Cardiac Sodium Channel Dysfunction and Dilated Cardiomyopathy: A Contemporary Reappraisal of Pathophysiological Concepts


{kind=link}
Abstract
1. Introduction
2. Clinical Evidence
3. Experimental Evidence
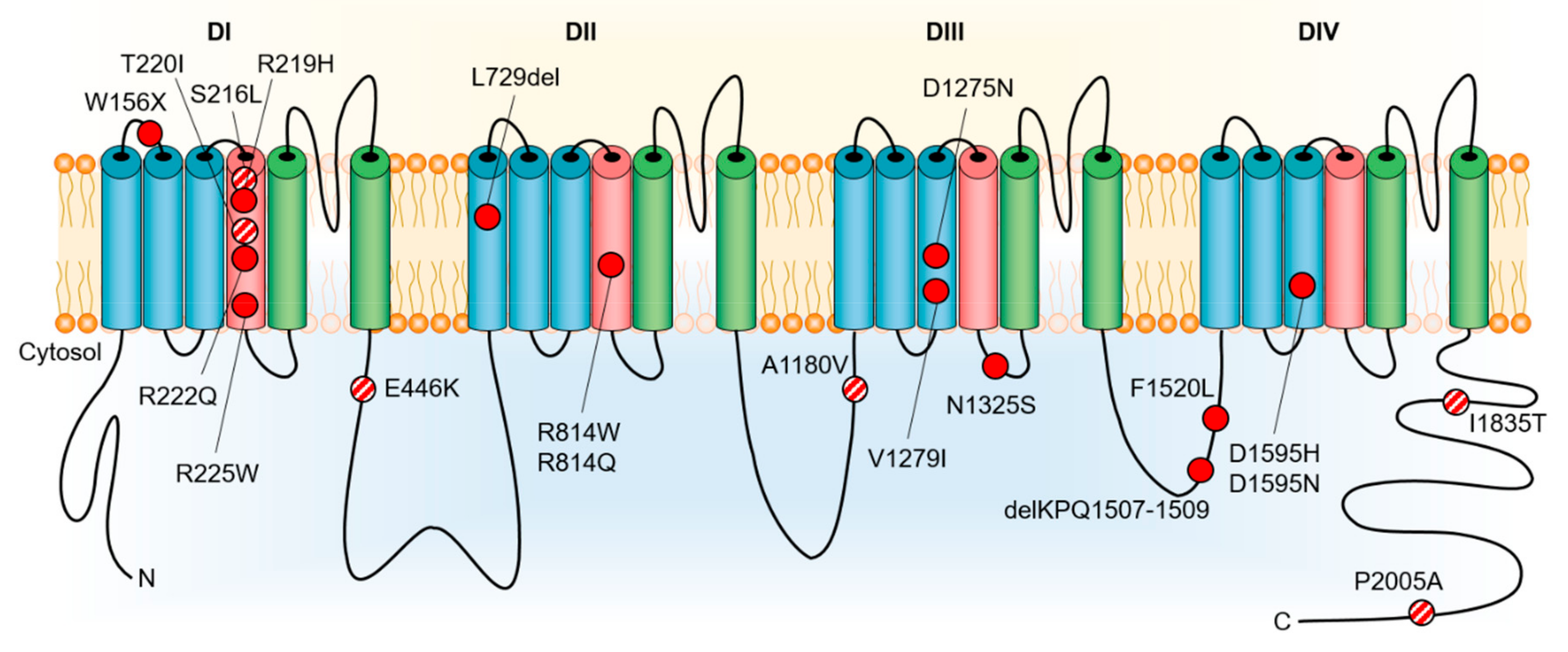
4. DCM as a Manifestation of Pure NaV1.5 Dysfunction
5. DCM Caused by Disrupted NaV1.5 Interaction with Partner Proteins
6. DCM Resulting from Long-Standing and Frequent Arrhythmias
7. DCM Secondary to Cardiac Conduction Disturbances
8. Mechanisms Involving Other Genetic Influences
9. Non-Genetic and Epigenetic Influences
10. Clinical Implications
11. Conclusions
Conflicts of Interest
References
- Wang, Q.; Shen, J.; Splawski, I.; Atkinson, D.; Li, Z.; Robinson, J.L.; Moss, A.J.; Towbin, J.A.; Keating, M.T. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995, 80, 805–811. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Priori, S.G.; Locati, E.H.; Napolitano, C.; Cantu, F.; Towbin, J.A.; Keating, M.T.; Hammoude, H.; Brown, A.M.; Chen, L.S.; et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation 1995, 92, 3381–3386. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G.; et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998, 392, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Rook, M.B.; Bezzina Alshinawi, C.; Groenewegen, W.A.; van Gelder, I.C.; van Ginneken, A.C.; Jongsma, H.J.; Mannens, M.M.; Wilde, A.A. Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovasc. Res. 1999, 44, 507–517. [Google Scholar] [CrossRef]
- Schott, J.J.; Alshinawi, C.; Kyndt, F.; Probst, V.; Hoorntje, T.M.; Hulsbeek, M.; Wilde, A.A.; Escande, D.; Mannens, M.M.; Le Marec, H. Cardiac conduction defects associate with mutations in SCN5A. Nat. Genet. 1999, 23, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.W.; Viswanathan, P.C.; Balser, J.R.; George, A.L., Jr.; Benson, D.W. Clinical, genetic, and biophysical characterization of SCN5A mutations associated with atrioventricular conduction block. Circulation 2002, 105, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.W.; Wang, D.W.; Dyment, M.; Knilans, T.K.; Fish, F.A.; Strieper, M.J.; Rhodes, T.H.; George, A.L., Jr. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Invest. 2003, 112, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Groenewegen, W.A.; Firouzi, M.; Bezzina, C.R.; Vliex, S.; van Langen, I.M.; Sandkuijl, L.; Smits, J.P.; Hulsbeek, M.; Rook, M.B.; Jongsma, H.J.; et al. A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ. Res. 2003, 92, 14–22. [Google Scholar] [CrossRef]
- Laurent, G.; Saal, S.; Amarouch, M.Y.; Beziau, D.M.; Marsman, R.F.; Faivre, L.; Barc, J.; Dina, C.; Bertaux, G.; Barthez, O.; et al. Multifocal ectopic Purkinje-related premature contractions: A new SCN5A-related cardiac channelopathy. J. Am. Coll. Cardiol. 2012, 60, 144–156. [Google Scholar] [CrossRef]
- Laitinen-Forsblom, P.J.; Makynen, P.; Makynen, H.; Yli-Mayry, S.; Virtanen, V.; Kontula, K.; Aalto-Setala, K. SCN5A mutation associated with cardiac conduction defect and atrial arrhythmias. J. Cardiovasc. Electrophysiol. 2006, 17, 480–485. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Giordano, U.; Collisani, G.; Memmi, M. Brugada syndrome and sudden cardiac death in children. Lancet 2000, 355, 808–809. [Google Scholar] [CrossRef]
