Implication for Cancer Stem Cells in Solid Cancer Chemo-Resistance: Promising Therapeutic Strategies Based on the Use of HDAC Inhibitors



Abstract
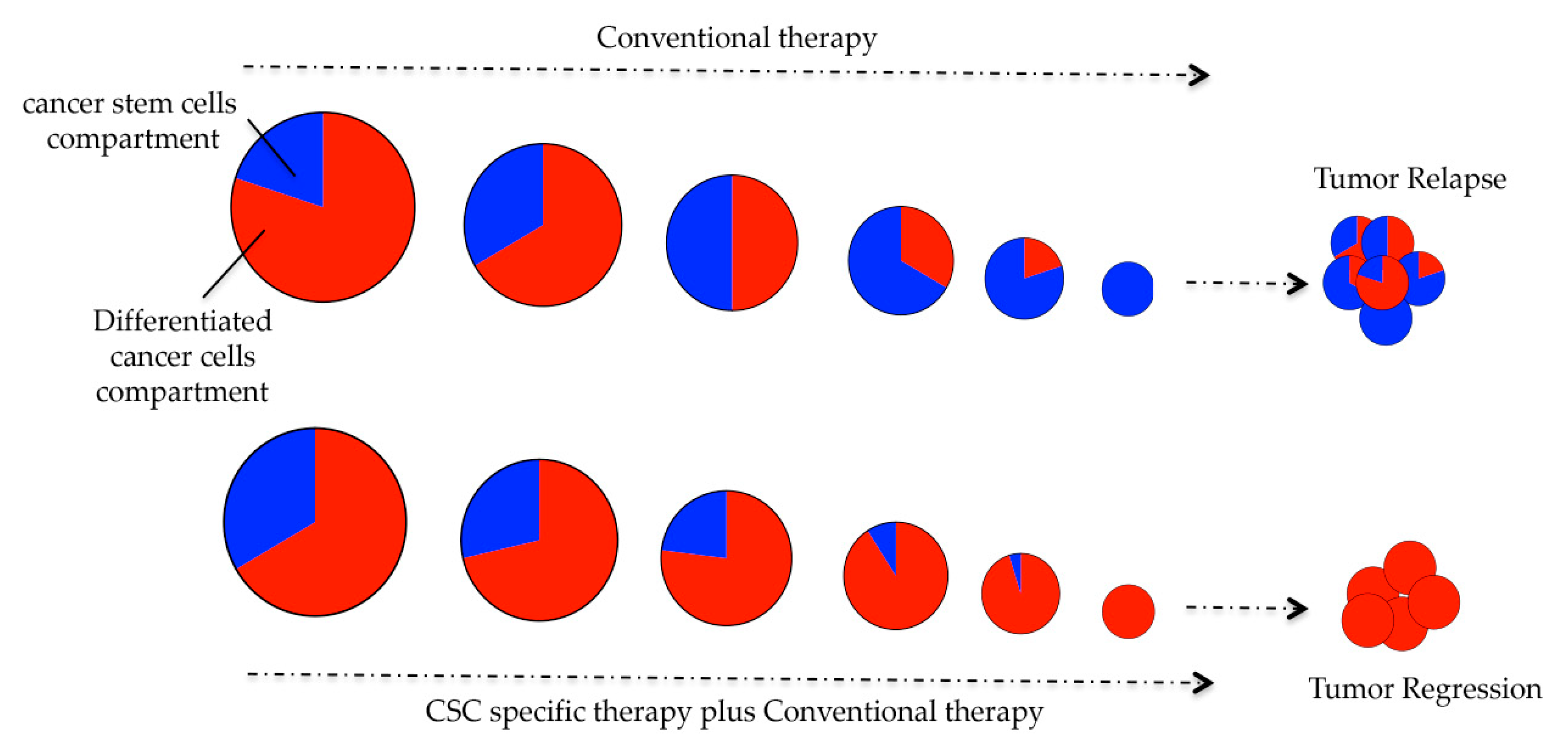
1. Introduction
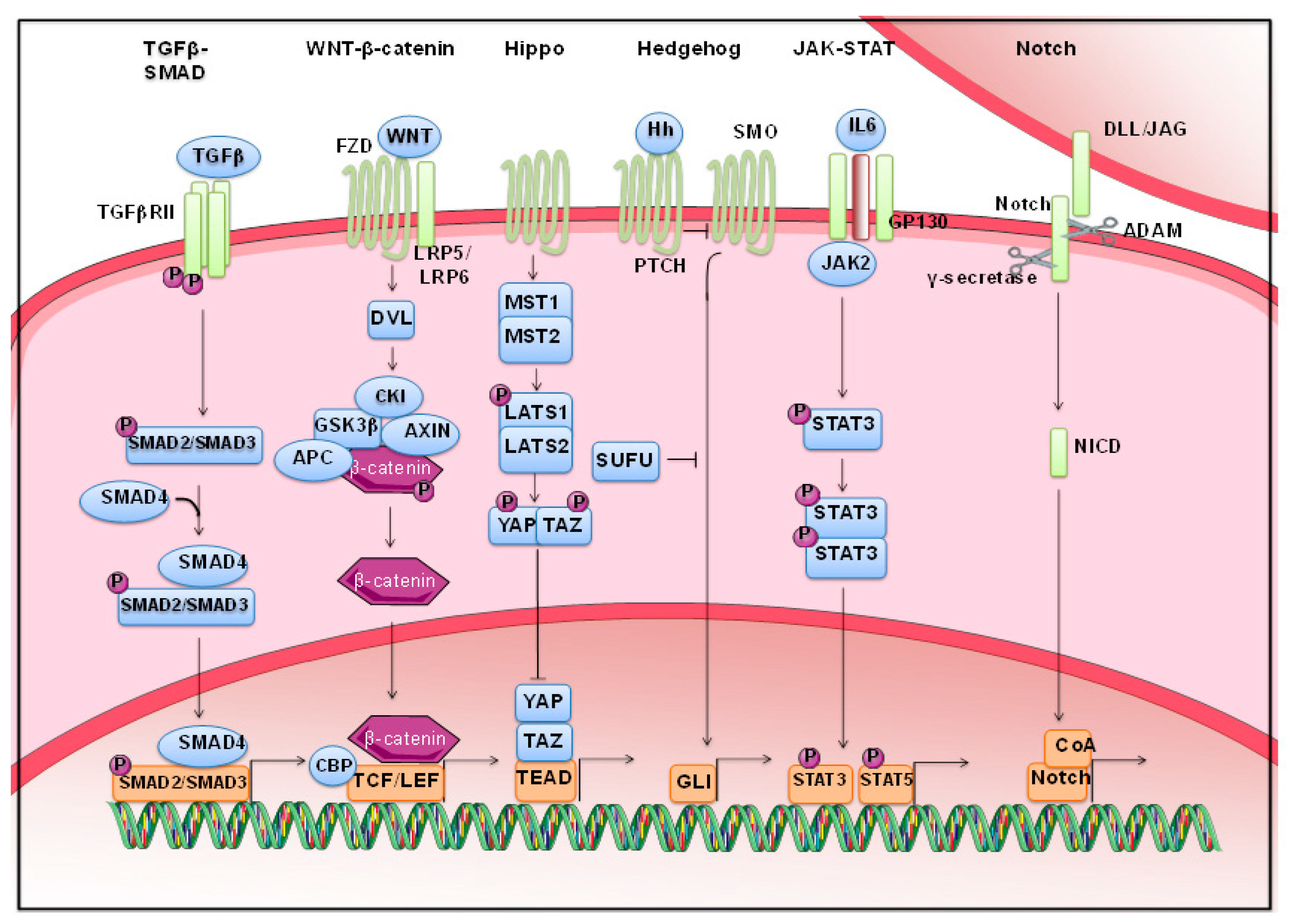
2. Morphogenetic Pathways Are Dysregulated in Solid Cancers
2.1. Wnt Signaling
2.2. Hippo Pathway
2.3. Stats
2.4. TGF-β Signaling
2.5. Notch Pathway
2.6. Hedgehog Pathway
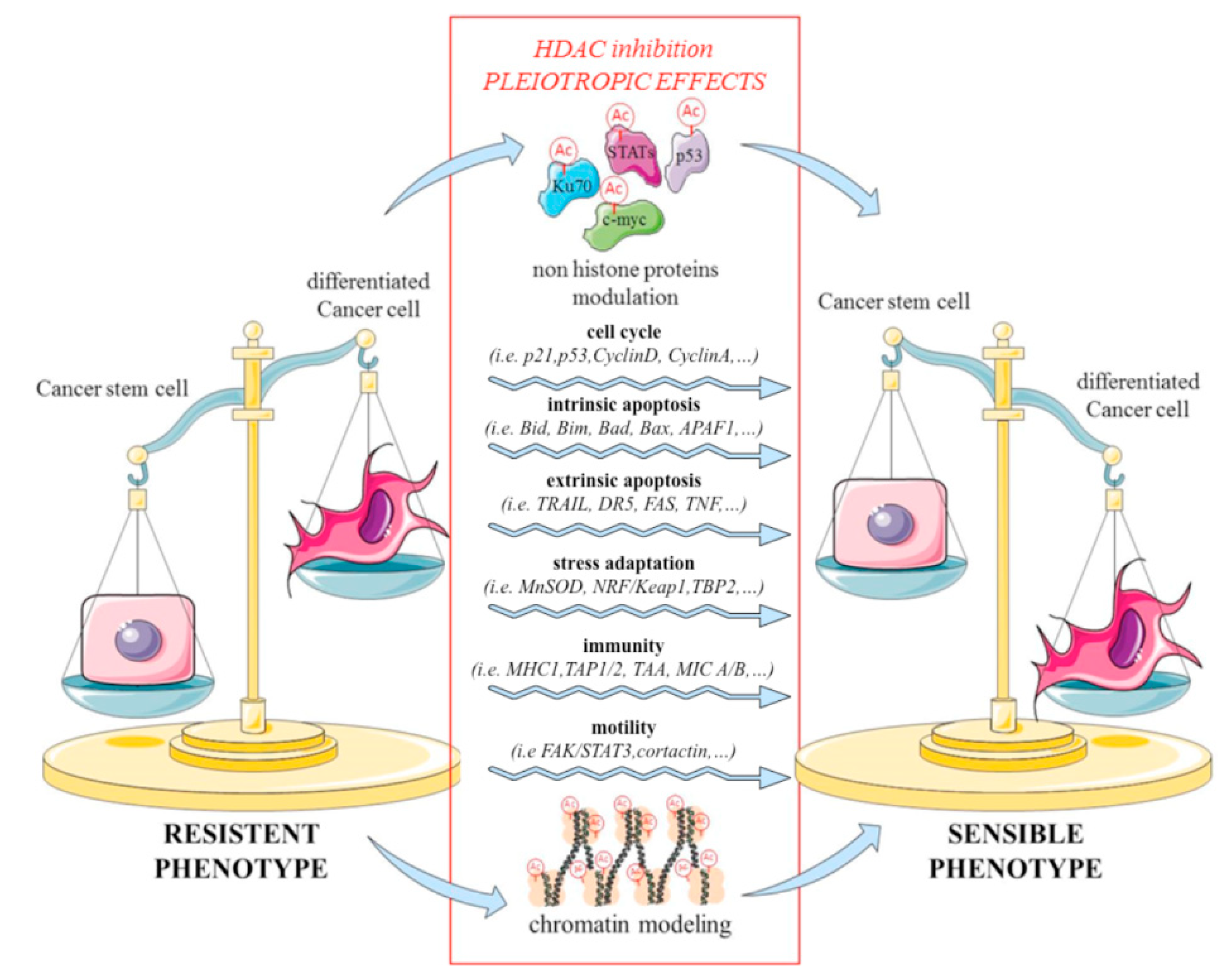
3. Histone Deacetylase (HDAC) Inhibitors Target Cancer Stem Cells (CSCs) and Morphogenetic Pathways
4. CSC Chemo-Toxicity Escape Mechanisms
4.1. Multidrug Resistance
4.2. Apoptosis
4.3. Alteration of DNA Damage Repair System
4.4. Quiescence State
4.5. Metabolism Adaptation
4.6. Immune Evasion
5. HDAC Inhibitors Are Able to Overcome Chemo-Resistance
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Burrell, R.A.; Swanton, C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol. Oncol. 2014, 8, 1095–1111. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.P.; Rose, C.J.; Waterton, J.C.; Carano, R.A.; Parker, G.J.; Jackson, A. Imaging intratumor heterogeneity: Role in therapy response, resistance, and clinical outcome. Clin. Cancer Res. 2015, 21, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef]
- Medemas, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344. [Google Scholar] [CrossRef]
- De Smedt, E.; Lui, H.; Maes, K.; De Veirman, K.; Menu, E.; Vanderkerken, K.; De Bruyne, E. The epigenome in multiple myeloma: Impact on tumor cell plasticity and drug response. Front. Oncol. 2018, 8, 566. [Google Scholar] [CrossRef]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef]
- Wainwright, E.N.; Scaffidi, P. Epigenetics and cancer stem cells: Unleashing, hijacking, and restricting cellular plasticity. Trends Cancer 2017, 3, 372–386. [Google Scholar] [CrossRef]
- Budillon, A.; Di Gennaro, E.; Bruzzese, F.; Rocco, M.; Manzo, G.; Caraglia, M. Histone deacetylase inhibitors: A new wave of molecular targeted anticancer agents. Recent Pat. Anticancer Drug Discov. 2007, 2, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Li, S.; Wu, N.; Cho, K.S. Acetylation and deacetylation in cancer stem-like cells. Oncotarget 2017, 8, 89315–89325. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, A.; Sulaiman, B.; Khouri, L.; McGarry, S.; Nessim, C.; Arnaout, A.; Li, X.; Addison, C.; Dimitroulakos, J.; Wang, L. Both bulk and cancer stem cell subpopulations in triple-negative breast cancer are susceptible to Wnt, HDAC, and ERα coinhibition. FEBS Lett. 2016, 590, 4606–4616. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Witt, A.E.; Lee, C.W.; Lee, T.I.; Azzam, D.J.; Wang, B.; Caslini, C.; Petrocca, F.; Grosso, J.; Jones, M.; Cohick, E.B.; et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene 2017, 36, 1707–1720. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.H.; Xu, X.G.; Yan, S.L.; Sun, Z.; Ying, Y.; Wang, B.K.; Tu, Y.X. Depletion of HDAC1, 7 and 8 by histone deacetylase inhibition confers elimination of pancreatic cancer stem cells in combination with gemcitabine. Sci. Rep. 2018, 8, 1621. [Google Scholar] [CrossRef]
- Chao, M.W.; Chu, P.C.; Chuang, H.C.; Shen, F.H.; Chou, C.C.; Hsu, E.C.; Himmel, L.E.; Huang, H.L.; Tu, H.J.; Kulp, S.K.; et al. Non-epigenetic function of HDAC8 in regulating breast cancer stem cells by maintaining Notch1 protein stability. Oncotarget 2016, 7, 1796–1807. [Google Scholar] [CrossRef]
