Early Local Inhibition of Club Cell Protein 16 Following Chest Trauma Reduces Late Sepsis-Induced Acute Lung Injury

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Section
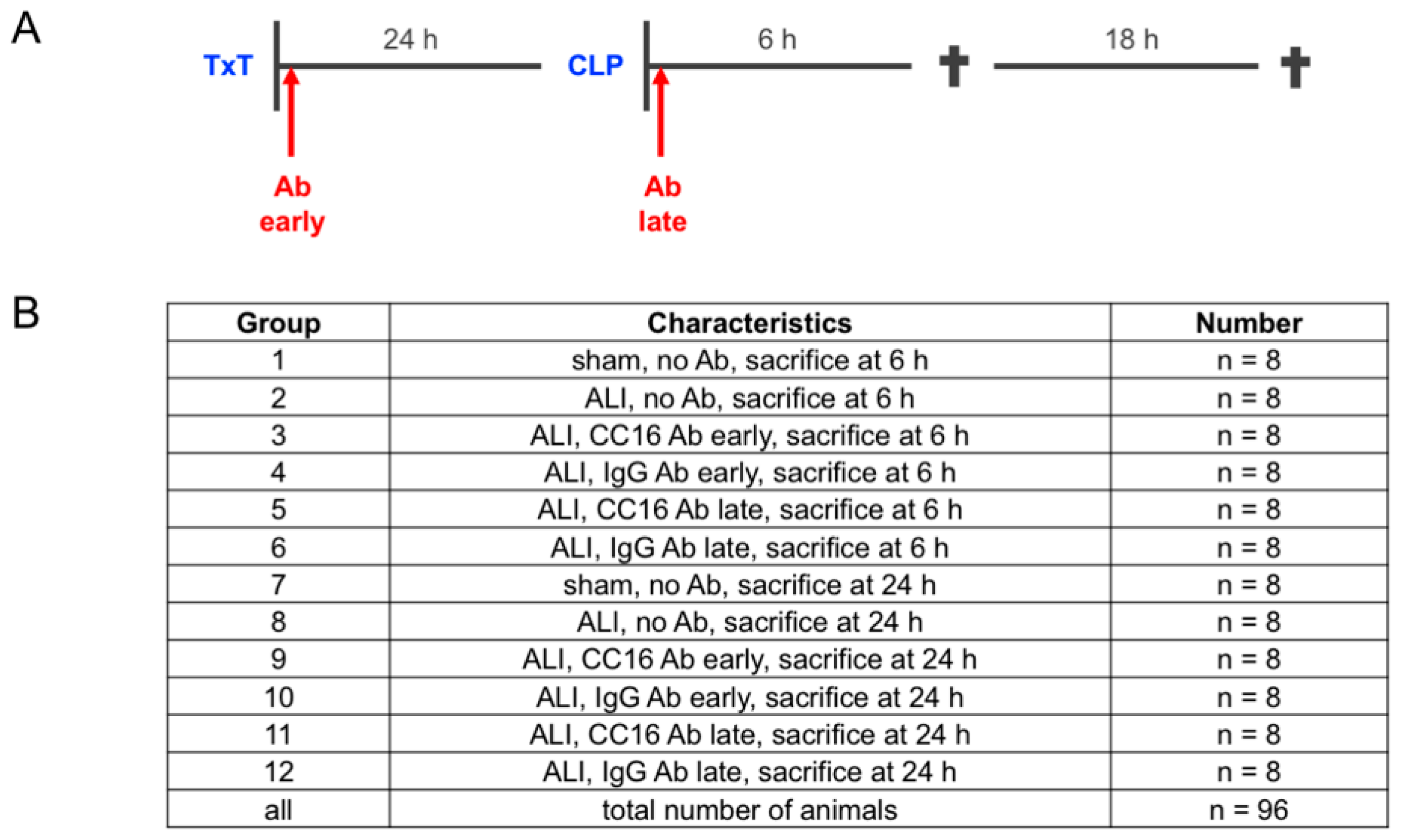
2.1. Animals and Experimental Model
2.2. Group Allocation According to the Application of Anti CC16-Antibody
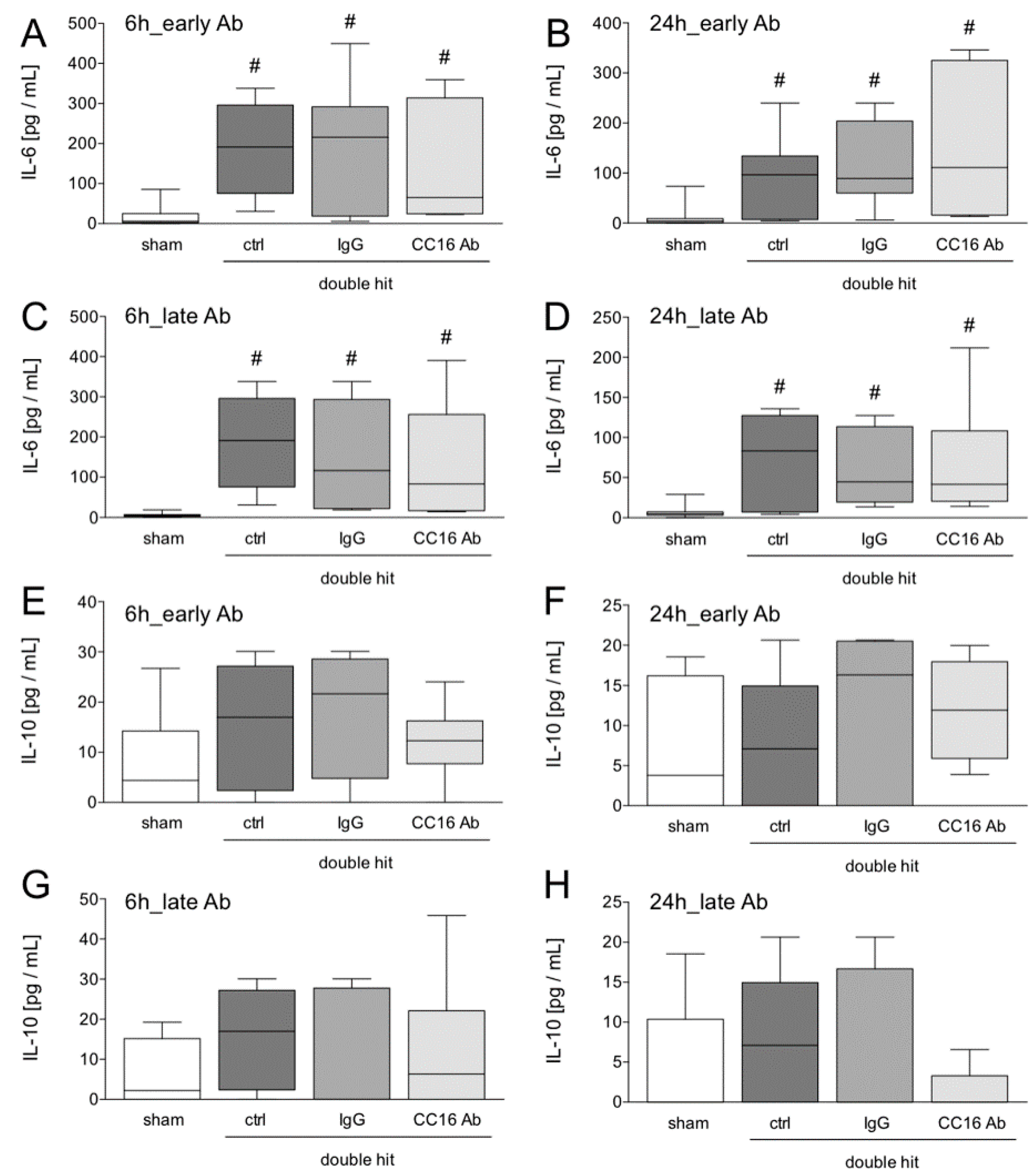
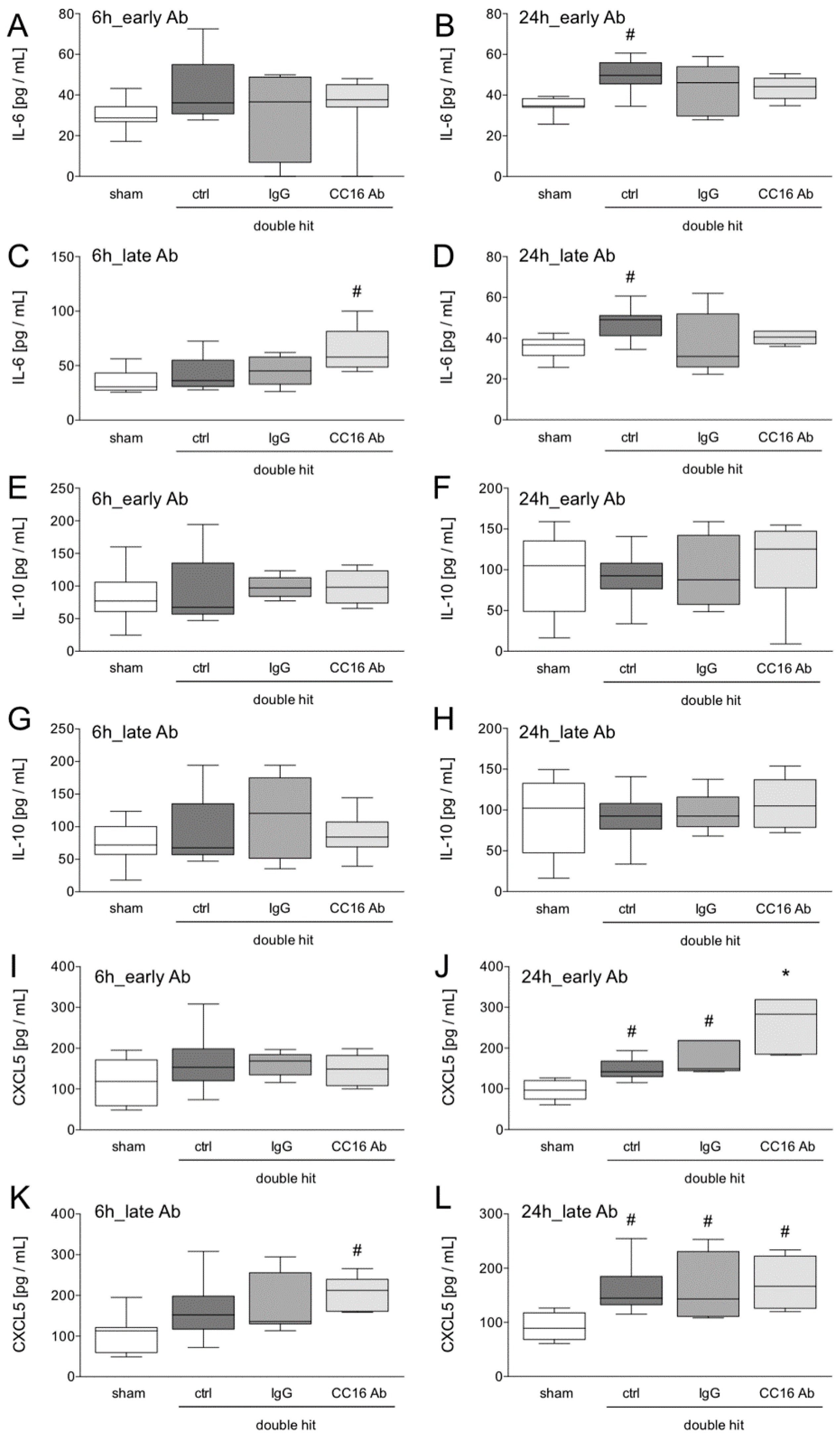
2.3. Cytometric Bead Array for IL-6 and IL-10 Determination
2.4. Quantification of Protein Expression Levels in the Lungs
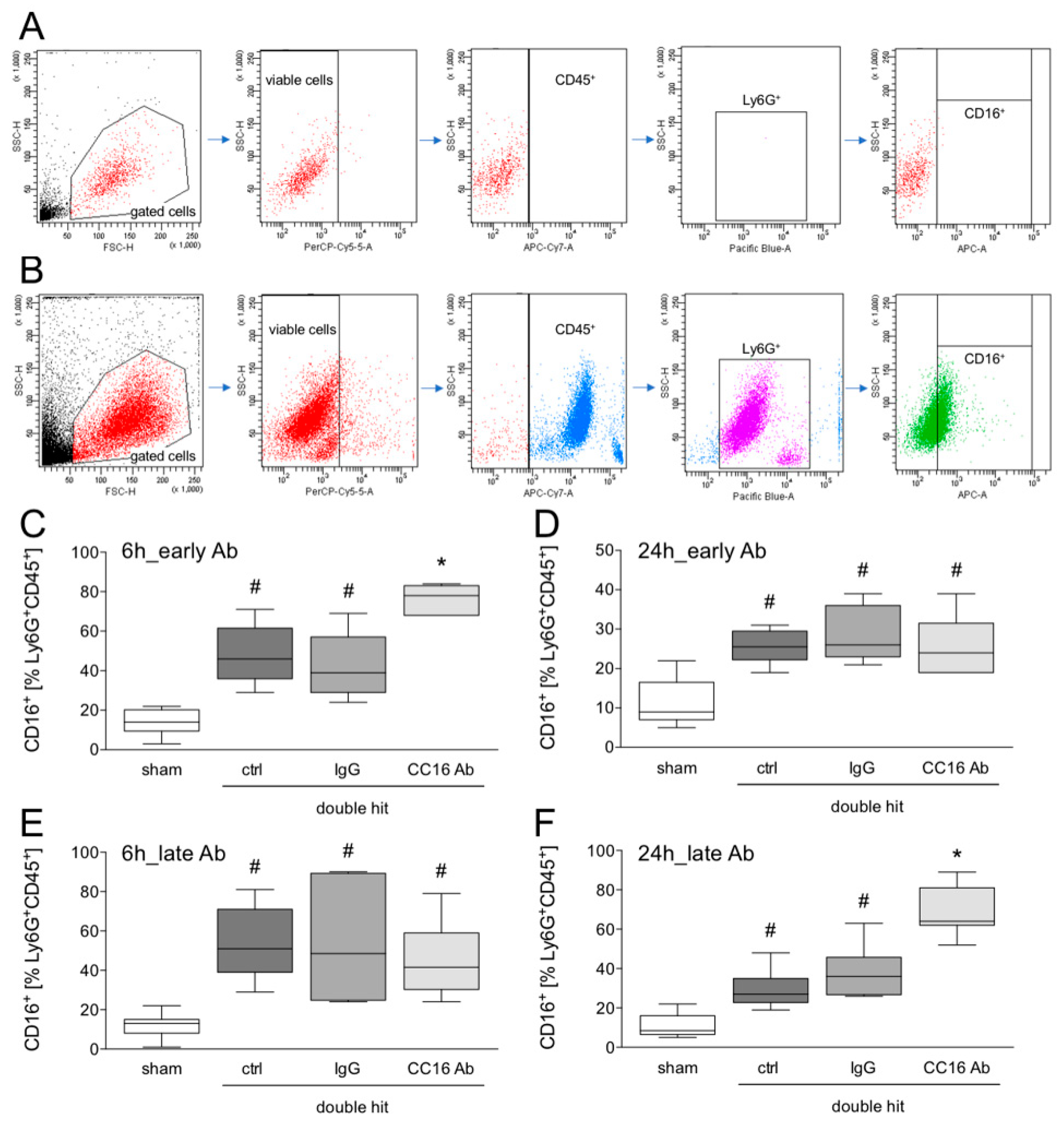
2.5. Detection of Granulocytes in the BAL by FLOW Cytometry
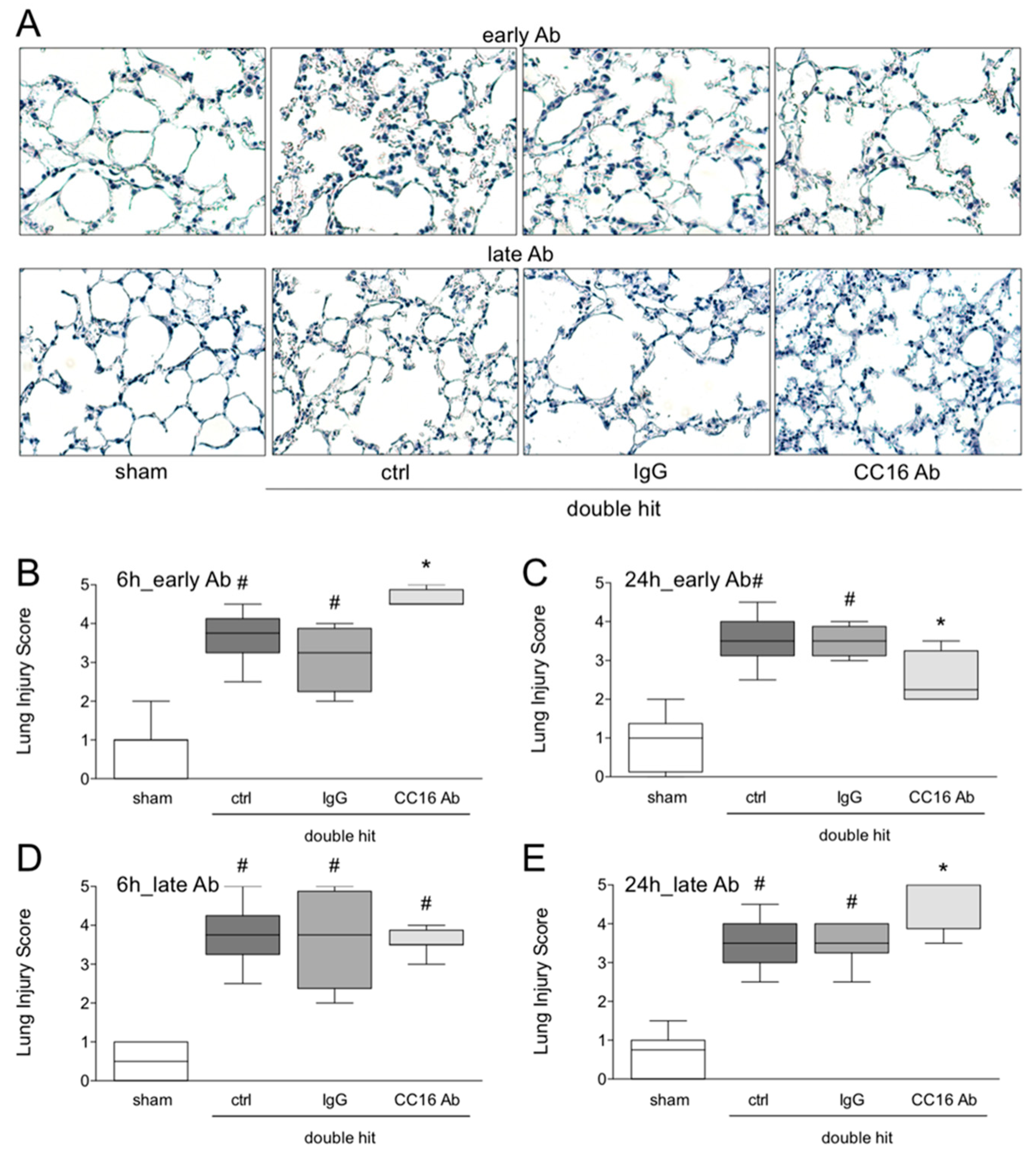
2.6. Histological Examination of Lung Injury
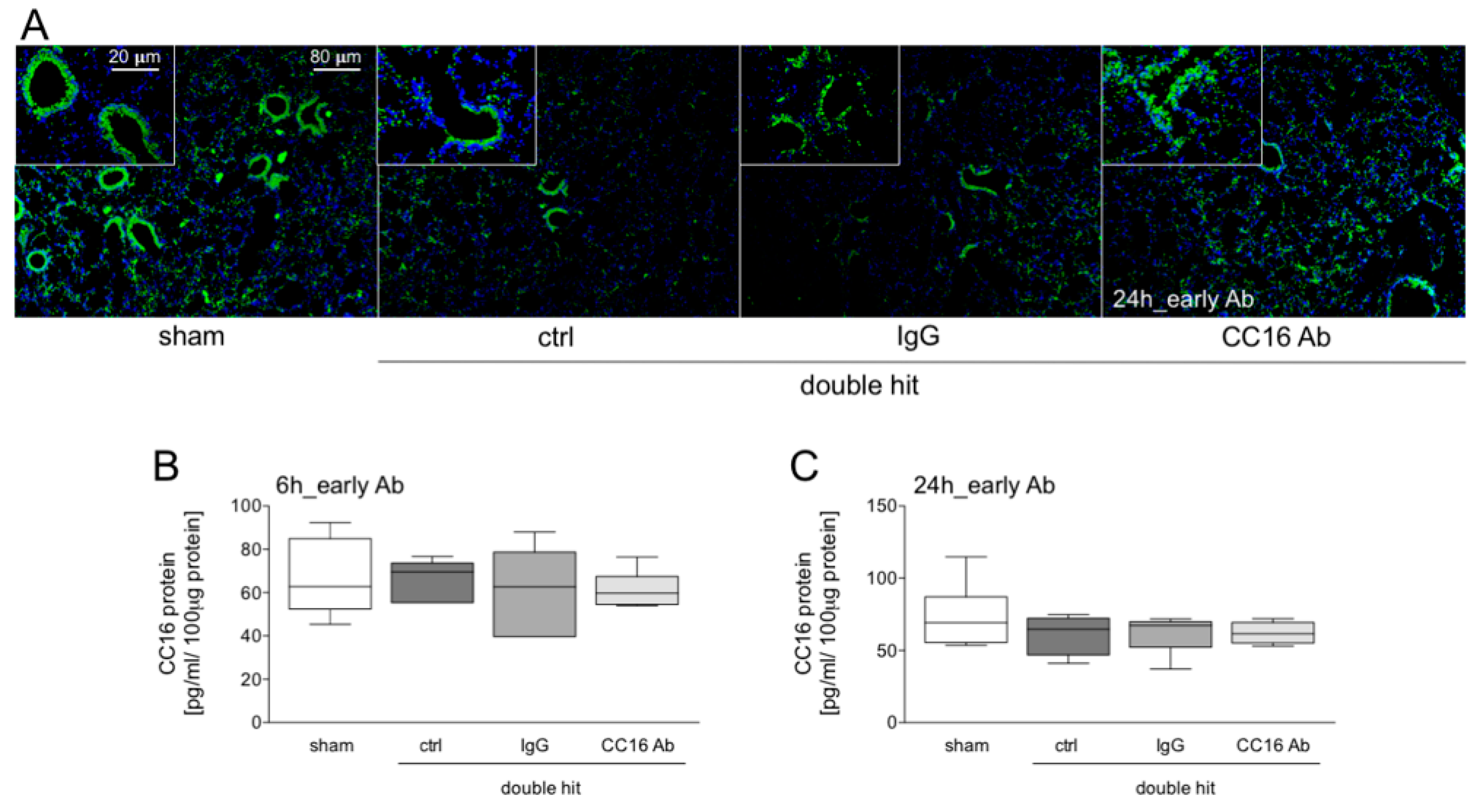
2.7. Staining of CC16
2.8. Statistical Analyses
3. Results
3.1. Study Data
3.1.1. Presence of CC16 in Lung Tissue
3.1.2. IL-6 and IL-10 Concentrations in the Blood
3.1.3. Quantification of Protein Expression Levels in the Lungs
3.1.4. Detection of CD16+Ly6G+CD45+ Neutrophils in the BAL by Flow Cytometry
3.1.5. Histological Examination of Lung Injury
4. Discussion
Limitations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chrysou, K.; Halat, G.; Hoksch, B.; Schmid, R.A.; Kocher, G.J. Lessons from a large trauma center: Impact of blunt chest trauma in polytrauma patients-still a relevant problem? Scand. J. Trauma Resusc. Emerg. Med. 2017, 25, 42. [Google Scholar] [CrossRef]
- Dennis, B.M.; Bellister, S.A.; Guillamondegui, O.D. Thoracic Trauma. Surg. Clin. N. Am. 2017, 97, 1047–1064. [Google Scholar] [CrossRef] [PubMed]
