Simvastatin Attenuates Cardiac Fibrosis via Regulation of Cardiomyocyte-Derived Exosome Secretion

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Section
2.1. Animals and Treatment
2.2. Immunostaining and Masson’s Trichrome Staining
2.3. Transmission Electron Microscopy
2.4. Scanning Electron Microscopy
2.5. Second-Harmonic Generation Microscopy
2.6. Cell Culture
2.7. Exosomes Isolation and CM-DiI Labelling
2.8. Western Blotting
2.9. Real-Time (RT)-PCR
2.10. Cell Migration and Motility Analysis
2.11. Nano Ultra-performance Liquid Chromatographic System with Tandem Mass Spectrometry
2.12. Statistical Analysis
3. Results
3.1. SIM Suppresses Ang II-Mediated Collagen Deposition and Cardiac Fibrosis In Vivo
3.2. Ultrastructure between Cardiomyocyte and Fibroblast of the Heart In Vivo
3.3. HCM-Secreted Exosomes Are Transported to HCFs In Vitro
3.4. SIM Suppresses Fibroblast to Myofibroblast Transformation, and Fibrosis-Associated Protein Expression In Vitro
3.5. SIM Suppresses HCF Migration Induced by CM-Derived Exosomes In Vitro
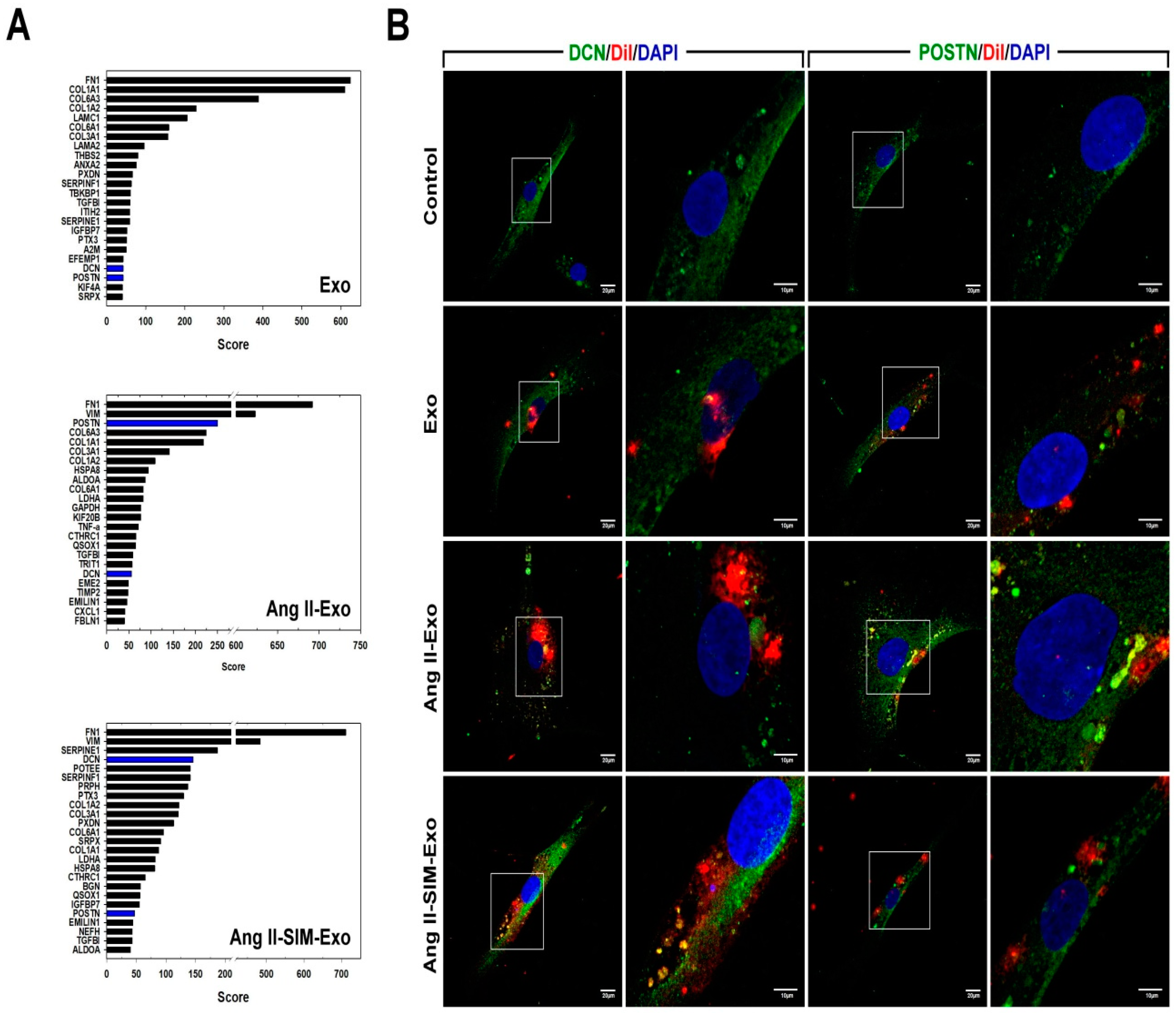
3.6. SIM Regulates HCM-Derived Exosome Protein Expression In Vitro
3.7. DCN Inhibits Collagen-Associated Gene Expression, Fibroblast Transformation, and Cell Motility In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schnee, J.M.; Hsueh, W.A. Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc. Res. 2000, 46, 264–268. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, Y.; Zhang, P.; Chen, Y.; Li, C.; Chen, J.; Wang, Y.; Li, Y. The crucial role of activin A/ALK4 pathway in the pathogenesis of Ang-II-induced atrial fibrosis and vulnerability to atrial fibrillation. Basic Res. Cardiol. 2017, 112, 47. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Guan, J. Antifibrotic therapies to control cardiac fibrosis. Biomater. Res. 2016, 20, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Su, J.; Mende, U. Cross talk between cardiac myocytes and fibroblasts: From multiscale investigative approaches to mechanisms and functional consequences. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1385–H1396. [Google Scholar] [CrossRef] [PubMed]
- Kofron, C.M.; Mende, U. In vitro models of the cardiac microenvironment to study myocyte and non-myocyte crosstalk: Bioinspired approaches beyond the polystyrene dish. J. Physiol. 2017, 595, 3891–3905. [Google Scholar] [CrossRef] [PubMed]