- An, P.; Li, J.; Lu, L.; Wu, Y.; Ling, Y.; Du, J.; Chen, Z.; Wang, H. Histone deacetylase 8 triggers the migration of triple negative breast cancer cells via regulation of YAP signals. Eur. J. Pharmacol. 2019, 845, 16–23. [Google Scholar] [CrossRef]
- Zimberlin, C.D.; Lancini, C.; Sno, R.; Rosekrans, S.L.; McLean, C.M.; Vlaming, H.; van den Brink, G.R.; Bots, M.; Medema, J.P.; Dannenberg, J.H. HDAC1 and HDAC2 collectively regulate intestinal stem cell homeostasis. FASEB J. 2015, 29, 2070–2080. [Google Scholar] [CrossRef]
- Jamaladdin, S.; Kelly, R.D.; O’Regan, L.; Dovey, O.M.; Hodson, G.E.; Millard, C.J.; Portolano, N.; Fry, A.M.; Schwabe, J.W.; Cowley, S.M. Histone deacetylase (HDAC) 1 and 2 are essential for accurate cell division and the pluripotency of embryonic stem cells. Proc. Natl. Acad Sci. USA 2014, 111, 9840–9845. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Yusoff, S.I.; Roman, M.; Lai, F.Y.; Eagle-Hemming, B.; Murphy, G.J.; Kumar, T.; Wozniak, M. Systematic review and meta-analysis of experimental studies evaluating the organ protective effects of histone deacetylase inhibitors. Transl. Res. 2019, 205, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018, 173, 400–416. [Google Scholar] [CrossRef] [PubMed]
- Fodde, R.; Brabletz, T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr. Opin. Cell Biol. 2007, 19, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Clevers, H. Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 997–1014. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Taha, Z.; Janse van Rensburg, H.J.; Yang, X. The Hippo pathway: Immunity and cancer. Cancers 2018, 10, 94. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Tretiakova, M.S.; Silvis, M.R.; Lucas, J.; Klezovitch, O.; Coleman, I.; Bolouri, H.; Kutyavin, V.I.; Morrissey, C.; True, L.D.; et al. ERG activates the YAP1 transcriptional program and induces the development of age-related prostate tumors. Cancer Cell 2015, 27, 797–808. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, S.; Chen, X.; Stauffer, S.; Yu, F.; Lele, S.M.; Fu, K.; Datta, K.; Palermo, N.; Chen, Y.; et al. The hippo pathway effector YAP regulates motility, invasion, and castration-resistant growth of prostate cancer cells. Mol. Cell Biol. 2015, 35, 1350–1362. [Google Scholar] [CrossRef]
- Bourgeais, J.; Gouilleux-Gruart, V.; Gouilleux, F. Oxidative metabolism in cancer: A STAT affair? Jak-Stat 2013, 2, e25764. [Google Scholar] [CrossRef]
- Ouedraogo, Z.G.; Biau, J.; Kemeny, J.L.; Morel, L.; Verrelle, P.; Chautard, E. Role of STAT3 in genesis and progression of human malignant gliomas. Mol. Neurobiol. 2017, 54, 5780–5797. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Fan, M.; Yang, C.H.; Zbytek, B.; Finkelstein, D.; Roussel, M.F.; Pfeffer, L.M. The critical role that STAT3 plays in glioma-initiating cells: STAT3 addiction in glioma. Oncotarget 2018, 9, 22095–22112. [Google Scholar] [CrossRef] [PubMed]
- West, A.J.; Tsui, V.; Stylli, S.S.; Nguyen, H.P.T.; Morokoff, A.P.; Kaye, A.H.; Luwor, R.B. The role of interleukin-6-STAT3 signalling in glioblastoma. Oncol. Lett. 2018, 16, 4095–4104. [Google Scholar] [CrossRef]