- Finkbeiner, R.; Krinner, S.; Langenbach, A.; Besendorfer, M.; Schulz-Drost, S. Age Distribution and Concomitant Injuries in Pulmonary Contusion: An Analysis Based on Routine Data. Thorac. Cardiovasc. Surg. 2018, 66, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Leenen, L.P. Focus on chest trauma. Eur. J. Trauma Emerg. Surg. 2017, 43, 153–154. [Google Scholar] [CrossRef]
- Wutzler, S.; Wafaisade, A.; Maegele, M.; Laurer, H.; Geiger, E.V.; Walcher, F.; Barker, J.; Lefering, R.; Marzi, I.; Trauma Registry of, D.G.U. Lung Organ Failure Score (LOFS): Probability of severe pulmonary organ failure after multiple injuries including chest trauma. Injury 2012, 43, 1507–1512. [Google Scholar] [CrossRef] [PubMed]
- Keel, M.; Meier, C. Chest injuries-what is new? Curr. Opin. Crit. Care 2007, 13, 674–679. [Google Scholar] [CrossRef]
- Proudfoot, A.G.; McAuley, D.F.; Griffiths, M.J.; Hind, M. Human models of acute lung injury. Dis. Model. Mech. 2011, 4, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Stormann, P.; Becker, N.; Kunnemeyer, L.; Wutzler, S.; Vollrath, J.T.; Lustenberger, T.; Hildebrand, F.; Marzi, I.; Relja, B. Contributing factors in the development of acute lung injury in a murine double hit model. Eur. J. Trauma Emerg. Surg. 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Perl, M.; Hohmann, C.; Denk, S.; Kellermann, P.; Lu, D.; Braumuller, S.; Bachem, M.G.; Thomas, J.; Knoferl, M.W.; Ayala, A.; et al. Role of activated neutrophils in chest trauma-induced septic acute lung injury. Shock 2012, 38, 98–106. [Google Scholar] [CrossRef]
