ross, J.A.; McLaren, D.B.; Parker, C.C.; Habib, F.K.; Riddick, A.C. The relevance of a hypoxic tumour microenvironment in prostate cancer. BJU Int. 2010, 105, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, T.; Iguchi, K.; Fukami, K.; Ishii, K.; Usui, S.; Sugimura, Y.; Hirano, K. Androgen receptor W741C and T877A mutations in AIDL cells, an androgen-independent subline of prostate cancer LNCaP cells. Tumour Biol. 2011, 32, 1097–1102. [Google Scholar] [CrossRef]
- Ishii, K.; Takahashi, S.; Sugimura, Y.; Watanabe, M. Role of Stromal Paracrine Signals in Proliferative Diseases of the Aging Human Prostate. J. Clin. Med. 2018, 7, 68. [Google Scholar] [CrossRef]
- Franco, O.E.; Jiang, M.; Strand, D.W.; Peacock, J.; Fernandez, S.; Jackson, R.S., 2nd; Revelo, M.P.; Bhowmick, N.A.; Hayward, S.W. Altered TGF-beta signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res. 2011, 71, 1272–1281. [Google Scholar] [CrossRef]
- Kiskowski, M.A.; Jackson, R.S., 2nd; Banerjee, J.; Li, X.; Kang, M.; Iturregui, J.M.; Franco, O.E.; Hayward, S.W.; Bhowmick, N.A. Role for stromal heterogeneity in prostate tumorigenesis. Cancer Res. 2011, 71, 3459–3470. [Google Scholar] [CrossRef] [PubMed]
- Cunha, G.R.; Hayward, S.W.; Wang, Y.Z.; Ricke, W.A. Role of the stromal microenvironment in carcinogenesis of the prostate. Int. J. Cancer 2003, 107, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yousef, G.M.; Diamandis, E.P. The new human tissue kallikrein gene family: Structure, function, and association to disease. Endocr. Rev. 2001, 22, 184–204. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Onishi, T.; Hoshina, A. Nadir PSA level and time to PSA nadir following primary androgen deprivation therapy are the early survival predictors for prostate cancer patients with bone metastasis. Prostate Cancer Prostatic Dis. 2011, 14, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Onishi, T.; Hoshina, A. Cutoff value of time to prostate-specific antigen nadir is inversely correlated with disease progression in advanced prostate cancer. Endocr. Relat. Cancer 2012, 19, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Flaberg, E.; Markasz, L.; Petranyi, G.; Stuber, G.; Dicso, F.; Alchihabi, N.; Olah, E.; Csizy, I.; Jozsa, T.; Andren, O.; et al. High-throughput live-cell imaging reveals differential inhibition of tumor cell proliferation by human fibroblasts. Int. J. Cancer 2011, 128, 2793–2802. [Google Scholar] [CrossRef] [PubMed]
- Alkasalias, T.; Flaberg, E.; Kashuba, V.; Alexeyenko, A.; Pavlova, T.; Savchenko, A.; Szekely, L.; Klein, G.; Guven, H. Inhibition of tumor cell proliferation and motility by fibroblasts is both contact and soluble factor dependent. Proc. Natl. Acad. Sci. USA 2014, 111, 17188–17193. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Yang, X.; Sun, F.; Jiang, R.; Linn, D.E.; Chen, H.; Chen, H.; Kong, X.; Melamed, J.; Tepper, C.G.; et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009, 69, 2305–2313. [Google Scholar] [CrossRef]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Invest. 2010, 120, 2715–2730. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Tamada, S.; Kato, M.; Hirayama, Y.; Takeyama, Y.; Iguchi, T.; Sadar, M.D.; Nakatani, T. Androgen Receptor Splice Variant 7 Drives the Growth of Castration Resistant Prostate Cancer without Being Involved in the Efficacy of Taxane Chemotherapy. J. Clin. Med. 2018, 7, 444. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; Jayaram, A.; Winquist, E.; McLaughlin, B.; Lu, D.; Fleisher, M.; Orr, S.; Lowes, L.; et al. Assessment of the Validity of Nuclear-Localized Androgen Receptor Splice Variant 7 in Circulating Tumor Cells as a Predictive Biomarker for Castration-Resistant Prostate Cancer. JAMA Oncol. 2018, 4, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Nevedomskaya, E.; Baumgart, S.J.; Haendler, B. Recent Advances in Prostate Cancer Treatment and Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1359. [Google Scholar] [CrossRef] [PubMed]
- Nuhn, P.; De Bono, J.S.; Fizazi, K.; Freedland, S.J.; Grilli, M.; Kantoff, P.W.; Sonpavde, G.; Sternberg, C.N.; Yegnasubramanian, S.; Antonarakis, E.S. Update on Systemic Prostate Cancer Therapies: Management of Metastatic Castration-resistant Prostate Cancer in the Era of Precision Oncology. Eur. Urol. 2018. [Google Scholar] [CrossRef]
- Zhao, S.G.; Chang, S.L.; Erho, N.; Yu, M.; Lehrer, J.; Alshalalfa, M.; Speers, C.; Cooperberg, M.R.; Kim, W.; Ryan, C.J.; et al. Associations of Luminal and Basal Subtyping of Prostate Cancer With Prognosis and Response to Androgen Deprivation Therapy. JAMA Oncol. 2017, 3, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, D.; Omlin, A.; Pezaro, C.; Shamseddine, A.; de Bono, J. Metastatic castration-resistant prostate cancer (CRPC): Preclinical and clinical evidence for the sequential use of novel therapeutics. Cancer Metastasis Rev. 2014, 33, 555–566. [Google Scholar] [CrossRef]
- Kita, Y.; Goto, T.; Akamatsu, S.; Yamasaki, T.; Inoue, T.; Ogawa, O.; Kobayashi, T. Castration-Resistant Prostate Cancer Refractory to Second-Generation Androgen Receptor Axis-Targeted Agents: Opportunities and Challenges. Cancers 2018, 10, 345. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishii, K.; Matsuoka, I.; Sasaki, T.; Nishikawa, K.; Kanda, H.; Imai, H.; Hirokawa, Y.; Iguchi, K.; Arima, K.; Sugimura, Y. Loss of Fibroblast-Dependent Androgen Receptor Activation in Prostate Cancer Cells is Involved in the Mechanism of Acquired Resistance to Castration. J. Clin. Med. 2019, 8, 1379. https://doi.org/10.3390/jcm8091379
Ishii K, Matsuoka I, Sasaki T, Nishikawa K, Kanda H, Imai H, Hirokawa Y, Iguchi K, Arima K, Sugimura Y. Loss of Fibroblast-Dependent Androgen Receptor Activation in Prostate Cancer Cells is Involved in the Mechanism of Acquired Resistance to Castration. Journal of Clinical Medicine. 2019; 8(9):1379. https://doi.org/10.3390/jcm8091379
Chicago/Turabian StyleIshii, Kenichiro, Izumi Matsuoka, Takeshi Sasaki, Kohei Nishikawa, Hideki Kanda, Hiroshi Imai, Yoshifumi Hirokawa, Kazuhiro Iguchi, Kiminobu Arima, and Yoshiki Sugimura. 2019. "Loss of Fibroblast-Dependent Androgen Receptor Activation in Prostate Cancer Cells is Involved in the Mechanism of Acquired Resistance to Castration" Journal of Clinical Medicine 8, no. 9: 1379. https://doi.org/10.3390/jcm8091379
APA StyleIshii, K., Matsuoka, I., Sasaki, T., Nishikawa, K., Kanda, H., Imai, H., Hirokawa, Y., Iguchi, K., Arima, K., & Sugimura, Y. (2019). Loss of Fibroblast-Dependent Androgen Receptor Activation in Prostate Cancer Cells is Involved in the Mechanism of Acquired Resistance to Castration. Journal of Clinical Medicine, 8(9), 1379. https://doi.org/10.3390/jcm8091379