- Kohsaka, S.; Wang, L.; Yachi, K.; Mahabir, R.; Narita, T.; Itoh, T.; Tanino, M.; Kimura, T.; Nishihara, H.; Tanaka, S. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol. Cancer Ther. 2012, 11, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.R.; Xiang, M.; Frank, D.A. STAT3 activity and function in cancer: Modulation by STAT5 and miR-146b. Cancers 2014, 6, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Yusra; Semba, S.; Yokozaki, H. Biological significance of tumor budding at the invasive front of human colorectal carcinoma cells. Int. J. Oncol. 2012, 41, 201–210. [Google Scholar]
- Yu, Y.; Kanwar, S.S.; Patel, B.B.; Oh, P.S.; Nautiyal, J.; Sarkar, F.H.; Majumdar, A.P. MicroRNA-21 induces stemness by downregulating transforming growth factor beta receptor 2 (TGFbetaR2) in colon cancer cells. Carcinogenesis 2012, 33, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, V.; Nataraj, R.; Thangaraj, G.S.; Karthikeyan, M.; Gnanasekaran, A.; Kaginelli, S.B.; Kuppanna, G.; Kallappa, C.G.; Basalingappa, K.M. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig. 2018, 5, 5. [Google Scholar] [CrossRef]
- Katoh, M. Networking of WNT, FGF, Notch, BMP, and Hedgehog signaling pathways during carcinogenesis. Stem Cell Rev. 2007, 3, 30–38. [Google Scholar] [CrossRef]
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Duan, Z.H.; Wang, H.C.; Zhao, D.M.; Ji, X.X.; Song, M.; Yang, X.J.; Cui, W. Cooperatively transcriptional and epigenetic regulation of sonic hedgehog overexpression drives malignant potential of breast cancer. Cancer Sci. 2015, 106, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.; Demuth, T.; Guthrie, T.; Wen, P.Y.; Mason, W.P.; Chinnaiyan, P.; Butowski, N.; Groves, M.D.; Kesari, S.; Freedman, S.J.; et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 2307–2313. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Munoz, P.; Iliou, M.S.; Esteller, M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol. Oncol. 2012, 6, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990, 265, 17174–17179. [Google Scholar]
- Ciardiello, C.; Roca, M.S.; Noto, A.; Bruzzese, F.; Moccia, T.; Vitagliano, C.; Di Gennaro, E.; Ciliberto, G.; Roscilli, G.; Aurisicchio, L.; et al. Synergistic antitumor activity of histone deacetylase inhibitors and anti-ErbB3 antibody in NSCLC primary cultures via modulation of ErbB receptors expression. Oncotarget 2016, 7, 19559–19574. [Google Scholar] [CrossRef]
- Terranova-Barberio, M.; Pecori, B.; Roca, M.S.; Imbimbo, S.; Bruzzese, F.; Leone, A.; Muto, P.; Delrio, P.; Avallone, A.; Budillon, A.; et al. Synergistic antitumor interaction between valproic acid, capecitabine and radiotherapy in colorectal cancer: Critical role of p53. J. Exp. Clin. Cancer Res. 2017, 36, 177. [Google Scholar] [CrossRef]
- Salvador, M.A.; Wicinski, J.; Cabaud, O.; Toiron, Y.; Finetti, P.; Josselin, E.; Lelievre, H.; Kraus-Berthier, L.; Depil, S.; Bertucci, F.; et al. The histone deacetylase inhibitor abexinostat induces cancer stem cells differentiation in breast cancer with low Xist expression. Clin. Cancer Res. 2013, 19, 6520–6531. [Google Scholar] [CrossRef]
- Roca, M.S.; Experimental Pharmacology Unit, Istituto Nazionale Tumori–IRCCS–Fondazione G. Pascale, Naples, Italy. Unpublished work. 2019.
