1. Introduction
An unfulfilled desire to have children is a serious medical and social phenomenon. More and more women are being treated for infertility and sterility. Approximately 10–15% of couples remain childless despite a persistent desire to have children and 1–3% of all couples experience recurrent miscarriages [
1,
2]. For the woman who is affected, this problem is a clear limitation on the quality of life, and a profound dilemma for the partnership [
3]. The diagnosis and therapy of couples with an unfulfilled desire to have children clearly confront the health professionals treating them with challenges, since although some causes are known, the majority of them remain undiscovered [
4]. However, infertility and pregnancy loss are common gynecological or reproductive problems caused by well-defined deficiencies associated with chromosomal, anatomic and hormonal abnormalities (progesterone, estrogens, diabetes, thyroid disease) [
5,
6]. The American Society of Reproductive Medicine (ASRM) defines infertility as failure to conceive after 1 year of appropriately timed unprotected intercourse. Further common causes of infertility are pelvic diseases (tubal disease, fibroids, endometriosis), male factor, disorders of ovulation, female reproductive aging and idiopathic causes. Infertility and pregnancy loss can be interdependent, often resulting in a combined disorder [
4]. In this context, the thrombotic hemostasis disorders associated with pregnancy loss include lupus anticoagulants and anticardiolipin antibodies, factor XII deficiency, dysfibrinogenemias associated with thrombosis, protein C defects, protein S defects, antithrombin deficiency, heparin cofactor II deficiency and fibrinolytic defects associated with thrombosis [
1,
7,
8,
9,
10,
11,
12,
13,
14]. Notably, pregnancy itself is a hypercoagulable state predisposing to uteroplacental thrombosis, owing at least in part to the changes in coagulation factors, their regulators and fibrinolytic systems [
6,
15]. In addition, low pressure and turbulent flow of the placental perfusion may also predispose to thrombosis [
16]. Thus, impaired placental circulation and hypercoagulability are involved in gynecological or reproductive complications such as recurrent pregnancy loss that occurs in 1–5% of pregnant women [
2,
6]. While platelets have very important roles in hemostasis [
17], excess activation of platelets or platelet hyperaggregability is involved in the pathophysiological processes underlying pregnancy loss or unexplained thrombosis, which have also been confirmed in diverse animal models [
6,
18,
19,
20,
21]. Thus, although not often screened for or recognized, perturbations of platelet function can be an etiology for prothrombotic conditions in pregnancy [
15]. As a consequence of platelet hyperaggregability, acetylsalicylic acid (ASA) reduces platelet aggregation by inhibiting the enzymes cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2), leading to the reduced formation of thromboxane A2 (TXA2), a potent stimulator of platelet activation, thereby reducing platelet secretion and aggregation [
22].
Al-Mefty et al. first described platelet hyperaggregability in a group of young adults with unexplained repetitive transient ischemic attacks who were noted to have hyperaggregable platelets when exposed to adenosine diphosphate and epinephrine [
23]. Nowadays, platelet function, particularly platelet hyperaggregability, is characterized by light transmission aggregometry and dose-independent platelet aggregation solely due to epinephrine and/or adenosine diphosphate (ADP) [
24]. In the early 1980s, Mammen and Bick suggested platelet hyperaggregation, or SPS, as the underlying cause of thrombophilia in patients with unexplained arterial and venous thromboembolism or recurrent spontaneous abortions [
6,
18]. They named platelet hyperaggregability, in response to either epinephrine or adenosine or both sticky platelet syndrome (SPS). This laboratory phenomenon has later been clinically described in children and young adults with no identifiable risk factors for thrombosis and an otherwise negative thrombophilia evaluation [
18]. Furthermore, SPS has been described by numerous clinical case reports associated with acute myocardial infarction, chronic kidney disease and thromboembolic kidney graft infarction [
6,
18,
24]. Further studies have revealed SPS as the second most common hereditary thrombophilia after resistance to activated protein C and the most common thrombophilia associated with arterial thrombosis, with a reported incidence of approximately 21% in unselected populations [
18,
25]. Although the incidence of SPS in pregnancy loss is not well known, it has been described in women with recurrent pregnancy loss without a history of thrombosis [
15]. Such cases are presently limited to small descriptive observations [
15,
24]. Therefore, SPS may be an underappreciated etiology for infertility and pregnancy loss [
15].
