Utility of Whole Blood Thiamine Pyrophosphate Evaluation in TPK1-Related Diseases

 ,
,  ,
, 
Abstract
1. Introduction
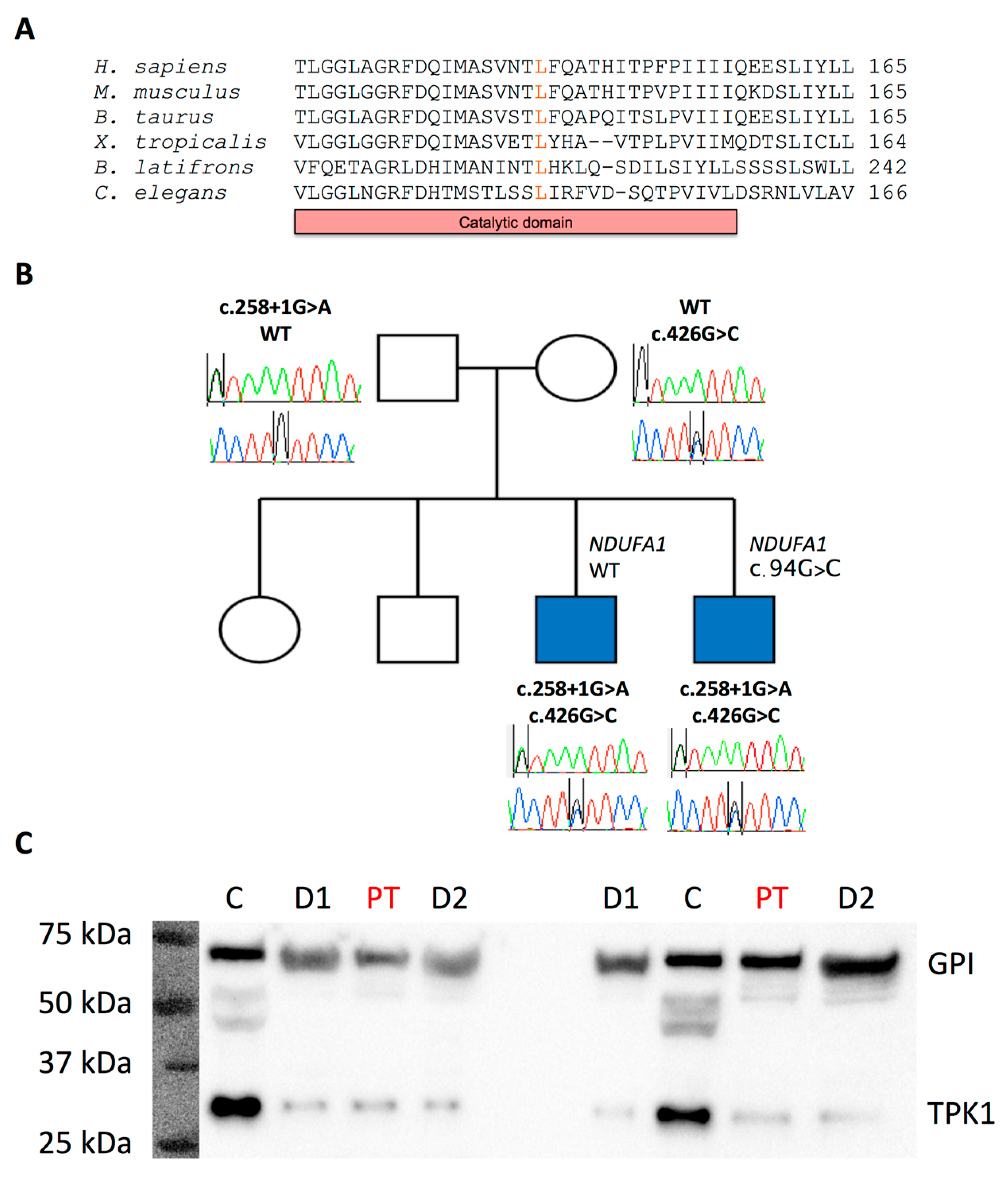
2. Case Presentation
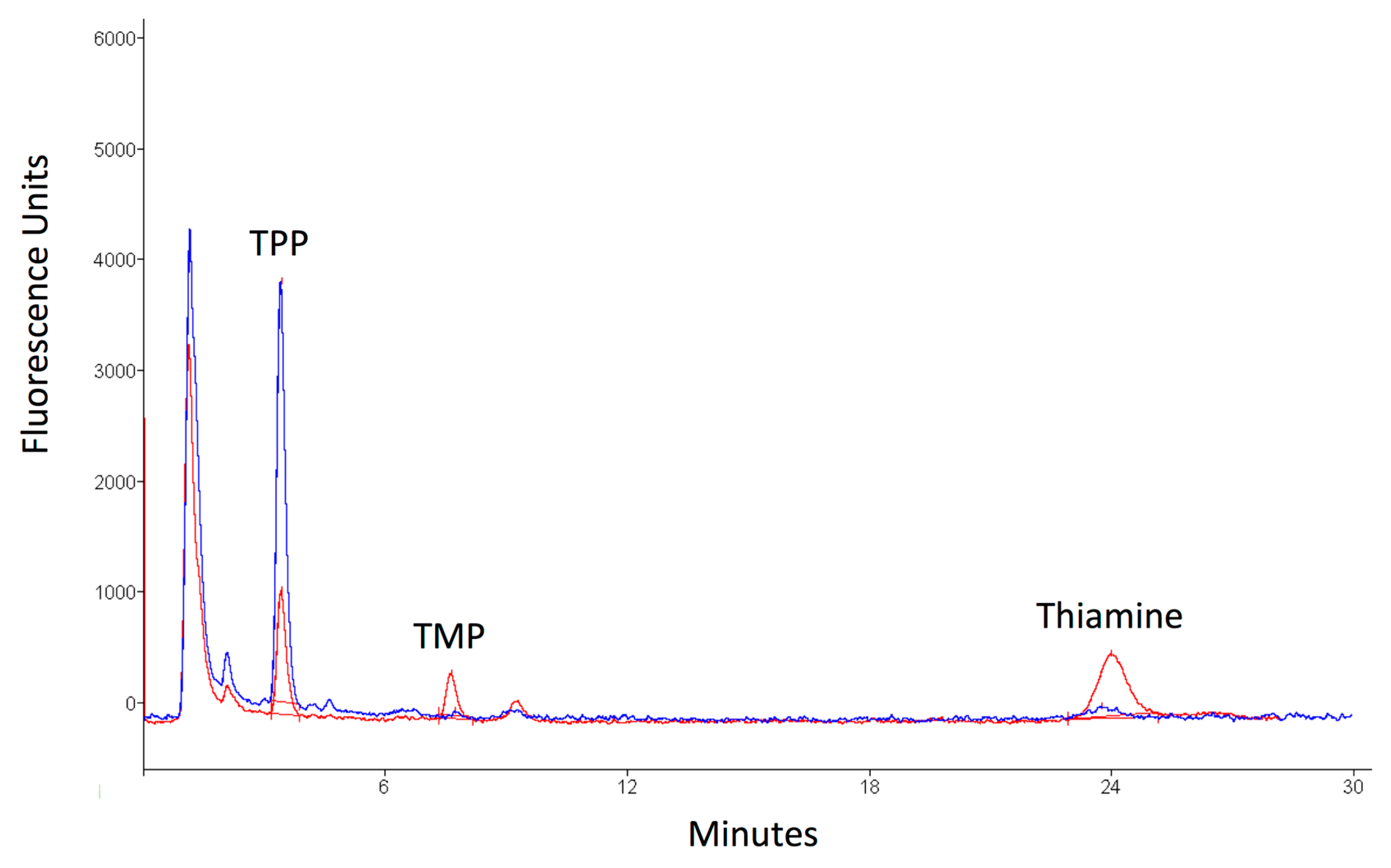
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Whole Exome Sequencing
Western Blot Analysis
References
- Brown, G. Defects of thiamine transport and metabolism. J. Inherit. Metab. Dis. 2014, 37, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed]
- Potluri, P.; Davila, A.; Ruiz-Pesini, E.; Mishmar, D.; O’Hearn, S.; Hancock, S.; Simon, M.; Scheffler, I.E.; Wallace, D.C.; Procaccio, V. A novel NDUFA1 mutation leads to a progressive mitochondrial complex I-specific neurodegenerative disease. Mol. Genet. Metab. 2009, 96, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Ortigoza-Escobar, J.D.; Alfadhel, M.; Molero-Luis, M.; Darin, N.; Spiegel, R.; de Coo, I.F.; Gerards, M.; Taylor, R.W.; Artuch, R.; Nashabat, M.; et al. Thiamine deficiency in childhood with attention to genetic causes: Survival and outcome predictors. Ann. Neurol. 2017, 82, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Mayr, J.A.; Freisinger, P.; Schlachter, K.; Rolinski, B.; Zimmermann, F.A.; Scheffner, T.; Haack, T.B.; Koch, J.; Ahting, U.; Prokisch, H.; et al. Thiamine pyrophosphokinase deficiency in encephalopathic children with defects in the pyruvate oxidation pathway. Am. J. Hum. Genet. 2011, 89, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Banka, S.; de Goede, C.; Yue, W.W.; Morris, A.A.; von Bremen, B.; Chandler, K.E.; Feichtinger, R.G.; Hart, C.; Khan, N.; Lunzer, V.; et al. Expanding the clinical and molecular spectrum of thiamine pyrophosphokinase deficiency: A treatable neurological disorder caused by TPK1 mutations. Mol. Genet. Metab. 2014, 113, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.L.; Vanderver, A.; Yang, S.; Chang, T.; Cramp, L.; Vezina, G.; Lichter-Konecki, U.; Cusmano-Ozog, K.P.; Smpokou, P.; Chapman, K.A.; et al. Thiamine pyrophosphokinase deficiency causes a Leigh Disease like phenotype in a sibling pair: Identification through whole exome sequencing and management strategies. Mol. Genet. Metab. Rep. 2014, 1, 66–70. [Google Scholar] [CrossRef]