- Wang, Y.; Liu, M.; Jin, Y.; Jiang, S.; Pan, J. In vitro and in vivo anti-uveal melanoma activity of JSL-1, a novel HDAC inhibitor. Cancer Lett. 2017, 400, 47–60. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.; Polakiewicz, R.D.; Yao, T.P.; Comb, M.J. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J. Biol. Chem. 2008, 283, 12686–12690. [Google Scholar] [CrossRef]
- Yu, S.; Cai, X.; Wu, C.; Liu, Y.; Zhang, J.; Gong, X.; Wang, X.; Wu, X.; Zhu, T.; Mo, L.; et al. Targeting HSP90-HDAC6 regulating network implicates precision treatment of breast cancer. Int. J. Biol. Sci. 2017, 13, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fang, R.; Liu, B.; Shi, H.; Wang, Y.; Zhang, W.; Zhang, X.; Ye, L. Deacetylation of tumor-suppressor MST1 in Hippo pathway induces its degradation through HBXIP-elevated HDAC6 in promotion of breast cancer growth. Oncogene 2016, 35, 4048–4057. [Google Scholar] [CrossRef] [PubMed]
- Shan, B.; Yao, T.P.; Nguyen, H.T.; Zhuo, Y.; Levy, D.R.; Klingsberg, R.C.; Tao, H.; Palmer, M.L.; Holder, K.N.; Lasky, J.A. Requirement of HDAC6 for transforming growth factor- beta1-induced epithelial-mesenchymal transition. J. Biol. Chem. 2008, 283, 21065–21073. [Google Scholar] [CrossRef] [PubMed]
- Sferra, R.; Pompili, S.; Festuccia, C.; Marampon, F.; Gravina, G.L.; Ventura, L.; Di Cesare, E.; Cicchinelli, S.; Gaudio, E.; Vetuschi, A. The possible prognostic role of histone deacetylase and transforming growth factor beta/Smad signaling in high grade gliomas treated by radio-chemotherapy: A preliminary immunohistochemical study. Eur. J. Histochem. 2017, 61, 2732. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, Y.; Gao, R.; Yu, H.; Sun, T. HDAC6 inhibition induces glioma stem cells differentiation and enhances cellular radiation sensitivity through the SHH/Gli1 signaling pathway. Cancer Lett. 2018, 415, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.N.; Ding, W.Q.; Guo, X.J.; Yuan, X.W.; Wang, D.M.; Song, J.G. Epigenetic regulation of Smad2 and Smad3 by profilin-2 promotes lung cancer growth and metastasis. Nat. Commun. 2015, 6, 8230. [Google Scholar] [CrossRef] [PubMed]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Mao, S.; Lu, G.; Li, L.; Lan, X.; Huang, Z.; Chen, Y.; Zhao, M.; Zhao, Y.; Xia, Q. Valproic acid sensitizes metformin-resistant human renal cell carcinoma cells by upregulating H3 acetylation and EMT reversal. BMC Cancer 2018, 18, 434. [Google Scholar] [CrossRef] [PubMed]
- Iannelli, F.; Experimental Pharmacology Unit, Istituto Nazionale Tumori–IRCCS–Fondazione G. Pascale, Naples, Italy. Unpublished work. 2019.
- Sun, G.; Mackey, L.V.; Coy, D.H.; Yu, C.Y.; Sun, L. The histone deacetylase inhibitor vaproic acid induces cell growth arrest in hepatocellular carcinoma cells via suppressing notch signaling. J. Cancer 2015, 6, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Ma, W.; Lei, F.; Li, Q.; Su, Y.; Lin, X.; Lin, C.; Zhang, X.; Ye, L.; Wu, S.; et al. Prostate tumour overexpressed-1 promotes tumourigenicity in human breast cancer via activation of Wnt/beta-catenin signalling. J. Pathol. 2016, 239, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Kobayashi, S.; Yamada, D.; Nagano, H.; Tomokuni, A.; Tomimaru, Y.; Noda, T.; Gotoh, K.; Asaoka, T.; Wada, H.; et al. A histone deacetylase inhibitor suppresses epithelial-mesenchymal transition and attenuates chemoresistance in biliary tract cancer. PLoS ONE 2016, 11, e0145985. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, L.; Liu, C.; Pan, J.; Lu, G.; Zhou, Z.; Chen, Z.; Qian, C. Notch3 overexpression enhances progression and chemoresistance of urothelial carcinoma. Oncotarget 2017, 8, 34362–34373. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.W.; Yarravarapu, N.; Shi, H.; Kulak, O.; Kim, J.; Chen, C.; Lum, L. A synthetic combinatorial approach to disabling deviant Hedgehog signaling. Sci. Rep. 2018, 8, 1133. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fan, B.; Zhang, L.H.; Xing, X.F.; Cheng, X.J.; Wang, X.H.; Guo, T.; Du, H.; Wen, X.Z.; Ji, J.F. Trichostatin A potentiates TRAIL-induced antitumor effects via inhibition of ERK/FOXM1 pathway in gastric cancer. Tumour Biol. 2016, 37, 10269–10278. [Google Scholar] [CrossRef] [PubMed]
- Gruber, W.; Peer, E.; Elmer, D.P.; Sternberg, C.; Tesanovic, S.; Del Burgo, P.; Coni, S.; Canettieri, G.; Neureiter, D.; Bartz, R.; et al. Targeting class I histone deacetylases by the novel small molecule inhibitor 4SC-202 blocks oncogenic hedgehog-GLI signaling and overcomes smoothened inhibitor resistance. Int. J. Cancer 2018, 142, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Debeb, B.G.; Lacerda, L.; Xu, W.; Larson, R.; Solley, T.; Atkinson, R.; Sulman, E.P.; Ueno, N.T.; Krishnamurthy, S.; Reuben, J.M.; et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/beta-catenin signaling. Stem Cells 2012, 30, 2366–2377. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A.; Ismail-Khan, R.R.; Melichar, B.; Lichinitser, M.; Munster, P.N.; Klein, P.M.; Cruickshank, S.; Miller, K.D.; Lee, M.J.; Trepel, J.B. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J. Clin. Oncol. 2013, 31, 2128–2135. [Google Scholar] [PubMed]
- Chan, E.; Chiorean, E.G.; O’Dwyer, P.J.; Gabrail, N.Y.; Alcindor, T.; Potvin, D.; Chao, R.; Hurwitz, H. Phase I/II study of mocetinostat in combination with gemcitabine for patients with advanced pancreatic cancer and other advanced solid tumors. Cancer Chemother. Pharmacol. 2018, 81, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Manic, G.; De Maria, R.; Kroemer, G.; Galluzzi, L. DNA damage in stem cells. Mol. Cell 2017, 66, 306–319. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Chau, W.K.; Ip, C.K.; Mak, A.S.; Lai, H.C.; Wong, A.S. c-Kit mediates chemoresistance and tumor-initiating capacity of ovarian cancer cells through activation of Wnt/beta-catenin-ATP-binding cassette G2 signaling. Oncogene 2013, 32, 2767–2781. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Grimmig, T.; Gonzalez, G.; Giobbie-Hurder, A.; Berg, G.; Carr, N.; Wilson, B.J.; Banerjee, P.; Ma, J.; Gold, J.S.; et al. ATP-binding cassette member B5 (ABCB5) promotes tumor cell invasiveness in human colorectal cancer. J. Biol. Chem. 2018, 293, 11166–11178. [Google Scholar] [CrossRef] [PubMed]