Although SPS is the second most common thrombophilia that causes recurrent spontaneous abortions or fetal loss syndrome [
15,
26], this potential association has not yet been investigated epidemiologically in the field of miscarriage diagnostics. In previous investigations of the causal associations of platelet hyperaggregability, miscarriages and SPS have frequently been reported together, but no investigation has so far looked more closely at the pregnancy outcome in patients with SPS in addition to prevalence analyses. Based on these considerations, we aimed to investigate retrospectively the platelet aggregability in a large patient cohort with infertility and pregnancy loss history, in comparison to healthy non-pregnant women of fertile age. In this explorative study, we evaluated the thrombocyte function diagnostics modified according to Mammen with regard to the following aspects: (a) investigation of the prevalence of SPS, (b) diagnostic value of thrombocyte function analysis modified according to Mammen, and (c) retrospective assessment of the pregnancy outcome with and without ASA therapy with respect to the presence of SPS.
4. Discussions
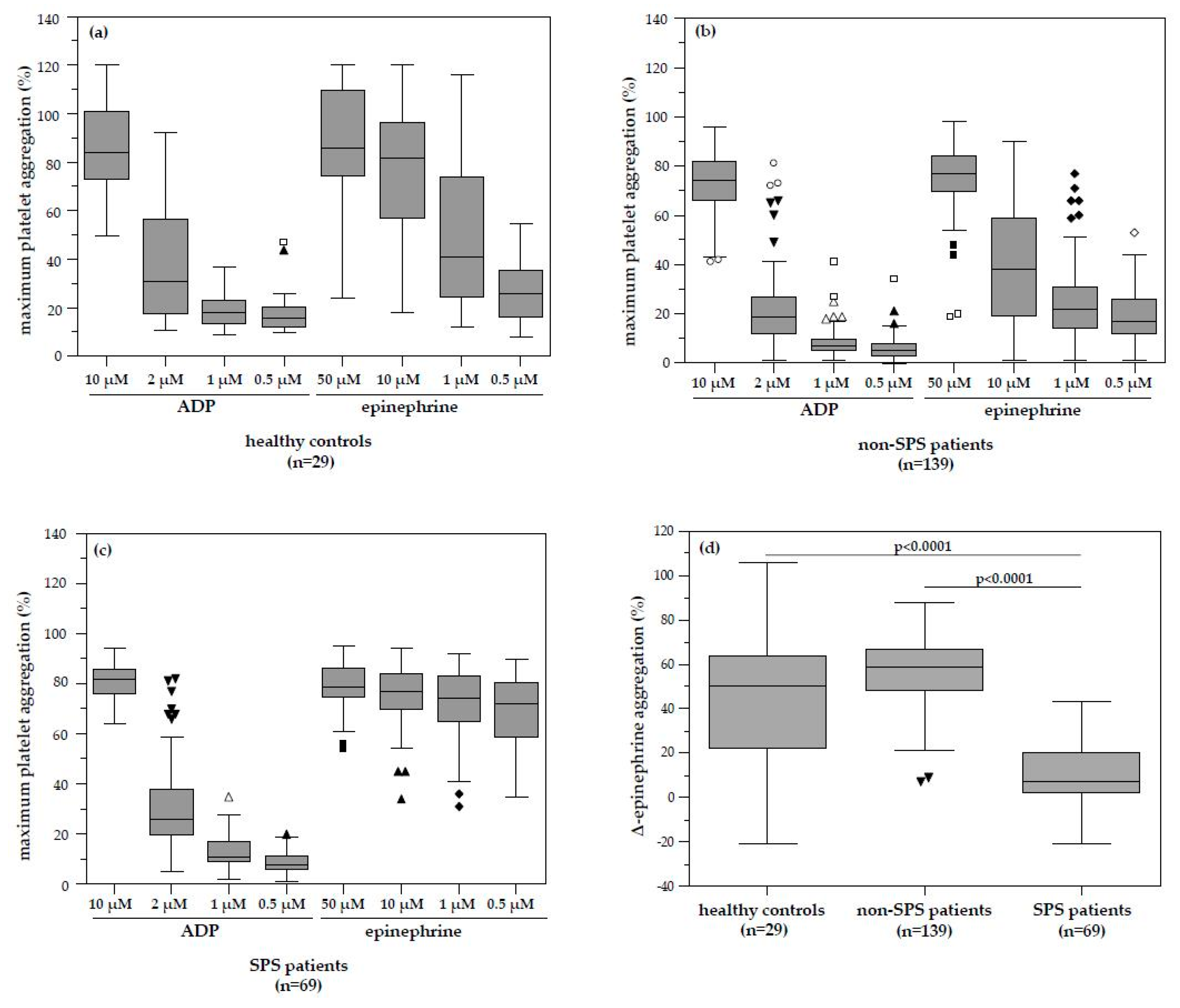
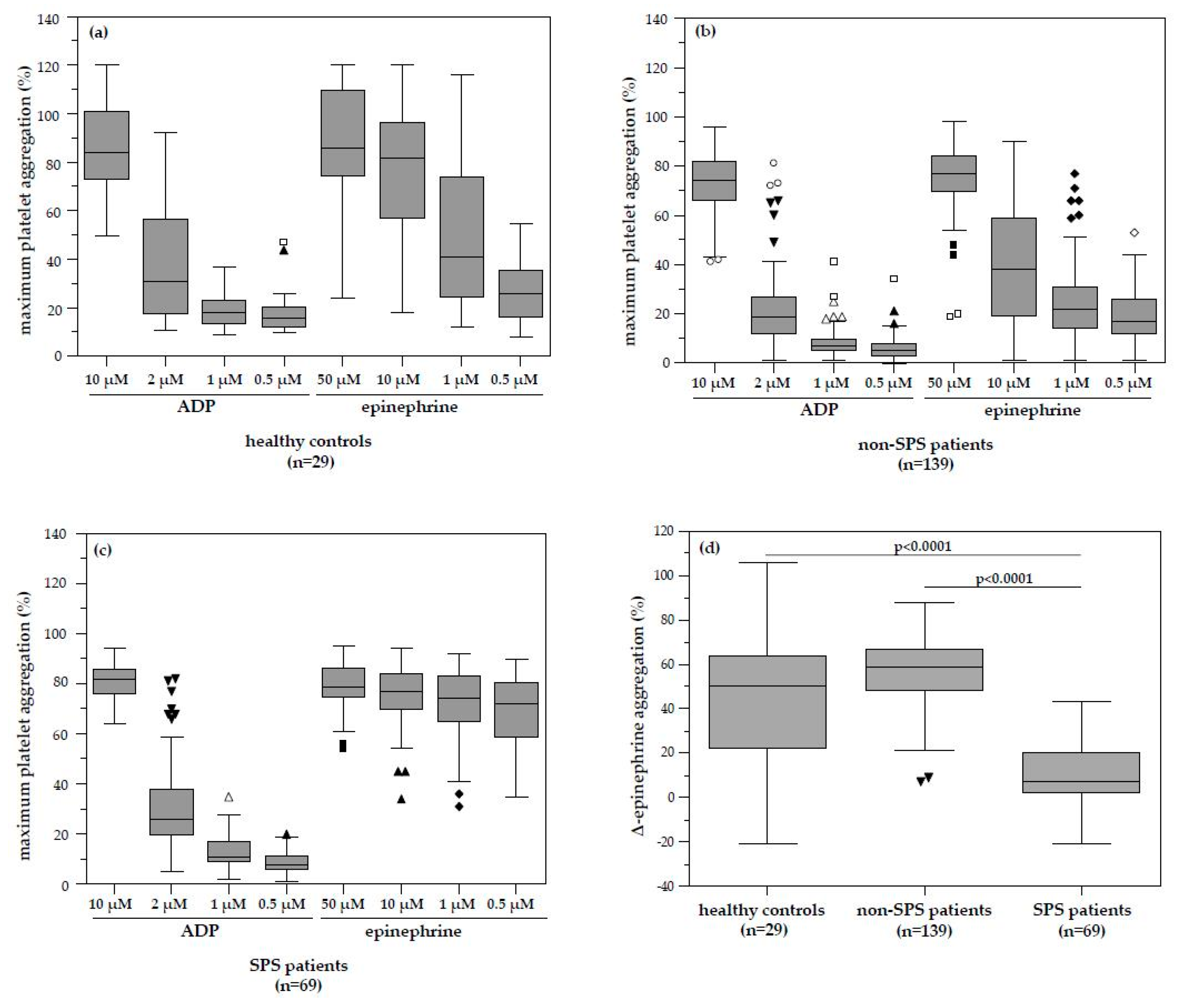
We herein report for the first time a surprisingly high prevalence of platelet hyperaggregability in patients with infertility and unexplained pregnancy loss history (33.2% for the 11-year period prevalence) when comparing to the study of Bick and Hoppensteadt [
6]. To our knowledge, this high prevalence is only exceeded in patients with chronic kidney disease (CKD), such as chronic hemodialysis (82%) or renal transplantation (67%) [