- Sung, T.Y.; You, H.J.; Cho, C.K.; Choi, H.R.; Kim, Y.B.; Shin, Y.S.; Yang, H.S. Effects of magnesium chloride on rocuronium-induced neuromuscular blockade and sugammadex reversal in an isolated rat phrenic nerve-hemidiaphragm preparation: An in-vitro study. Eur. J. Anaesthesiol. 2018, 35, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Kervadec, A.; Bellamy, V.; El Harane, N.; Arakelian, L.; Vanneaux, V.; Cacciapuoti, I.; Nemetalla, H.; Perier, M.C.; Toeg, H.D.; Richart, A.; et al. Cardiovascular progenitor-derived extracellular vesicles recapitulate the beneficial effects of their parent cells in the treatment of chronic heart failure. J. Heart Lung Transpl. 2016, 35, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Indolfi, C.; Curcio, A. Stargazing microRNA maps a new miR-21 star for cardiac hypertrophy. J. Clin Invest. 2014, 124, 1896–1898. [Google Scholar] [CrossRef]
- Shao, L.; Zhang, Y.; Lan, B.; Wang, J.; Zhang, Z.; Zhang, L.; Xiao, P.; Meng, Q.; Geng, Y.J.; Yu, X.Y.; et al. MiRNA-sequence indicates that mesenchymal stem cells and exosomes have similar mechanism to enhance cardiac repair. Biomed Res. Int. 2017, 2017, 4150705. [Google Scholar] [CrossRef]
- Dougherty, J.A.; Mergaye, M.; Kumar, N.; Chen, C.A.; Angelos, M.G.; Khan, M. Potential role of exosomes in mending a broken heart: Nanoshuttles propelling future clinical therapeutics forward. Stem Cells Int. 2017, 2017, 5785436. [Google Scholar] [CrossRef]
- Weber, K.T.; Diez, J. Targeting the Cardiac Myofibroblast Secretome to Treat Myocardial Fibrosis in Heart Failure. Circ. Heart Fail. 2016, 9, e003315. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Duan, Y.; Adkins, G.B.; Pan, S.; Wang, H.; Liu, Y.; Zhong, W. Highly efficient exosome isolation and protein analysis by an integrated nanomaterial-based platform. Anal. Chem. 2018, 90, 2787–2795. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Araya, J.; Ito, S.; Kobayashi, K.; Kosaka, N.; Yoshioka, Y.; Kadota, T.; Hara, H.; Kuwano, K.; Ochiya, T. Suppression of autophagy by extracellular vesicles promotes myofibroblast differentiation in COPD pathogenesis. J. Extracell. Vesicles 2015, 4, 28388. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Molkentin, J.D. Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell Cardiol. 2014, 70, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Chen, Z.; Huang, R.; Yao, Y.; Ma, G. Transforming growth factor beta1 induces the expression of collagen type I by DNA methylation in cardiac fibroblasts. PLoS ONE 2013, 8, e60335. [Google Scholar] [CrossRef]
- Yuan, M.J.; Maghsoudi, T.; Wang, T. Exosomes mediate the intercellular communication after myocardial infarction. Int. J. Med. Sci. 2016, 13, 113–116. [Google Scholar] [CrossRef]
- Bai, D.; Ge, L.; Gao, Y.; Lu, X.; Wang, H.; Yang, G. Cytoplasmic translocation of HuR contributes to angiotensin II induced cardiac fibrosis. Biochem. Biophys. Res. Commun. 2015, 463, 1273–1277. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.B.; Falk, E. Primary prevention with statins in the elderly. J. Am. Coll. Cardiol. 2018, 71, 85–94. [Google Scholar] [CrossRef]