- Shervington, A.; Lu, C. Expression of multidrug resistance genes in normal and cancer stem cells. Cancer Investig. 2008, 26, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef]
- Fulda, S. Regulation of apoptosis pathways in cancer stem cells. Cancer Lett. 2013, 338, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Madjd, Z.; Mehrjerdi, A.Z.; Sharifi, A.M.; Molanaei, S.; Shahzadi, S.Z.; Asadi-Lari, M. CD44+ cancer cells express higher levels of the anti-apoptotic protein Bcl-2 in breast tumours. Cancer Immun. 2009, 9, 4. [Google Scholar]
- Konopleva, M.; Zhao, S.; Hu, W.; Jiang, S.; Snell, V.; Weidner, D.; Jackson, C.E.; Zhang, X.; Champlin, R.; Estey, E.; et al. The anti-apoptotic genes Bcl-X(L) and Bcl-2 are over-expressed and contribute to chemoresistance of non-proliferating leukaemic CD34+ cells. Br. J. Haematol. 2002, 118, 521–534. [Google Scholar] [CrossRef]
- Colak, S.; Zimberlin, C.D.; Fessler, E.; Hogdal, L.; Prasetyanti, P.R.; Grandela, C.M.; Letai, A.; Medema, J.P. Decreased mitochondrial priming determines chemoresistance of colon cancer stem cells. Cell Death Differ. 2014, 21, 1170–1177. [Google Scholar] [CrossRef]
- Todaro, M.; Alea, M.P.; Di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Iovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 2007, 1, 389–402. [Google Scholar] [CrossRef]
- Xie, Q.; Wang, S.; Zhao, Y.; Zhang, Z.; Qin, C.; Yang, X. MiR-519d impedes cisplatin-resistance in breast cancer stem cells by down-regulating the expression of MCL-1. Oncotarget 2017, 8, 22003–22013. [Google Scholar] [CrossRef] [PubMed]
- Rouhrazi, H.; Turgan, N.; Oktem, G. Zoledronic acid overcomes chemoresistance by sensitizing cancer stem cells to apoptosis. Biotech. Histochem. 2018, 93, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.R.; Pucci, B.; Bifulco, K.; Iannelli, F.; Lombardi, R.; Ciardiello, C.; Bruzzese, F.; Carriero, M.V.; Budillon, A. Proteomic analysis of zoledronic-acid resistant prostate cancer cells unveils novel pathways characterizing an invasive phenotype. Oncotarget 2015, 6, 5324–5341. [Google Scholar] [PubMed]
- Milone, M.R.; Pucci, B.; Bruzzese, F.; Carbone, C.; Piro, G.; Costantini, S.; Capone, F.; Leone, A.; Di Gennaro, E.; Caraglia, M.; et al. Acquired resistance to zoledronic acid and the parallel acquisition of an aggressive phenotype are mediated by p38-MAP kinase activation in prostate cancer cells. Cell Death Dis. 2013, 4, e641. [Google Scholar] [CrossRef] [PubMed]
- Van Houdt, W.J.; Emmink, B.L.; Pham, T.V.; Piersma, S.R.; Verheem, A.; Vries, R.G.; Fratantoni, S.A.; Pronk, A.; Clevers, H.; Borel Rinkes, I.H.; et al. Comparative proteomics of colon cancer stem cells and differentiated tumor cells identifies BIRC6 as a potential therapeutic target. Mol. Cell Proteomics 2011, 10, M111.011353. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, R.; Ahmed, S.U.; Strathdee, K.; Gomez-Roman, N.; Amoah-Buahin, E.; Watts, C.; Chalmers, A.J. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol. Oncol. 2015, 9, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wei, Y.; Wang, L.; Debeb, B.G.; Yuan, Y.; Zhang, J.; Yuan, J.; Wang, M.; Chen, D.; Sun, Y.; et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 2014, 16, 864–875. [Google Scholar] [CrossRef]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed]
- Soeda, A.; Lathia, J.; Williams, B.J.; Wu, Q.; Gallagher, J.; Androutsellis-Theotokis, A.; Giles, A.J.; Yang, C.; Zhuang, Z.; Gilbert, M.R.; et al. The p38 signaling pathway mediates quiescence of glioma stem cells by regulating epidermal growth factor receptor trafficking. Oncotarget 2017, 8, 33316–33328. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Honma, D.; Adachi, N.; Araki, K.; Takamatsu, E.; Katsumoto, T.; Yamagata, K.; Akashi, K.; Aoyama, K.; Iwama, A.; et al. Dual inhibition of EZH1/2 breaks the quiescence of leukemia stem cells in acute myeloid leukemia. Leukemia 2018, 32, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Joshi, K.; Ezhilarasan, R.; Myers, T.R.; Siu, J.; Gu, C.; Nakano-Okuno, M.; Taylor, D.; Minata, M.; Sulman, E.P.; et al. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem Cell Rep. 2015, 4, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Yang, J.Y.; Xia, W.; Chen, C.T.; Xie, X.; Chao, C.H.; Woodward, W.A.; Hsu, J.M.; Hortobagyi, G.N.; Hung, M.C. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-β-catenin signaling. Cancer Cell 2011, 19, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Leone, A.; Roca, M.S.; Ciardiello, C.; Costantini, S.; Budillon, A. Oxidative stress gene expression profile correlates with cancer patient poor prognosis: Identification of crucial pathways might select novel therapeutic approaches. Oxid. Med. Cell Longev. 2017, 2017, 2597581. [Google Scholar] [CrossRef]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef]
- Prieto, J.; Leon, M.; Ponsoda, X.; Sendra, R.; Bort, R.; Ferrer-Lorente, R.; Raya, A.; Lopez-Garcia, C.; Torres, J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 2016, 7, 11124. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.E.; Villanova, L.; De Maria, R. Cancer stem cells: At the forefront of personalized medicine and immunotherapy. Curr. Opin. Pharmacol. 2017, 35, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Maccalli, C.; Volonte, A.; Cimminiello, C.; Parmiani, G. Immunology of cancer stem cells in solid tumours. A review. Eur. J. Cancer 2014, 50, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/beta-catenin pathway activation correlates with immune exclusion across human cancers. Clin. Cancer Res. 2019, 25, 3074–3083. [Google Scholar] [CrossRef]