- Invernizzi, F.; Panteghini, C.; Chiapparini, L.; Moroni, I.; Nardocci, N.; Garavaglia, B.; Tonduti, D. Thiamine-responsive disease due to mutation of tpk1: Importance of avoiding misdiagnosis. Neurology 2017, 89, 870–871. [Google Scholar] [CrossRef]
- Mahajan, A.; Sidiropoulos, C. TPK1 mutation induced childhood onset idiopathic generalized dystonia: Report of a rare mutation and effect of deep brain stimulation. J. Neurol. Sci. 2017, 376, 42–43. [Google Scholar] [CrossRef] [PubMed]
- Mayr, J.A.; Bodamer, O.; Haack, T.B.; Zimmermann, F.A.; Madignier, F.; Prokisch, H.; Rauscher, C.; Koch, J.; Sperl, W. Heterozygous mutation in the X chromosomal NDUFA1 gene in a girl with complex I deficiency. Mol. Genet. Metab. 2011, 103, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.J.; Ostergaard, E. Correspondence to: Heterozygous mutation in the X chromosomal NDUFA1 gene in a girl with complex I deficiency and A novel NDUFA1 mutation leads to progressive mitochondrial complex I-specific neurodegenerative disease. Mol. Genet. Metab. Rep. 2017, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.A.; Tu-Maung, N.; Cheng, K.; Wang, B.; Baeumner, A.J.; Kraft, C.E. Thiamine Assays—Advances, challenges, and caveats. ChemistryOpen 2017, 6, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Frank, E.L. Rapid HPLC measurement of thiamine and its phosphate esters in whole blood. Clin. Chem. 2008, 54, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Collie, J.T.B.; Greaves, R.F.; Jones, O.A.H.; Lam, Q.; Eastwood, G.M.; Bellomo, R. Vitamin B1 in critically ill patients: Needs and challenges. Clin. Chem. Lab. Med. 2017, 55, 1652–1668. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Ma, D.; Fei, G.; Ma, Z.; Xiao, F.; Yu, Q.; Pan, X.; Zhou, F.; Zhao, L.; Zhong, C. A single-step method for simultaneous quantification of thiamine and its phosphate esters in whole blood sample by ultra-performance liquid chromatography-mass spectrometry. J. Chromatogr. B 2018, 1095, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Ortigoza-Escobar, J.D.; Molero-Luis, M.; Arias, A.; Oyarzabal, A.; Darin, N.; Serrano, M.; Garcia-Cazorla, A.; Tondo, M.; Hernandez, M.; Garcia-Villoria, J.; et al. Free-thiamine is a potential biomarker of thiamine transporter-2 deficiency: A treatable cause of Leigh syndrome. Brain 2016, 139, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Roelofsen-de Beer, R.; van Zelst, B.D.; Wardle, R.; Kooij, P.G.; de Rijke, Y.B. Simultaneous measurement of whole blood vitamin B1 and vitamin B6 using LC-ESI-MS/MS. J. Chromatogr. B 2017, 1063, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Desvignes, J.P.; Bartoli, M.; Delague, V.; Krahn, M.; Miltgen, M.; Beroud, C.; Salgado, D. VarAFT: A variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res. 2018, 46, W545–W553. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | TPK1 Mutations | Predicted Effect on Protein | TPP Levels | Western Blot | Reference |
|---|---|---|---|---|---|
| P1 | c.[148A>C];[501+4A>T] | p.[Asn50His];[Val119_Pro167del] | N/A | N/A | Mayr et al. [6] |
| P2 | c.[148A>C];[501+4A>T] | p.[Asn50His];[Val119_Pro167del] | 68 * (B) | ↓hTPK1 | Mayr et al. [6] |
| P3 | c.[119T>C];[119T>C] | p.[Leu40Pro)];[Leu40Pro] | 50.4 * (B) | ↓hTPK1 | Mayr et al. [6] |
| P4 | c.[119T>C];[119T>C] | p.[Leu40Pro)];[(Leu40Pro] | N/A | N/A | Mayr et al. [6] |
| P5 | c.[179_182delGAGA];[656A>G] | p.[Arg60LysfsTer52];[Asn219Ser] | 96.9 * (B) | ↓hTPK1 | Mayr et al. [6] |
| P1 (P76 in [5]) | c.[604T>G];[604T>G] | p.[Trp202Gly];[Trp202Gly] | N/A | N/A | Fraser et al. [8] |
| P2 (P77 in [5]) | c.[604T>G];[604T>G] | p.[Trp202Gly];[Trp202Gly] | N/A | N/A | Fraser et al. [8] |
| P1 (P79 in [5]) | c.[479C>T];[479C>T] | p.[Ser160Leu];[Ser160Leu] | 60.9 ^ (B) | Normal | Banka et al. [7] |