- Weckbach, S.; Hohmann, C.; Denk, S.; Kellermann, P.; Huber-Lang, M.S.; Baumann, B.; Wirth, T.; Gebhard, F.; Bachem, M.; Perl, M. Apoptotic and inflammatory signaling via Fas and tumor necrosis factor receptor I contribute to the development of chest trauma-induced septic acute lung injury. J. Trauma Acute Care Surg. 2013, 74, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Stormann, P.; Wagner, N.; Kohler, K.; Auner, B.; Simon, T.P.; Pfeifer, R.; Horst, K.; Pape, H.C.; Hildebrand, F.; Wutzler, S.; et al. Monotrauma is associated with enhanced remote inflammatory response and organ damage, while polytrauma intensifies both in porcine trauma model. Eur. J. Trauma Emerg. Surg. 2019, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.B.; Pugin, J.; Lee, J.S.; Matthay, M.A. Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev. 2003, 14, 523–535. [Google Scholar] [CrossRef]
- Buendia-Roldan, I.; Ruiz, V.; Sierra, P.; Montes, E.; Ramirez, R.; Vega, A.; Salgado, A.; Vargas, M.H.; Mejia, M.; Pardo, A.; et al. Increased Expression of CC16 in Patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 2016, 11, e0168552. [Google Scholar] [CrossRef] [PubMed]
- Laucho-Contreras, M.E.; Polverino, F.; Gupta, K.; Taylor, K.L.; Kelly, E.; Pinto-Plata, V.; Divo, M.; Ashfaq, N.; Petersen, H.; Stripp, B.; et al. Protective role for club cell secretory protein-16 (CC16) in the development of COPD. Eur. Respir. J. 2015, 45, 1544–1556. [Google Scholar] [CrossRef]