- Agudo, J.; Park, E.S.; Rose, S.A.; Alibo, E.; Sweeney, R.; Dhainaut, M.; Kobayashi, K.S.; Sachidanandam, R.; Baccarini, A.; Merad, M.; et al. Quiescent tissue stem cells evade immune surveillance. Immunity 2018, 48, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.H.; Mu, L.; Li, X.L.; Hu, Y.B.; Liu, H.; Han, L.T.; Gong, J.P. Characterization and functional analysis of a slow-cycling subpopulation in colorectal cancer enriched by cell cycle inducer combined chemotherapy. Oncotarget 2017, 8, 78466–78479. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Tanikawa, T.; Li, W.; Zhao, L.; Vatan, L.; Szeliga, W.; Wan, S.; Wei, S.; Wang, Y.; Liu, Y.; et al. Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH cross-talk signaling. Cancer Res. 2016, 76, 3156–3165. [Google Scholar] [CrossRef]
- Huang, X.P.; Li, X.; Situ, M.Y.; Huang, L.Y.; Wang, J.Y.; He, T.C.; Yan, Q.H.; Xie, X.Y.; Zhang, Y.J.; Gao, Y.H.; et al. Entinostat reverses cisplatin resistance in esophageal squamous cell carcinoma via down-regulation of multidrug resistance gene 1. Cancer Lett. 2018, 414, 294–300. [Google Scholar] [CrossRef]
- Zhao, G.; Wang, G.; Bai, H.; Li, T.; Gong, F.; Yang, H.; Wen, J.; Wang, W. Targeted inhibition of HDAC8 increases the doxorubicin sensitivity of neuroblastoma cells via up regulation of miR-137. Eur. J. Pharmacol. 2017, 802, 20–26. [Google Scholar] [CrossRef]
- To, K.K.; Tong, W.S.; Fu, L.W. Reversal of platinum drug resistance by the histone deacetylase inhibitor belinostat. Lung Cancer 2017, 103, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huang, C.; Zhao, L.; Zhang, H.; Yang, J.M.; Luo, P.; Zhan, B.X.; Pan, Q.; Li, J.; Wang, B.L. Histone deacetylase inhibitors regulate P-gp expression in colorectal cancer via transcriptional activation and mRNA stabilization. Oncotarget 2016, 7, 49848–49858. [Google Scholar] [CrossRef]
- Tomono, T.; Machida, T.; Kamioka, H.; Shibasaki, Y.; Yano, K.; Ogihara, T. Entinostat reverses P-glycoprotein activation in snail-overexpressing adenocarcinoma HCC827 cells. PLoS ONE 2018, 13, e0200015. [Google Scholar] [CrossRef] [PubMed]
- Chikamatsu, K.; Ishii, H.; Murata, T.; Sakakura, K.; Shino, M.; Toyoda, M.; Takahashi, K.; Masuyama, K. Alteration of cancer stem cell-like phenotype by histone deacetylase inhibitors in squamous cell carcinoma of the head and neck. Cancer Sci. 2013, 104, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Aztopal, N.; Erkisa, M.; Erturk, E.; Ulukaya, E.; Tokullugil, A.H.; Ari, F. Valproic acid, a histone deacetylase inhibitor, induces apoptosis in breast cancer stem cells. Chem. Biol. Interact. 2018, 280, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Di Pompo, G.; Salerno, M.; Rotili, D.; Valente, S.; Zwergel, C.; Avnet, S.; Lattanzi, G.; Baldini, N.; Mai, A. Novel histone deacetylase inhibitors induce growth arrest, apoptosis, and differentiation in sarcoma cancer stem cells. J. Med. Chem. 2015, 58, 4073–4079. [Google Scholar] [CrossRef] [PubMed]
- Nalls, D.; Tang, S.N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef]
- Song, K.H.; Choi, C.H.; Lee, H.J.; Oh, S.J.; Woo, S.R.; Hong, S.O.; Noh, K.H.; Cho, H.; Chung, E.J.; Kim, J.H.; et al. HDAC1 upregulation by NANOG promotes multidrug resistance and a stem-like phenotype in immune edited tumor cells. Cancer Res. 2017, 77, 5039–5053. [Google Scholar] [CrossRef]
- Di Gennaro, E.; Bruzzese, F.; Pepe, S.; Leone, A.; Delrio, P.; Subbarayan, P.R.; Avallone, A.; Budillon, A. Modulation of thymidilate synthase and p53 expression by HDAC inhibitor vorinostat resulted in synergistic antitumor effect in combination with 5FU or raltitrexed. Cancer Biol. Ther. 2009, 8, 782–791. [Google Scholar] [CrossRef]
- Di Gennaro, E.; Piro, G.; Chianese, M.I.; Franco, R.; Di Cintio, A.; Moccia, T.; Luciano, A.; de Ruggiero, I.; Bruzzese, F.; Avallone, A.; et al. Vorinostat synergises with capecitabine through upregulation of thymidine phosphorylase. Br. J. Cancer 2010, 103, 1680–1691. [Google Scholar] [CrossRef]
- Terranova-Barberio, M.; Roca, M.S.; Zotti, A.I.; Leone, A.; Bruzzese, F.; Vitagliano, C.; Scogliamiglio, G.; Russo, D.; D’Angelo, G.; Franco, R.; et al. Valproic acid potentiates the anticancer activity of capecitabine in vitro and in vivo in breast cancer models via induction of thymidine phosphorylase expression. Oncotarget 2016, 7, 7715–7731. [Google Scholar] [CrossRef] [PubMed]
- Fazzone, W.; Wilson, P.M.; Labonte, M.J.; Lenz, H.J.; Ladner, R.D. Histone deacetylase inhibitors suppress thymidylate synthase gene expression and synergize with the fluoropyrimidines in colon cancer cells. Int. J. Cancer 2009, 125, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Piro, G.; Roca, M.S.; Bruzzese, F.; Carbone, C.; Iannelli, F.; Leone, A.; Volpe, M.G.; Budillon, A.; Di Gennaro, E. Vorinostat potentiates cisplatin-5-fluorouracil combination by inhibiting chemotherapy-induced EGFR nuclear translocation and increasing cisplatin uptake. Mol. Cancer Ther. 2019, in press. [Google Scholar]
- Muller, B.M.; Jana, L.; Kasajima, A.; Lehmann, A.; Prinzler, J.; Budczies, J.; Winzer, K.J.; Dietel, M.; Weichert, W.; Denkert, C. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer—Overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 2013, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Spurling, C.C.; Godman, C.A.; Noonan, E.J.; Rasmussen, T.P.; Rosenberg, D.W.; Giardina, C. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol. Carcinog. 2008, 47, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Minami, J.; Suzuki, R.; Mazitschek, R.; Gorgun, G.; Ghosh, B.; Cirstea, D.; Hu, Y.; Mimura, N.; Ohguchi, H.; Cottini, F.; et al. Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia 2014, 28, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Ackland, S.P.; Clarke, S.J.; Beale, P.; Peters, G.J. Thymidylate synthase inhibitors. Cancer Chemother. Biol. Response Modif. 2002, 20, 1–36. [Google Scholar] [PubMed]
- Avallone, A.; Piccirillo, M.C.; Delrio, P.; Pecori, B.; Di Gennaro, E.; Aloj, L.; Tatangelo, F.; D’Angelo, V.; Granata, C.; Cavalcanti, E.; et al. Phase 1/2 study of valproic acid and short-course radiotherapy plus capecitabine as preoperative treatment in low-moderate risk rectal cancer-V-shoRT-R3 (Valproic acid—Short Radiotherapy—Rectum 3rd trial). BMC Cancer 2014, 14, 875. [Google Scholar] [CrossRef] [PubMed]
- Avallone, A.; Istituto Nazionale Tumori–IRCCS–Fondazione G. Pascale, 80131, Naples, Italy. Unpublished work. 2019.