- McNair, W.P.; Ku, L.; Taylor, M.R.; Fain, P.R.; Dao, D.; Wolfel, E.; Mestroni, L.; Familial Cardiomyopathy Registry Research Group. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 2004, 110, 2163–2167. [Google Scholar] [CrossRef] [PubMed]
- Te Riele, A.S.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Sobreira, N.L.; Amat-Alarcon, N.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.M.; Michels, V.V.; Ballew, J.D.; Reyna, S.P.; Karst, M.L.; Herron, K.J.; Horton, S.C.; Rodeheffer, R.J.; Anderson, J.L. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 2005, 293, 447–454. [Google Scholar] [CrossRef] [PubMed]
- McNair, W.P.; Sinagra, G.; Taylor, M.R.; Di Lenarda, A.; Ferguson, D.A.; Salcedo, E.E.; Slavov, D.; Zhu, X.; Caldwell, J.H.; Mestroni, L.; et al. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J. Am. Coll. Cardiol. 2011, 57, 2160–2168. [Google Scholar] [CrossRef]
- Mann, S.A.; Castro, M.L.; Ohanian, M.; Guo, G.; Zodgekar, P.; Sheu, A.; Stockhammer, K.; Thompson, T.; Playford, D.; Subbiah, R.; et al. R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J. Am. Coll. Cardiol. 2012, 60, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef]
- Nikolova, V.; Leimena, C.; McMahon, A.C.; Tan, J.C.; Chandar, S.; Jogia, D.; Kesteven, S.H.; Michalicek, J.; Otway, R.; Verheyen, F.; et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Invest. 2004, 113, 357–369. [Google Scholar] [CrossRef]
- Hesse, M.; Kondo, C.S.; Clark, R.B.; Su, L.; Allen, F.L.; Geary-Joo, C.T.; Kunnathu, S.; Severson, D.L.; Nygren, A.; Giles, W.R.; et al. Dilated cardiomyopathy is associated with reduced expression of the cardiac sodium channel Scn5a. Cardiovasc. Res. 2007, 75, 498–509. [Google Scholar] [CrossRef]
- Nguyen, T.P.; Wang, D.W.; Rhodes, T.H.; George, A.L., Jr. Divergent biophysical defects caused by mutant sodium channels in dilated cardiomyopathy with arrhythmia. Circ. Res. 2008, 102, 364–371. [Google Scholar] [CrossRef]
- Shi, R.; Zhang, Y.; Yang, C.; Huang, C.; Zhou, X.; Qiang, H.; Grace, A.A.; Huang, C.L.; Ma, A. The cardiac sodium channel mutation delQKP 1507-1509 is associated with the expanding phenotypic spectrum of LQT3, conduction disorder, dilated cardiomyopathy, and high incidence of youth sudden death. Europace 2008, 10, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Moreau, A.; Gosselin-Badaroudine, P.; Boutjdir, M.; Chahine, M. Mutations in the Voltage Sensors of Domains I and II of Nav1.5 that are Associated with Arrhythmias and Dilated Cardiomyopathy Generate Gating Pore Currents. Front. Pharmacol. 2015, 6, 301. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Remme, C.A. Dilated cardiomyopathy due to sodium channel dysfunction: What is the connection? Circ. Arrhythm Electrophysiol. 2008, 1, 80–82. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Amin, A.S. SCN5A-related dilated cardiomyopathy: What do we know? Heart Rhythm. 2014, 11, 1454–1455. [Google Scholar] [CrossRef] [PubMed]