- Miele, L.; Cordella-Miele, E.; Mantile, G.; Peri, A.; Mukherjee, A.B. Uteroglobin and uteroglobin-like proteins: The uteroglobin family of proteins. J. Endocrinol. Investig. 1994, 17, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Negrin, L.L.; Halat, G.; Kettner, S.; Gregori, M.; Ristl, R.; Hajdu, S.; Heinz, T. Club cell protein 16 and cytokeratin fragment 21-1 as early predictors of pulmonary complications in polytraumatized patients with severe chest trauma. PLoS ONE 2017, 12, e0175303. [Google Scholar] [CrossRef] [PubMed]
- Wutzler, S.; Backhaus, L.; Henrich, D.; Geiger, E.; Barker, J.; Marzi, I.; Laurer, H. Clara cell protein 16: A biomarker for detecting secondary respiratory complications in patients with multiple injuries. J. Trauma Acute Care Surg. 2012, 73, 838–842. [Google Scholar] [CrossRef]
- Wutzler, S.; Lehnert, T.; Laurer, H.; Lehnert, M.; Becker, M.; Henrich, D.; Vogl, T.; Marzi, I. Circulating levels of Clara cell protein 16 but not surfactant protein D identify and quantify lung damage in patients with multiple injuries. J. Trauma 2011, 71, E31–E36. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Ding, B.; Yang, X.; Ma, D.; Zhang, C.; Hua, C. Club cell protein 16 as a biomarker in pulmonary contusion. Biomed. Rep. 2016, 5, 251–253. [Google Scholar] [CrossRef][Green Version]
- Pang, M.; Yuan, Y.; Wang, D.; Li, T.; Wang, D.; Shi, X.; Guo, M.; Wang, C.; Zhang, X.; Zheng, G.; et al. Recombinant CC16 protein inhibits the production of pro-inflammatory cytokines via NF-kappaB and p38 MAPK pathways in LPS-activated RAW264.7 macrophages. Acta Biochim. Biophys. Sin. 2017, 49, 435–443. [Google Scholar] [CrossRef]