- Choi, S.Y.; Park, J.S.; Roh, M.S.; Kim, C.R.; Kim, M.H.; Serebruany, V. Inhibition of angiotensin II-induced cardiac fibrosis by atorvastatin in adiponectin knockout mice. Lipids 2017, 52, 415–422. [Google Scholar] [CrossRef]
- Xu, S.; Zhi, H.; Hou, X.; Cohen, R.A.; Jiang, B. IkappaBbeta attenuates angiotensin II-induced cardiovascular inflammation and fibrosis in mice. Hypertension 2011, 58, 310–316. [Google Scholar] [CrossRef]
- Kuo, H.F.; Liu, P.L.; Chong, I.W.; Liu, Y.P.; Chen, Y.H.; Ku, P.M.; Li, C.Y.; Chen, H.H.; Chiang, H.C.; Wang, C.L.; et al. Pigment epithelium-derived factor mediates autophagy and apoptosis in myocardial hypoxia/reoxygenation injury. PLoS ONE 2016, 11, e0156059. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.L.; Liu, W.L.; Chang, J.M.; Chen, Y.H.; Liu, Y.P.; Kuo, H.F.; Hsieh, C.C.; Ding, Y.S.; Chen, W.W.; Chong, I.W. MicroRNA-200c inhibits epithelial-mesenchymal transition, invasion, and migration of lung cancer by targeting HMGB1. PLoS ONE 2017, 12, e0180844. [Google Scholar] [CrossRef]
- Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Invest. Derm. 2007, 127, 526–537. [Google Scholar] [CrossRef]
- Zouein, F.A.; Kurdi, M.; Booz, G.W.; Fuseler, J.W. Applying fractal dimension and image analysis to quantify fibrotic collagen deposition and organization in the normal and hypertensive heart. Microsc. Microanal. 2014, 20, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Duan, W.; Zhang, Y.; Zhang, L.; Qile, M.; Liu, Z.; Qiu, F.; Zhao, D.; Lu, Y.; Chu, W. Simvastatin alleviates cardiac fibrosis induced by infarction via up-regulation of TGF-beta receptor III expression. Br. J. Pharm. 2015, 172, 3779–3792. [Google Scholar] [CrossRef] [PubMed]
- Barile, L.; Moccetti, T.; Marban, E.; Vassalli, G. Roles of exosomes in cardioprotection. Eur. Heart J. 2017, 38, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Marini, M.; Ibba-Manneschi, L.; Manetti, M. Cardiac telocyte-derived exosomes and their possible implications in cardiovascular pathophysiology. Adv. Exp. Med. Biol. 2017, 998, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Savvatis, K.; Kang, J.S.; Fan, P.; Zhong, H.; Schwartz, K.; Barry, V.; Mikels-Vigdal, A.; Karpinski, S.; Kornyeyev, D.; et al. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat. Commun. 2016, 7, 13710. [Google Scholar] [CrossRef]
- Siddesha, J.M.; Valente, A.J.; Sakamuri, S.S.; Yoshida, T.; Gardner, J.D.; Somanna, N.; Takahashi, C.; Noda, M.; Chandrasekar, B. Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK. J. Mol. Cell. Cardiol. 2013, 65, 9–18. [Google Scholar] [CrossRef]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Naik, P.K.; Bozyk, P.D.; Bentley, J.K.; Popova, A.P.; Birch, C.M.; Wilke, C.A.; Fry, C.D.; White, E.S.; Sisson, T.H.; Tayob, N.; et al. Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L1046–L1056. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, D.N.; Moore, B.B. The role of periostin in lung fibrosis and airway remodeling. Cell. Mol. Life Sci. 2017, 74, 4305–4314. [Google Scholar] [CrossRef] [PubMed]