24]. Remarkably, no pathological platelet aggregation could be found in healthy, non-pregnant female individuals of fertile age. However, the definite prevalence of the SPS in the unselected total population has not yet been fully clarified [
37]. Due to the limited number of patients studied, the exact prevalence and incidence of the SPS in the general population is not answered in detail by this study. It is considered that the possibility of SPS-diagnosis is less recognized in patients with unexplained or unprovoked thromboembolic diseases and in women with repeated miscarriages [
6].
In our study, platelet hyperaggregability after both ADP and epinephrine (type 1 SPS) and platelet hyperaggregability after ADP only (type 3 SPS) were not found. According to Kubisz et al. [
34], type 2 (platelet hyperaggregability after epinephrine only) is most common, followed by type 1, whereas type 3 is rare. However, it is important that this classification is based on laboratory criteria. In addition, no relation or differences to the clinical manifestation, treatment, or prognosis of patients were observed among the types so far. Interestingly, type 2 is the most frequent variant of SPS in white populations, whereas type 1 is the most frequent variant in Mexican mestizos [
35]. However, despite the clear clinical definition and strong evidence of familiar occurrence, published results so far failed to identify a single genetic defect responsible for SPS. In sum, the laboratory heterogeneity of the syndrome with three clearly distinct types might suggest that SPS might have multifactorial genetics. Furthermore, because SPS diagnosis is made solely on the clinical and laboratory criteria and not on genetic testing, inherited and acquired changes of platelet aggregation cannot be ruled out in the currently diagnosed patients. It is therefore also likely that the SPS itself does not cause thromboembolic events, similar to protein C resistance or deficiency, but only predisposes to them. It is suspected that an additional factor is required for the development of a clinically relevant thromboembolic event [