- Relja, B.; Mors, K.; Marzi, I. Danger signals in trauma. Eur. J. Trauma Emerg. Surg. 2018, 44, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Wutzler, S.; Lustenberger, T.; Relja, B.; Lehnert, M.; Marzi, I. Pathophysiology of multiple trauma: Intensive care medicine and timing of treatment. Chirurg 2013, 84, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Zhu, Y.H.; Liu, D.X.; Li, J.; Zhao, P.C.; Zhong, Y.P.; Chen, Y.Q.; Xu, W.; Zhu, Z.Q. Intranasal Application of Budesonide Attenuates Lipopolysaccharide-Induced Acute Lung Injury by Suppressing Nucleotide-Binding Oligomerization Domain-Like Receptor Family, Pyrin Domain-Containing 3 Inflammasome Activation in Mice. J. Immunol. Res. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.B.; Krispinsky, L.T.; Stark, R.J. Late immune consequences of combat trauma: A review of trauma-related immune dysfunction and potential therapies. Mil. Med. Res. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Vourc’h, M.; Roquilly, A.; Asehnoune, K. Trauma-Induced Damage-Associated Molecular Patterns-Mediated Remote Organ Injury and Immunosuppression in the Acutely Ill Patient. Front. Immunol. 2018, 9, 1330. [Google Scholar] [CrossRef]