- Gosselin-Badaroudine, P.; Keller, D.I.; Huang, H.; Pouliot, V.; Chatelier, A.; Osswald, S.; Brink, M.; Chahine, M. A proton leak current through the cardiac sodium channel is linked to mixed arrhythmia and the dilated cardiomyopathy phenotype. PLoS ONE 2012, 7, e38331. [Google Scholar] [CrossRef]
- Ge, J.; Sun, A.; Paajanen, V.; Wang, S.; Su, C.; Yang, Z.; Li, Y.; Wang, S.; Jia, J.; Wang, K.; et al. Molecular and clinical characterization of a novel SCN5A mutation associated with atrioventricular block and dilated cardiomyopathy. Circ. Arrhythm Electrophysiol. 2008, 1, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Clatot, J.; Hoshi, M.; Wan, X.; Liu, H.; Jain, A.; Shinlapawittayatorn, K.; Marionneau, C.; Ficker, E.; Ha, T.; Deschenes, I. Voltage-gated sodium channels assemble and gate as dimers. Nat. Commun. 2017, 8, 2077. [Google Scholar] [CrossRef] [PubMed]
- Abriel, H.; Rougier, J.S.; Jalife, J. Ion channel macromolecular complexes in cardiomyocytes: Roles in sudden cardiac death. Circ. Res. 2015, 116, 1971–1988. [Google Scholar] [CrossRef]
- Shan, L.; Makita, N.; Xing, Y.; Watanabe, S.; Futatani, T.; Ye, F.; Saito, K.; Ibuki, K.; Watanabe, K.; Hirono, K.; et al. SCN5A variants in Japanese patients with left ventricular noncompaction and arrhythmia. Mol. Genet. Metab. 2008, 93, 468–474. [Google Scholar] [CrossRef]
- Coronel, R.; Casini, S.; Koopmann, T.T.; Wilms-Schopman, F.J.; Verkerk, A.O.; de Groot, J.R.; Bhuiyan, Z.; Bezzina, C.R.; Veldkamp, M.W.; Linnenbank, A.C.; et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: A combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005, 112, 2769–2777. [Google Scholar] [CrossRef]
- Frustaci, A.; Priori, S.G.; Pieroni, M.; Chimenti, C.; Napolitano, C.; Rivolta, I.; Sanna, T.; Bellocci, F.; Russo, M.A. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 2005, 112, 3680–3687. [Google Scholar] [CrossRef] [PubMed]
- Nademanee, K.; Raju, H.; de Noronha, S.V.; Papadakis, M.; Robinson, L.; Rothery, S.; Makita, N.; Kowase, S.; Boonmee, N.; Vitayakritsirikul, V.; et al. Fibrosis, Connexin-43, and Conduction Abnormalities in the Brugada Syndrome. J. Am. Coll. Cardiol. 2015, 66, 1976–1986. [Google Scholar] [CrossRef]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Abriel, H. Cardiac sodium channel Na(v)1.5 and interacting proteins: Physiology and pathophysiology. J. Mol. Cell Cardiol. 2010, 48, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Sato, A.; Marcou, C.A.; Tester, D.J.; Ackerman, M.J.; Crotti, L.; Schwartz, P.J.; On, Y.K.; Park, J.E.; Nakamura, K.; et al. A novel disease gene for Brugada syndrome: Sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ. Arrhythm Electrophysiol. 2012, 5, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Mlynarova, J.; Trentin-Sonoda, M.; Gaisler da Silva, F.; Major, J.L.; Salih, M.; Carneiro-Ramos, M.S.; Tuana, B.S. SLMAP3 isoform modulates cardiac gene expression and function. PLoS ONE 2019, 14, e0214669. [Google Scholar] [CrossRef]
- Hao, X.; Zhang, Y.; Zhang, X.; Nirmalan, M.; Davies, L.; Konstantinou, D.; Yin, F.; Dobrzynski, H.; Wang, X.; Grace, A.; et al. TGF-beta1-mediated fibrosis and ion channel remodeling are key mechanisms in producing the sinus node dysfunction associated with SCN5A deficiency and aging. Circ. Arrhythm Electrophysiol. 2011, 4, 397–406. [Google Scholar] [CrossRef]