5].
Furthermore, SPS patients with low-dose ASA therapy showed a trend to improved pregnancy outcome compared to SPS patients without low-dose ASA. This finding is in line with the fact that SPS could be responsible for a large number of unexplained thromboembolic events of unknown causes in selected populations such as patients with pregnancy loss or diseases associated with increased vascular morbidity such as CKD [
5,
24]. Due to the low number of patients both without ASA therapy but successful pregnancy, the diagnostic and clinical relevance of less successful pregnancy in these patients might be premature at this stage of this explorative statistical analysis. However, further research should aim at investigating more SPS patients without ASA in regard to pregnancy outcome and to clarify the exact pathogenic and therapeutic role of platelet hyperaggregability in clinical setting. Bick described that SPS could be diagnosed in 21% of patients with miscarriages as well as coagulation disorders [
5]. Only the antiphospholipid syndrome occurring with a frequency of 67% was more frequent [
6]. Other reports also suggest similar frequencies. According to Kubisz et al., SPS has been identified in approximately 21% of unexplained arterial thrombotic episodes, regarded to be the most common thrombophilia in arterial thrombosis and 13.2% of unexplained venous thromboembolism [
38]. Therefore, SPS is described as the most frequent hereditary prothrombotic platelet disorder in vascular morbidity [
34].