- Horiguchi, H.; Loftus, T.J.; Hawkins, R.B.; Raymond, S.L.; Stortz, J.A.; Hollen, M.K.; Weiss, B.P.; Miller, E.S.; Bihorac, A.; Larson, S.D.; et al. Innate Immunity in the Persistent Inflammation, Immunosuppression, and Catabolism Syndrome and Its Implications for Therapy. Front. Immunol. 2018, 9, 595. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Knoferl, M.W.; Liener, U.C.; Seitz, D.H.; Perl, M.; Bruckner, U.B.; Kinzl, L.; Gebhard, F. Cardiopulmonary, histological, and inflammatory alterations after lung contusion in a novel mouse model of blunt chest trauma. Shock 2003, 19, 519–525. [Google Scholar] [CrossRef]
- Stormann, P.; Auner, B.; Schimunek, L.; Serve, R.; Horst, K.; Simon, T.P.; Pfeifer, R.; Kohler, K.; Hildebrand, F.; Wutzler, S.; et al. Leukotriene B4 indicates lung injury and on-going inflammatory changes after severe trauma in a porcine long-term model. Prostaglandins Leukot. Essent. Fatty Acids 2017, 127, 25–31. [Google Scholar] [CrossRef]
- Raymond, S.L.; Holden, D.C.; Mira, J.C.; Stortz, J.A.; Loftus, T.J.; Mohr, A.M.; Moldawer, L.L.; Moore, F.A.; Larson, S.D.; Efron, P.A. Microbial recognition and danger signals in sepsis and trauma. Biochim. Biophys. Acta 2017, 1863, 2564–2573. [Google Scholar] [CrossRef]
- Puneet, P.; Moochhala, S.; Bhatia, M. Chemokines in acute respiratory distress syndrome. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L3–L15. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ahyi, A.N.; Pepper-Cunningham, Z.A.; Ferrari, J.D.; Wilson, A.A.; Jones, M.R.; Quinton, L.J.; Mizgerd, J.P. Roles of lung epithelium in neutrophil recruitment during pneumococcal pneumonia. Am. J. Respir. Cell Mol. Biol. 2014, 50, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, K.; Nagano, T.; Kobayashi, K.; Dokuni, R.; Katsurada, M.; Yamamoto, M.; Yoshikawa, Y.; Kataoka, T.; Nishimura, Y. Phospholipase Cepsilon plays a crucial role in neutrophilic inflammation accompanying acute lung injury through augmentation of CXC chemokine production from alveolar epithelial cells. Respir. Res. 2019, 20, 9. [Google Scholar] [CrossRef] [PubMed]
- Rousselle, A.; Qadri, F.; Leukel, L.; Yilmaz, R.; Fontaine, J.F.; Sihn, G.; Bader, M.; Ahluwalia, A.; Duchene, J. CXCL5 limits macrophage foam cell formation in atherosclerosis. J. Clin. Investig. 2013, 123, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Michel, O.; Murdoch, R.; Bernard, A. Inhaled LPS induces blood release of Clara cell specific protein (CC16) in human beings. J. Allergy Clin. Immunol. 2005, 115, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Bedreag, O.H.; Rogobete, A.F.; Sandesc, D.; Cradigati, C.A.; Sarandan, M.; Popovici, S.E.; Dumache, R.; Horhat, F.G.; Vernic, C.; Sima, L.V.; et al. Modulation of the Redox Expression and Inflammation Response in the Critically Ill Polytrauma Patient with Thoracic Injury. Statistical Correlations between Antioxidant Therapy. Clin. Lab. 2016, 62, 1747–1759. [Google Scholar] [CrossRef]
- Pang, M.; Liu, H.Y.; Li, T.; Wang, D.; Hu, X.Y.; Zhang, X.R.; Yu, B.F.; Guo, R.; Wang, H.L. Recombinant club cell protein 16 (CC16) ameliorates cigarette smokeinduced lung inflammation in a murine disease model of COPD. Mol. Med. Rep. 2018, 18, 2198–2206. [Google Scholar] [PubMed]