- Zakrzewska-Koperska, J.; Franaszczyk, M.; Bilinska, Z.; Truszkowska, G.; Karczmarz, M.; Szumowski, L.; Zielinski, T.; Ploski, R.; Bilinska, M. Rapid and effective response of the R222Q SCN5A to quinidine treatment in a patient with Purkinje-related ventricular arrhythmia and familial dilated cardiomyopathy: A case report. BMC Med. Genet. 2018, 19, 94. [Google Scholar] [CrossRef]
- Beckermann, T.M.; McLeod, K.; Murday, V.; Potet, F.; George, A.L., Jr. Novel SCN5A mutation in amiodarone-responsive multifocal ventricular ectopy-associated cardiomyopathy. Heart Rhythm. 2014, 11, 1446–1453. [Google Scholar] [CrossRef]
- Swan, H.; Amarouch, M.Y.; Leinonen, J.; Marjamaa, A.; Kucera, J.P.; Laitinen-Forsblom, P.J.; Lahtinen, A.M.; Palotie, A.; Kontula, K.; Toivonen, L.; et al. Gain-of-function mutation of the SCN5A gene causes exercise-induced polymorphic ventricular arrhythmias. Circ. Cardiovasc. Genet. 2014, 7, 771–781. [Google Scholar] [CrossRef]
- Calloe, K.; Broendberg, A.K.; Christensen, A.H.; Pedersen, L.N.; Olesen, M.S.; de Los Angeles Tejada, M.; Friis, S.; Thomsen, M.B.; Bundgaard, H.; Jensen, H.K. Multifocal atrial and ventricular premature contractions with an increased risk of dilated cardiomyopathy caused by a Nav1.5 gain-of-function mutation (G213D). Int. J. Cardiol. 2018, 257, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Yang, T.; Stroud, D.M.; Lowe, J.S.; Harris, L.; Atack, T.C.; Wang, D.W.; Hipkens, S.B.; Leake, B.; Hall, L.; et al. Striking In vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circulation 2011, 124, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Morales, A.; Siegfried, J.D.; Li, D.; Norton, N.; Song, J.; Gonzalez-Quintana, J.; Makielski, J.C.; Hershberger, R.E. SCN5A rare variants in familial dilated cardiomyopathy decrease peak sodium current depending on the common polymorphism H558R and common splice variant Q1077del. Clin. Transl. Sci. 2010, 3, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.W.; Mistry, A.M.; Kahlig, K.M.; Kearney, J.A.; Xiang, J.; George, A.L., Jr. Propranolol blocks cardiac and neuronal voltage-gated sodium channels. Front. Pharmacol. 2010, 1, 144. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asatryan, B. Cardiac Sodium Channel Dysfunction and Dilated Cardiomyopathy: A Contemporary Reappraisal of Pathophysiological Concepts. J. Clin. Med. 2019, 8, 1029. https://doi.org/10.3390/jcm8071029
Asatryan B. Cardiac Sodium Channel Dysfunction and Dilated Cardiomyopathy: A Contemporary Reappraisal of Pathophysiological Concepts. Journal of Clinical Medicine. 2019; 8(7):1029. https://doi.org/10.3390/jcm8071029
Chicago/Turabian StyleAsatryan, Babken. 2019. "Cardiac Sodium Channel Dysfunction and Dilated Cardiomyopathy: A Contemporary Reappraisal of Pathophysiological Concepts" Journal of Clinical Medicine 8, no. 7: 1029. https://doi.org/10.3390/jcm8071029
APA StyleAsatryan, B. (2019). Cardiac Sodium Channel Dysfunction and Dilated Cardiomyopathy: A Contemporary Reappraisal of Pathophysiological Concepts. Journal of Clinical Medicine, 8(7), 1029. https://doi.org/10.3390/jcm8071029