The investigation of platelet function analysis in our retrospectively evaluated patients showed a pathologic platelet aggregation pattern in 69 patients. The median ∆-epinephrine aggregation response in these patients with identified dose-independent platelet hyperaggregation was significantly lower compared to patients without SPS and healthy controls. This pattern is typical for sticky platelet syndrome type II. Although some of these patients with SPS had MTHFR, Factor–V–Leiden or Prothrombin mutations, none had any recorded history of clinical thromboembolism other than the gynaecological disease. Although there was a high number of successful pregnancy outcomes in patients with SPS and even more in SPS plus low-dose ASA therapy compared to patients without SPS or ASA therapy, it did not reach statistical significance. It is likely that platelet hyperaggregability itself does not cause thromboembolic events, similar to activated Protein C resistance or Protein C deficiency, but only predisposes to them and a second hit may be required to develop overt clinical disease. Our finding is in accordance with the fact that Mammen and Bick suggested platelet hyperaggregability in the context of SPS as the cause of thromboembolic events, which could not be explained by any of the established risk factors (Protein C/S deficiency, Factor V Leiden mutation, antiphospholipid syndrome, etc.) [
5,
6]. In the course of time, this change in platelet function was associated with a variety of clinical pictures (myocardial infarction without congenital heart defect, recurrent apoplexy despite optimal oral anticoagulation, ischemic optic neuropathy, thromboembolic complications after kidney transplantation [
1,
8,
9,
10,
11]). The thrombotic defects associated with fetal loss are thought to occur due to thrombosis of early placental vessels, with a peak in the first trimester, but small peaks also occur in the second and third trimesters [
5,
6]. It appears that the earlier the pregnancy, the smaller the placental and uterine vessels and, therefore, the greater the propensity to undergo partial or total occlusion by thrombus formation. Thrombotic occlusion of placental vessels, both venous and arterial, preclude adequate nutrition and, thus, viability of the fetus [
5,
6].
The therapy of SPS is based on the inhibition of platelet aggregation with low-dose ASA. However, standardized guidelines do not yet exist and the decision for treatment is considered individual [
37]. In most patients, a low dosage of ASA of 80–100 mg per day is sufficient to normalize platelet hyperaggregation [
39]. ASA is effective in both therapy and the prevention of thrombosis. When ASA is discontinued, however, pathological platelet function can be measured again. Alternatively, therapy with ADP inhibitors is also possible. In 2013, Velázquez–Sànchez–de–Cima et al. observed a very good therapeutic effect in SPS patients both with ASA and with a combination therapy with ASA and clopidogrel [
40]. Other anticoagulants that act on plasmatic coagulation [oral anticoagulants] are ineffective and do not prevent the occurrence of thromboembolic events in SPS [
35].
In addition, low-dose ASA treatment significantly improves ovarian responsiveness, uterine and ovarian blood flow velocity, and implantation and increases pregnancy rates and birth rates in IVF patients [
41,
42]. However, despite the better pregnancy outcome of SPS patients with ASA therapy, we could not find any statistically significant associations between ASA therapy and pregnancy outcome in our cohort. Similar conclusions were found in evaluations of ASA therapy in women undergoing IVF treatment. ASA did not support but showed a trend of improvement of clinical pregnancy outcome [
43,
44].
Mammen et al. had already suspected a hereditary cause of SPS by family testing at that time [
15]. Recently, SPS is characterized as an autosomal dominant trait [
37]. However, no distinct molecular genetic cause for SPS has been found to date [
37]. Nevertheless, in several studies different genes were investigated, but no exact mutation could be detected which is responsible for the development of SPS. Kotuličová et al. investigated the glycoprotein 6 (GP6) polymorphism in SPS patients. It was found that there was a significant association between GP6 gene polymorphism and thromboembolic events in SPS patients [
45]. Sokol et al. showed a significantly increased incidence of these GP6 polymorphisms in SPS patients with miscarriages [
20]. Furthermore, Ruiz–Argüelles et al. analyzed the glycoprotein IIIa PI A1/A2 polymorphism in conjunction with the SPS, but no significant association was found [
46]. Sokol et al. investigated SPS patients with miscarriages and controls for genetic polymorphism and Gas6/PEAR1 gene polymorphisms could be described [
16]. Yee et al. showed in healthy individuals with platelet hyperaggregability an association with GNB3 polymorphisms [
47].