- Landry, N.M.; Cohen, S.; Dixon, I.M.C. Periostin in cardiovascular disease and development: A tale of two distinct roles. Basic Res. Cardiol. 2017, 113, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ge, Y.; Cheng, Q.; Zhang, Q.; Fang, L.; Zheng, J. Decorin is a pivotal effector in the extracellular matrix and tumour microenvironment. Oncotarget 2018, 9, 5480–5491. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.W.; Sun, B.; Ma, P.P.; Gai, Y.S.; Sun, W.Z.; Yu, H.Q.; Li, J. Rosuvastatin inhibits inflammatory response and resists fibrosis after myocardial infarction. Eur. Rev. Med. Pharm. Sci. 2018, 22, 238–245. [Google Scholar] [CrossRef]
- Bei, W.J.; Chen, S.Q.; Li, H.L.; Wu, D.X.; Duan, C.; Chen, P.Y.; Chen, J.Y.; Tan, N.; Xie, N.J.; Liu, Y. Comparing common doses (double-dose vs usual-dose) of atorvastatin for preventing contrast-induced acute kidney injury and mortality after coronary angiography. Medicine (Baltimore) 2017, 96, e7501. [Google Scholar] [CrossRef]
- Pedersen, T.R.; Tobert, J.A. Simvastatin: A review. Expert Opin. Pharm. 2004, 5, 2583–2596. [Google Scholar] [CrossRef]
- Kim, M.J.; Bible, K.L.; Regnier, M.; Adams, M.E.; Froehner, S.C.; Whitehead, N.P. Simvastatin provides long-term improvement of left ventricular function and prevents cardiac fibrosis in muscular dystrophy. Physiol. Rep. 2019, 7, e14018. [Google Scholar] [CrossRef]
- Xinwei, J.; Xianghua, F.; Jing, Z.; Xinshun, G.; Ling, X.; Weize, F.; Guozhen, H.; Yunfa, J.; Weili, W.; Shiqiang, L. Comparison of usefulness of simvastatin 20 mg versus 80 mg in preventing contrast-induced nephropathy in patients with acute coronary syndrome undergoing percutaneous coronary intervention. Am. J. Cardiol. 2009, 104, 519–524. [Google Scholar] [CrossRef]
- Bei, Y.; Das, S.; Rodosthenous, R.S.; Holvoet, P.; Vanhaverbeke, M.; Monteiro, M.C.; Monteiro, V.V.S.; Radosinska, J.; Bartekova, M.; Jansen, F.; et al. Extracellular vesicles in cardiovascular theranostics. Theranostics 2017, 7, 4168–4182. [Google Scholar] [CrossRef]
- Huang, Q.; Cai, B. Exosomes as new intercellular mediators in development and therapeutics of cardiomyocyte hypertrophy. Adv. Exp. Med. Biol. 2017, 998, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Poe, A.J.; Knowlton, A.A. Exosomes as agents of change in the cardiovascular system. J. Mol. Cell. Cardiol. 2017, 111, 40–50. [Google Scholar] [CrossRef]
- Lyu, L.; Wang, H.; Li, B.; Qin, Q.; Qi, L.; Nagarkatti, M.; Nagarkatti, P.; Janicki, J.S.; Wang, X.L.; Cui, T. A critical role of cardiac fibroblast-derived exosomes in activating renin angiotensin system in cardiomyocytes. J. Mol. Cell. Cardiol. 2015, 89, 268–279. [Google Scholar] [CrossRef]