| P2 | c.[664G>C];[664G>C] | p.[Asp222His];[Asp222His] | 85.4 ^ (B) | ↓hTPK1 | Banka et al. [7] |
| N/A | c.[656A>G];deletion of exons 3 and 4 on mRNA studies | p.[Asn219Ser];[?] | N/A | N/A | Invernizzi et al. [9] |
| Proband | c.[119T>C];[119T>C] | p.[Leu40Pro];[Leu40Pro] | 32 ~ (P) | N/A | Mahajan et al. [10] |
| Sister | c.[119T>C];[119T>C] | p.[Leu40Pro];[Leu40Pro] | N/A | N/A | Mahajan et al. [10] |
| P78 | c.[365T>C];[365T>C] | p.[Ile122Thr];[Ile122Thr] | N/A | N/A | Ortigoza-Escobar et al. [5] |
| Proband | c.[258+1G>A];[426G>C] | p.[Leu142Phe];[?] | 35 † (B) | ↓hTPK1 | Present study |
| Brother | c.[258+1G>A];[426G>C] | p.[Leu142Phe];[?] | N/A | N/A | Present study |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bugiardini, E.; Pope, S.; Feichtinger, R.G.; Poole, O.V.; Pittman, A.M.; Woodward, C.E.; Heales, S.; Quinlivan, R.; Houlden, H.; Mayr, J.A.; et al. Utility of Whole Blood Thiamine Pyrophosphate Evaluation in TPK1-Related Diseases. J. Clin. Med. 2019, 8, 991. https://doi.org/10.3390/jcm8070991
Bugiardini E, Pope S, Feichtinger RG, Poole OV, Pittman AM, Woodward CE, Heales S, Quinlivan R, Houlden H, Mayr JA, et al. Utility of Whole Blood Thiamine Pyrophosphate Evaluation in TPK1-Related Diseases. Journal of Clinical Medicine. 2019; 8(7):991. https://doi.org/10.3390/jcm8070991
Chicago/Turabian StyleBugiardini, Enrico, Simon Pope, René G. Feichtinger, Olivia V. Poole, Alan M. Pittman, Cathy E. Woodward, Simon Heales, Rosaline Quinlivan, Henry Houlden, Johannes A. Mayr, and et al. 2019. "Utility of Whole Blood Thiamine Pyrophosphate Evaluation in TPK1-Related Diseases" Journal of Clinical Medicine 8, no. 7: 991. https://doi.org/10.3390/jcm8070991
APA StyleBugiardini, E., Pope, S., Feichtinger, R. G., Poole, O. V., Pittman, A. M., Woodward, C. E., Heales, S., Quinlivan, R., Houlden, H., Mayr, J. A., Hanna, M. G., & Pitceathly, R. D. S. (2019). Utility of Whole Blood Thiamine Pyrophosphate Evaluation in TPK1-Related Diseases. Journal of Clinical Medicine, 8(7), 991. https://doi.org/10.3390/jcm8070991