- Huang, T.H.; Wu, S.Y.; Huang, Y.J.; Wei, P.L.; Wu, A.T.; Chao, T.Y. The identification and validation of Trichosstatin A as a potential inhibitor of colon tumorigenesis and colon cancer stem-like cells. Am. J. Cancer Res. 2017, 7, 1227–1237. [Google Scholar]
- Kumar, B.; Yadav, A.; Lang, J.C.; Teknos, T.N.; Kumar, P. Suberoylanilide hydroxamic acid (SAHA) reverses chemoresistance in head and neck cancer cells by targeting cancer stem cells via the downregulation of nanog. Genes Cancer 2015, 6, 169–181. [Google Scholar]
- Wang, L.; Liu, X.; Ren, Y.; Zhang, J.; Chen, J.; Zhou, W.; Guo, W.; Wang, X.; Chen, H.; Li, M.; et al. Cisplatin-enriching cancer stem cells confer multidrug resistance in non-small cell lung cancer via enhancing TRIB1/HDAC activity. Cell Death Dis. 2017, 8, e2746. [Google Scholar] [CrossRef]
- Miyajima, C.; Inoue, Y.; Hayashi, H. Pseudokinase tribbles 1 (TRB1) negatively regulates tumor-suppressor activity of p53 through p53 deacetylation. Biol. Pharm. Bull. 2015, 38, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, N.; Pang, B.; Tong, D.; Sun, D.; Sun, H.; Zhang, C.; Sun, W.; Meng, X.; Bai, J.; et al. TRIB1 promotes colorectal cancer cell migration and invasion through activation MMP-2 via FAK/Src and ERK pathways. Oncotarget 2017, 8, 47931–47942. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.X.; Hu, J.J.; Fang, Y.; Wang, Z.T.; Xie, J.J.; Zhan, Q.; Deng, X.X.; Chen, H.; Jin, J.B.; Peng, C.H.; et al. A case-control study indicates that the TRIB1 gene is associated with pancreatic cancer. Genet. Mol. Res. 2014, 13, 6142–6147. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Park, S.B.; Kim, S.A.; Kwon, S.K.; Cha, H.; Lee, D.Y.; Ro, S.; Cho, J.M.; Song, S.Y. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci. Rep. 2017, 7, 41615. [Google Scholar] [CrossRef] [PubMed]
- Ouaissi, M.; Sielezneff, I.; Silvestre, R.; Sastre, B.; Bernard, J.P.; Lafontaine, J.S.; Payan, M.J.; Dahan, L.; Pirro, N.; Seitz, J.F.; et al. High histone deacetylase 7 (HDAC7) expression is significantly associated with adenocarcinomas of the pancreas. Ann. Surg. Oncol. 2008, 15, 2318–2328. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.E.; Park, S.B.; Kim, K.; Kim, C.; Song, S.Y. CG200745, an HDAC inhibitor, induces anti-tumour effects in cholangiocarcinoma cell lines via miRNAs targeting the Hippo pathway. Sci. Rep. 2017, 7, 10921. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Lee, E.J.; Ji, M.; Park, S.M. HDAC inhibitors, trichostatin A and valproic acid, increase Ecadherin and vimentin expression but inhibit migration and invasion of cholangiocarcinoma cells. Oncol. Rep. 2018, 40, 346–354. [Google Scholar]
- Lopez, G.; Braggio, D.; Zewdu, A.; Casadei, L.; Batte, K.; Bid, H.K.; Koller, D.; Yu, P.; Iwenofu, O.H.; Strohecker, A.; et al. Mocetinostat combined with gemcitabine for the treatment of leiomyosarcoma: Preclinical correlates. PLoS ONE 2017, 12, e0188859. [Google Scholar] [CrossRef]
- Matulonis, U.; Berlin, S.; Lee, H.; Whalen, C.; Obermayer, E.; Penson, R.; Liu, J.; Campos, S.; Krasner, C.; Horowitz, N. Phase I study of combination of vorinostat, carboplatin, and gemcitabine in women with recurrent, platinum-sensitive epithelial ovarian, fallopian tube, or peritoneal cancer. Cancer Chemother. Pharmacol. 2015, 76, 417–423. [Google Scholar] [CrossRef]
- Bruzzese, F.; Leone, A.; Rocco, M.; Carbone, C.; Piro, G.; Caraglia, M.; Di Gennaro, E.; Budillon, A. HDAC inhibitor vorinostat enhances the antitumor effect of gefitinib in squamous cell carcinoma of head and neck by modulating ErbB receptor expression and reverting EMT. J. Cell Physiol. 2011, 226, 2378–2390. [Google Scholar] [CrossRef]
- Leone, A.; Roca, M.S.; Ciardiello, C.; Terranova-Barberio, M.; Vitagliano, C.; Ciliberto, G.; Mancini, R.; Di Gennaro, E.; Bruzzese, F.; Budillon, A. Vorinostat synergizes with EGFR inhibitors in NSCLC cells by increasing ROS via up-regulation of the major mitochondrial porin VDAC1 and modulation of the c-Myc-NRF2-KEAP1 pathway. Free Radic. Biol. Med. 2015, 89, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, F.; Hu, P.; Wang, Y.; Gong, J.; Sun, S.; Xie, C. HDAC6 promotes cell proliferation and confers resistance to gefitinib in lung adenocarcinoma. Oncol. Rep. 2016, 36, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, H.; Ren, Y.; Zou, S.; Fang, W.; Jiang, X.; Jia, L.; Li, M.; Liu, X.; Yuan, X.; et al. Targeting HDAC with a novel inhibitor effectively reverses paclitaxel resistance in non-small cell lung cancer via multiple mechanisms. Cell Death Dis. 2016, 7, e2063. [Google Scholar] [CrossRef] [PubMed]
- Galloway, T.J.; Wirth, L.J.; Colevas, A.D.; Gilbert, J.; Bauman, J.E.; Saba, N.F.; Raben, D.; Mehra, R.; Ma, A.W.; Atoyan, R.; et al. A phase I study of CUDC-101, a multitarget inhibitor of HDACs, EGFR, and HER2, in combination with chemoradiation in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H. The immune revolution: A case for priming, not checkpoint. Cancer Cell 2018, 33, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Terranova-Barberio, M.; Thomas, S.; Munster, P.N. Epigenetic modifiers in immunotherapy: A focus on checkpoint inhibitors. Immunotherapy 2016, 8, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Terranova-Barberio, M.; Thomas, S.; Ali, N.; Pawlowska, N.; Park, J.; Krings, G.; Rosenblum, M.D.; Budillon, A.; Munster, P.N. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 2017, 8, 114156–114172. [Google Scholar] [CrossRef]
- Miyashita, T.; Miki, K.; Kamigaki, T.; Makino, I.; Tajima, H.; Nakanuma, S.; Hayashi, H.; Takamura, H.; Fushida, S.; Ahmed, A.K.; et al. Low-dose valproic acid with low-dose gemcitabine augments MHC class I-related chain A/B expression without inducing the release of soluble MHC class I-related chain A/B. Oncol. Lett. 2017, 14, 5918–5926. [Google Scholar] [CrossRef]
- Krug, L.M.; Kindler, H.L.; Calvert, H.; Manegold, C.; Tsao, A.S.; Fennell, D.; Ohman, R.; Plummer, R.; Eberhardt, W.E.; Fukuoka, K.; et al. Vorinostat in patients with advanced malignant pleural mesothelioma who have progressed on previous chemotherapy (VANTAGE-014): A phase 3, double-blind, randomised, placebo-controlled trial. Lancet Oncol. 2015, 16, 447–456. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: Achieving the full therapeutic potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Maitland, M.L.; Frankel, P.; Argiris, A.E.; Koczywas, M.; Gitlitz, B.; Thomas, S.; Espinoza-Delgado, I.; Vokes, E.E.; Gandara, D.R.; et al. Carboplatin and Paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Wilting, R.H.; Yanover, E.; Heideman, M.R.; Jacobs, H.; Horner, J.; van der Torre, J.; DePinho, R.A.; Dannenberg, J.H. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 2010, 29, 2586–2597. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Status | CSCs-Activated Signaling Pathways | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Solid Cancer | TCGA_Id | Whole Sample | Alive | Dead | Hippo | Wnt | Jak/STAT | TGF | Notch | Hedgehog |