Some methodical limitations regarding the platelet function analysis provided need to be taken into account. PRP does not contain all the platelets. Activated platelets or large platelets may be lost during the enrichment. The PRP analysis always takes place in an artificial environment. For example, erythrocytes and leukocytes are missing as endogenous sources for ADP and adenosine triphosphate (ATP) [
48]. Furthermore, the definition of platelet hyperaggregability and the laboratory methods used are not standardized. Furthermore, platelets ex vivo and during phlebotomy could be heavily affected by preanalytical errors [
12,
30]. However, our laboratory practices were in line with recognized guidelines regarding the laboratory investigation of heritable disorders of platelet function and published recommendations for the standardization of light transmission aggregometry [
31,
32].
Light transmission aggregometry (LTA) measures the transmission of light through a sample of platelets in various suspensions such as platelet-rich plasma (PRP), washed platelets or gel-filtrated platelets. What complicates this methodology is that LTA results can be strongly influenced by the time between blood collection and analysis, platelet count, but also size, hematocrit, storage and measurement temperature, depending on the test system. Despite the complexity of platelet stabilization before testing, however, blood samples for LTA should be drawn into sodium citrate, buffered anticoagulant. In various standardization guidelines, venous citrated plasma and PRP preparation is recommended as an anticoagulant [
49]. Nevertheless, this circumstance is contrary to the fact that stability of blood sample may be improved if the blood is anticoagulated without citrate, as citrate complexes calcium and platelets need calcium ions to function normally. Therefore, other anticoagulants can be similarly used and sometimes should be preferred, e.g., hirudin. Hirudinized blood contains the normal concentration of Ca
2+ and Mg
2+ [
50]. However, citrate anticoagulated blood is still used in most test methods. Ultimately, the aggregation formation as it is registered in aggregometers is an artefact and only very indirectly corresponds to the complex in vivo process of platelet function. Thus, there is still no generally accepted ideal measure of platelet activation [
50].
According to Cattaneo and coworkers, the platelet count of PRP samples should not be adjusted to a standardized value with autologous PPP [
32]. Thus, recent studies demonstrated that platelet counts in PRP within the range that is observed in PRP samples from subjects with normal platelet count in whole blood do not affect the results of LTA studies [
32]. In this line, abnormalities of platelet aggregation were more frequent using adjusted platelet count both in controls and patients [
28]. Overall, it must therefore be stated that there is still controversy concerning whether the platelet count should be adjusted or not [
51,
52]. On the one hand, it has been argued that in vitro aggregation is basically influenced by the platelet count in PRP and thus platelet count adjustment is recommended. On the other hand, PPP may contain substances affecting platelet function that are released by platelets or other blood cells during centrifugation of blood samples, which is necessary to obtain PPP [
50]. However, citrate plasma is not the optimal medium for platelet function testing. Therefore, further studies should be conducted to investigate the diagnostic value of other high-quality analytical strategies based on gel-filtered platelets or/and plasma switch in LTA analysis. Moreover, specialized scientific assessments such as enzymes of homocysteine metabolism, soluble P-selectin, E-selectin or pentraxin 3, plasminogen activator inhibitor (PAI)-1 4G/5G insertion–deletion mutations and coagulation factor XIII Val34Leu polymorphism or flow cytometry for detection of platelet-specific activation markers (such as P-selectin and fibrinogen binding or others like marker antibodies CD62, CD41, PAC1 (activated GP IIb/IIIa) and CD154) may potentially be explored. The analysis of these parameters might provide more objectivity and stability of results in an analysis of platelet function.