- Lam, D.C.; Kwok, H.H.; Yu, W.C.; Ko, F.W.; Tam, C.Y.; Lau, A.C.; Fong, D.Y.; Ip, M.S. CC16 levels correlate with cigarette smoke exposure in bronchial epithelial cells and with lung function decline in smokers. BMC Pulm. Med. 2018, 18, 47. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Störmann, P.; Becker, N.; Vollrath, J.T.; Köhler, K.; Janicova, A.; Wutzler, S.; Hildebrand, F.; Marzi, I.; Relja, B. Early Local Inhibition of Club Cell Protein 16 Following Chest Trauma Reduces Late Sepsis-Induced Acute Lung Injury. J. Clin. Med. 2019, 8, 896. https://doi.org/10.3390/jcm8060896
Störmann P, Becker N, Vollrath JT, Köhler K, Janicova A, Wutzler S, Hildebrand F, Marzi I, Relja B. Early Local Inhibition of Club Cell Protein 16 Following Chest Trauma Reduces Late Sepsis-Induced Acute Lung Injury. Journal of Clinical Medicine. 2019; 8(6):896. https://doi.org/10.3390/jcm8060896
Chicago/Turabian StyleStörmann, Philipp, Nils Becker, Jan Tilmann Vollrath, Kernt Köhler, Andrea Janicova, Sebastian Wutzler, Frank Hildebrand, Ingo Marzi, and Borna Relja. 2019. "Early Local Inhibition of Club Cell Protein 16 Following Chest Trauma Reduces Late Sepsis-Induced Acute Lung Injury" Journal of Clinical Medicine 8, no. 6: 896. https://doi.org/10.3390/jcm8060896
APA StyleStörmann, P., Becker, N., Vollrath, J. T., Köhler, K., Janicova, A., Wutzler, S., Hildebrand, F., Marzi, I., & Relja, B. (2019). Early Local Inhibition of Club Cell Protein 16 Following Chest Trauma Reduces Late Sepsis-Induced Acute Lung Injury. Journal of Clinical Medicine, 8(6), 896. https://doi.org/10.3390/jcm8060896