| Mixed Colon Adenocarcinoma | COAD | 174 | 140 | 15 | 0.880 | 0.310 | 0.470 | 0.640 | 0.950 | 0.910 |
| Rectum adenocarcinoma | READ | 95 | 87 | 7 | 0.360 | 0.800 | 0.00041 | 0.150 | 1.000 | 0.360 |
| Pancreatic adenocarcinoma | PAAD | 178 | 119 | 59 | 0.270 | 0.330 | 0.430 | 0.550 | 0.390 | 0.520 |
| Lung Adenocarcinoma | LUAD | 515 | 389 | 126 | 0.900 | 0.300 | 0.400 | 0.210 | 0.800 | 0.420 |
| Lung Squamous Cell Carcinoma | LUSC | 81 | 61 | 19 | 0.390 | 0.530 | 0.610 | 0.440 | 0.580 | 0.620 |
| Prostate Adenocarcinoma | PRAD | 497 | 489 | 8 | 0.016 | 0.300 | 0.670 | 0.210 | 0.360 | 0.360 |
| Stomach adenocarcinoma | STAD | 415 | 336 | 79 | 0.260 | 0.410 | 0.500 | 0.310 | 0.460 | 0.520 |
| Liver Hepatocellular Carcinoma | LIHC | 371 | 282 | 89 | 0.840 | 0.510 | 0.580 | 0.680 | 0.550 | 0.680 |
| Kidney Renal Clear Cell Carcinoma | KIRC | 533 | 363 | 160 | 0.780 | 0.620 | 0.810 | 0.120 | 0.080 | 0.950 |
| Head Neck Squamous Cell Carcinoma | HNSC | 520 | 353 | 167 | 0.140 | 0.006 | 0.030 | 0.930 | 0.440 | 0.500 |
| Cervical Squamous Cell Carcinoma | CESC | 305 | 244 | 60 | 0.400 | 0.610 | 0.520 | 0.630 | 0.480 | 0.530 |
| Bladder Urothelial Carcinoma | BLCA | 408 | 300 | 108 | 0.260 | 0.100 | 0.500 | 0.310 | 0.440 | 0.520 |
| Sarcoma | SARC | 259 | 184 | 75 | 0.580 | 0.680 | 0.740 | 0.620 | 0.720 | 0.750 |
| Breast Invasive Carcinoma | BRCA | 1097 | 992 | 104 | 0.100 | 0.300 | 0.400 | 0.670 | 0.800 | 0.000008 |
| Glioblastoma | TARGET_NBL | 153 | 52 | 99 | 0.840 | 0.400 | 0.002 | 0.410 | 0.550 | 0.160 |
| Skin Cutaneous Melanoma | SKCM | 470 | 313 | 156 | 0.360 | 0.500 | 0.580 | 0.410 | 0.550 | 0.600 |
| Recruiting | Active, Not Recruiting | Not Yet Recruiting | Completed | Terminated | Suspended | Withdrawn | Unknown Status | Sum | |
|---|---|---|---|---|---|---|---|---|---|
| Early Phase 1 | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 4 |
| Phase 1 | 24 | 34 | 8 | 109 | 45 | 2 | 5 | 3 | 230 |
| Phase 2 | 15 | 16 | 3 | 55 | 37 | 0 | 1 | 3 | 130 |
| Phase 3 | 1 | 2 | 0 | 1 | 1 | 0 | 0 | 2 | 7 |
| Not Applicable | 0 | 1 | 0 | 4 | 0 | 0 | 1 | 0 | 6 |
| Phase | Title | Status | Completion Date | Description | Condition | Url |
|---|---|---|---|---|---|---|
| Phase 3 | Hydralazine Valproate for Ovarian Cancer | Unknown status | December 2009 | Randomized, double-blind phase III trial. A total of 211 patients (alpha 0.5, power 0.8) with cisplatin-resistant recurrent or persistent cancer will be randomized to topotecan + placebo or topotecan + hydralazine + valproate for 6 courses every 4 weeks. | Ovarian Cancer | https://ClinicalTrials.gov/show/NCT00533299 |
| Phase 3 | Hydralazine Valproate for Cervical Cancer | Unknown status | December 2010 | Randomized, double-blind phase III trial. A total of 143 patients (alpha 0.5, power 0.8) with metastatic, persistent or recurrent cervical cancer without previous systemic treatment will be randomized to cisplatin topotecan + placebo or cisplatin topotecan hydralazine valproate for 6 courses every 3 weeks. | Metastatic Cervical Cancer | https://ClinicalTrials.gov/show/NCT00532818 |
| Phase 3 | Anticancer Activity of Nicotinamide on Lung Cancer | Active, not recruiting | June 2020 | Randomized Double-blinded Comparative Trial to study the Add-on Activity of Combination Treatment of Nicotinamide on Progression Free Survival for EGFR Mutated Lung Cancer Terminal Stage Patients Being Treated With Gefitinib or Erlotinib. | Non-Small-Cell Lung Carcinoma | https://ClinicalTrials.gov/show/NCT02416739 |
| Phase 3 | Exemestane With or Without Entinostat in Treating Patients With Recurrent Hormone Receptor-Positive Breast Cancer That is Locally Advanced or Metastatic | Active, not recruiting | - | Randomized phase III trial studies exemestane and entinostat to see how well they work compared to exemestane alone in treating patients with hormone receptor-positive breast cancer that has spread to nearby tissue or lymph nodes or another place in the body. | Breast Adenocarcinoma | https://ClinicalTrials.gov/show/NCT02115282 |
| Phase 3 | Exemestane With or Without Entinostat in Chinese Patients With Hormone Receptor-Positive, Locally Advanced or Metastatic Breast Cancer | Recruiting | August 2021 | A Randomized Phase III Clinical Study of Entinostat/Placebo in Combination With Exemestane in Chinese Patients With Hormone Receptor-positive Advanced Breast Cancer. | Advanced Breast Carcinoma | https://ClinicalTrials.gov/show/NCT03538171 |
| Phase 3 | A Clinical Trial of Vorinostat (MK0683, SAHA) in Combination With FDA Approved Cancer Drugs in Patients With Advanced Non-Small Cell Lung Cancer (NSCLC)(0683-056) | Terminated | December 2008 | A Phase II/III Randomized, Double-Blind Study of Paclitaxel Plus Carboplatin in Combination With Vorinostat or Placebo in Patients With Stage IIIB (With Pleural Effusion) or Stage IV Non-Small-Cell Lung Cancer (NSCLC). | Stage IIIB or IV Non-Small Cell Lung Cancer | https://ClinicalTrials.gov/show/NCT00473889 |
| Phase 3 | Suberoylanilide Hydroxamic Acid (Vorinostat, MK-0683) Versus Placebo in Advanced Malignant Pleural Mesothelioma (0683-014 AM5, EXT1) | Completed | November 2011 | A Phase III, Randomized, Double-Blind, Placebo-Controlled Trial of Oral Suberoylanilide Hydroxamic Acid (Vorinostat, MK-0683) in Patients With Advanced Malignant Pleural Mesothelioma Previously Treated With Systemic Chemotherapy. | Mesothelioma/Lung Cancer | https://ClinicalTrials.gov/show/NCT00128102 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roca, M.S.; Di Gennaro, E.; Budillon, A. Implication for Cancer Stem Cells in Solid Cancer Chemo-Resistance: Promising Therapeutic Strategies Based on the Use of HDAC Inhibitors. J. Clin. Med. 2019, 8, 912. https://doi.org/10.3390/jcm8070912
Roca MS, Di Gennaro E, Budillon A. Implication for Cancer Stem Cells in Solid Cancer Chemo-Resistance: Promising Therapeutic Strategies Based on the Use of HDAC Inhibitors. Journal of Clinical Medicine. 2019; 8(7):912. https://doi.org/10.3390/jcm8070912
Chicago/Turabian StyleRoca, Maria Serena, Elena Di Gennaro, and Alfredo Budillon. 2019. "Implication for Cancer Stem Cells in Solid Cancer Chemo-Resistance: Promising Therapeutic Strategies Based on the Use of HDAC Inhibitors" Journal of Clinical Medicine 8, no. 7: 912. https://doi.org/10.3390/jcm8070912
APA StyleRoca, M. S., Di Gennaro, E., & Budillon, A. (2019). Implication for Cancer Stem Cells in Solid Cancer Chemo-Resistance: Promising Therapeutic Strategies Based on the Use of HDAC Inhibitors. Journal of Clinical Medicine, 8(7), 912. https://doi.org/10.3390/jcm8070912