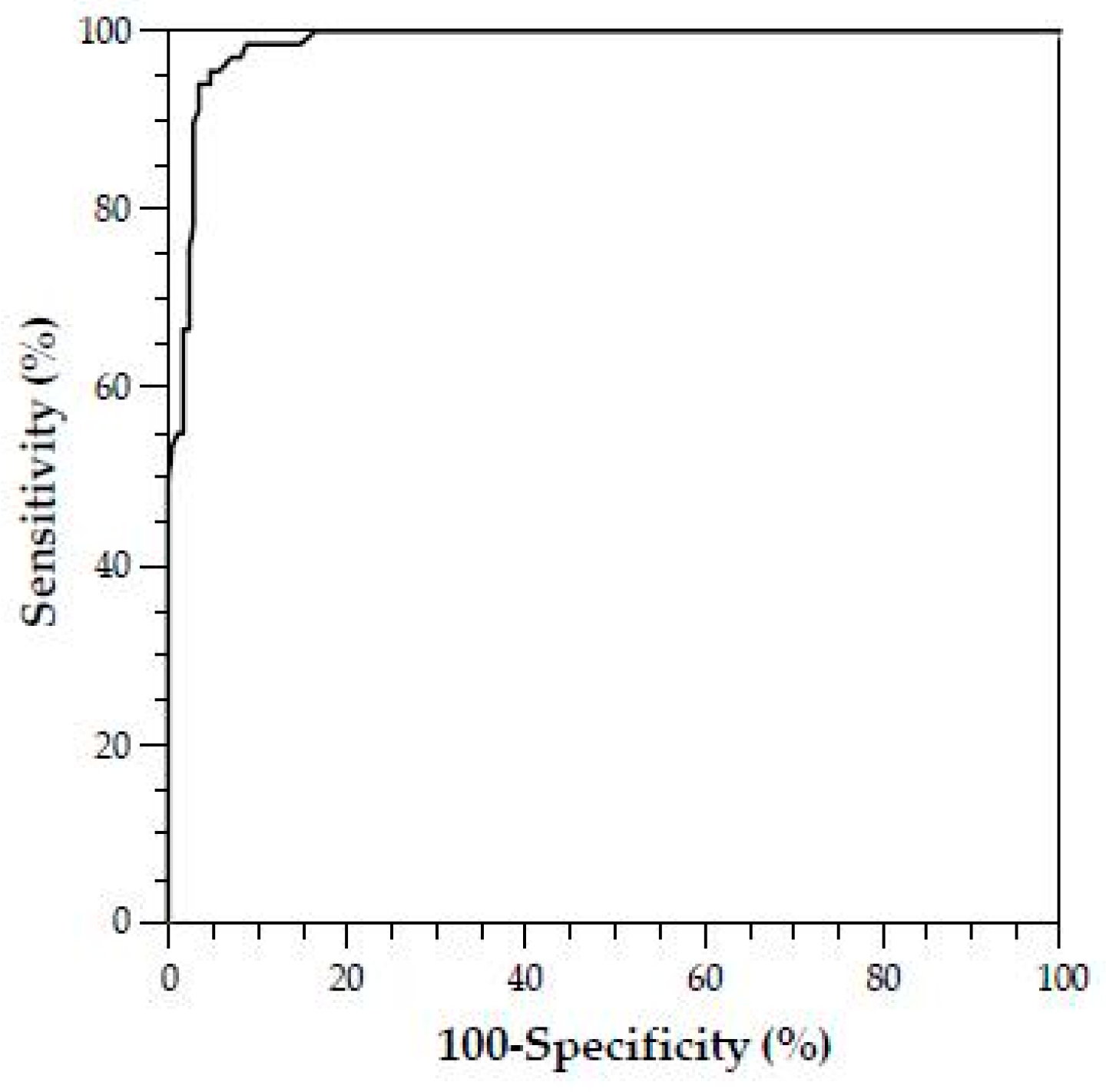
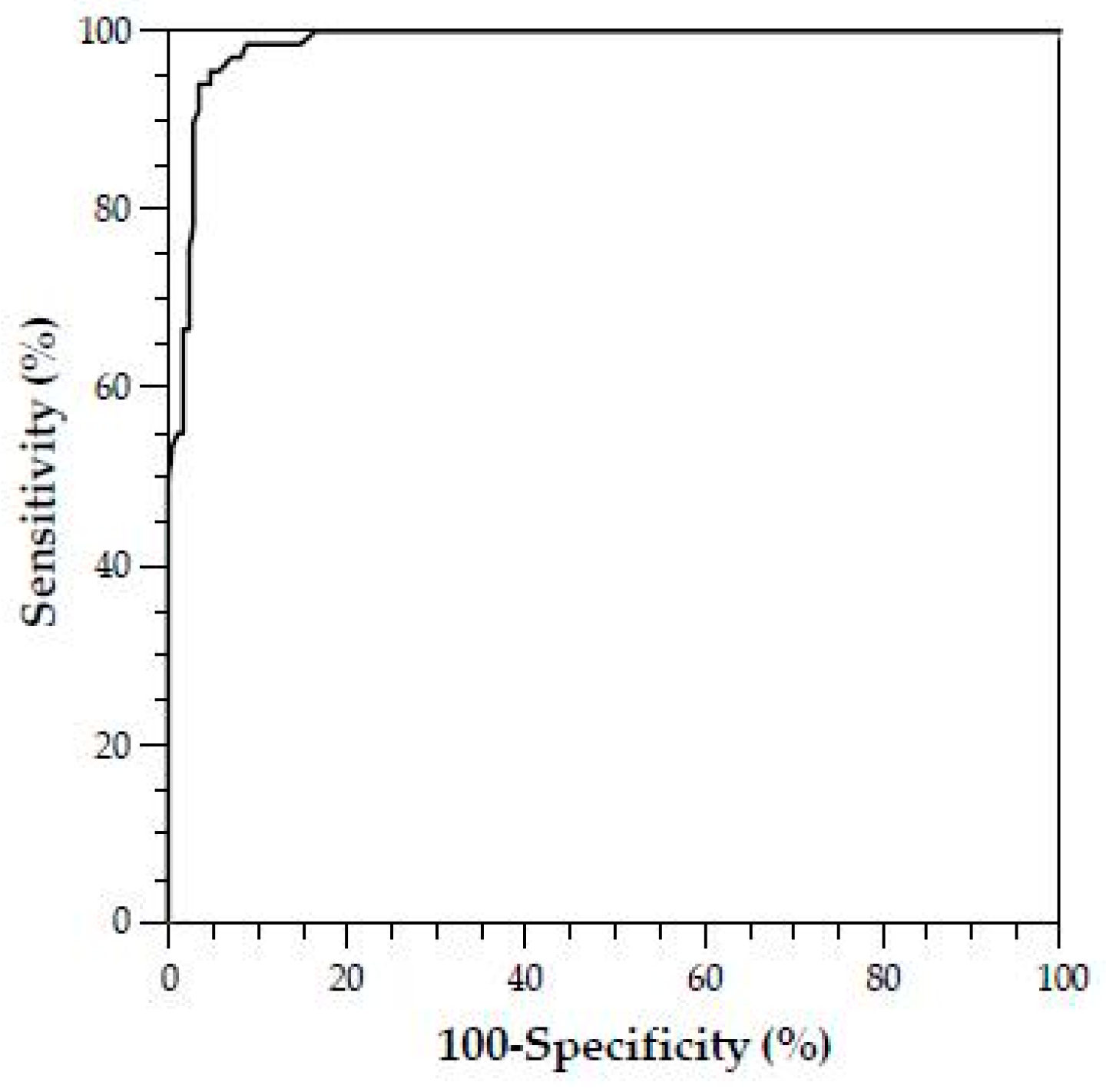
In addition, there are no studies to date that investigate the prevalence of SPS in the unselected total population; it is probable that the frequency of SPS is lower here than in the patient groups with thromboembolic events investigated to date [
34]. For example, Kubisz et al. reported an SPS prevalence of 14% based on other patient cohorts with general thromboembolic events [
25]. In contrast, Ruiz–Arguelles found SPS in up to 60% of primarily hypercoagulable patients [
53,
54].

 ,
, 

{kind=link}
{kind=link}