- Bang, C.; Batkai, S.; Dangwal, S.; Gupta, S.K.; Foinquinos, A.; Holzmann, A.; Just, A.; Remke, J.; Zimmer, K.; Zeug, A.; et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Invest. 2014, 124, 2136–2146. [Google Scholar] [CrossRef]
- Rizvi, F.; Siddiqui, R.; DeFranco, A.; Homar, P.; Emelyanova, L.; Holmuhamedov, E.; Ross, G.; Tajik, A.J.; Jahangir, A. Simvastatin reduces TGF-beta1-induced SMAD2/3-dependent human ventricular fibroblasts differentiation: Role of protein phosphatase activation. Int. J. Cardiol. 2018, 270, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.I.; Koo, S.H.; Cha, T.J.; Heo, J.H.; Kim, H.S.; Jo, G.B.; Lee, J.W. Simvastatin attenuates the oxidative stress, endothelial thrombogenicity and the inducibility of atrial fibrillation in a rat model of ischemic heart failure. Int. J. Mol. Sci. 2014, 15, 14803–14818. [Google Scholar] [CrossRef]
- Kruzynska-Frejtag, A.; Machnicki, M.; Rogers, R.; Markwald, R.R.; Conway, S.J. Periostin (an osteoblast-specific factor) is expressed within the embryonic mouse heart during valve formation. Mech. Dev. 2001, 103, 183–188. [Google Scholar] [CrossRef]
- Guo, X.; Yan, F.; Li, J.; Zhang, C.; Bu, P. SIRT3 attenuates AngII-induced cardiac fibrosis by inhibiting myofibroblasts transdifferentiation via STAT3-NFATc2 pathway. Am. J. Transl. Res. 2017, 9, 3258–3269. [Google Scholar]
- Li, Q.; Liu, X.; Wei, J. Ageing related periostin expression increase from cardiac fibroblasts promotes cardiomyocytes senescent. Biochem. Biophys. Res. Commun. 2014, 452, 497–502. [Google Scholar] [CrossRef]
- Zhao, S.; Wu, H.; Xia, W.; Chen, X.; Zhu, S.; Zhang, S.; Shao, Y.; Ma, W.; Yang, D.; Zhang, J. Periostin expression is upregulated and associated with myocardial fibrosis in human failing hearts. J. Cardiol. 2014, 63, 373–378. [Google Scholar] [CrossRef]
- Paolillo, M.; Schinelli, S. Integrins and exosomes, a dangerous liaison in cancer progression. Cancers 2017, 9, 95. [Google Scholar] [CrossRef]
- Liu, A. Stimulated Brillouin scattering in single-frequency fiber amplifiers with delivery fibers. Opt. Express 2009, 17, 15201–15209. [Google Scholar] [CrossRef]
- Marzoll, A.; Melchior-Becker, A.; Cipollone, F.; Fischer, J.W. Small leucine-rich proteoglycans in atherosclerotic lesions: Novel targets of chronic statin treatment? J Cell. Mol. Med. 2011, 15, 232–243. [Google Scholar] [CrossRef]
- Dobaczewski, M.; de Haan, J.J.; Frangogiannis, N.G. The extracellular matrix modulates fibroblast phenotype and function in the infarcted myocardium. J. Cardiovasc. Transl. Res. 2012, 5, 837–847. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac fibrosis: The fibroblast awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Ladage, D.; Yaniz-Galende, E.; Rapti, K.; Ishikawa, K.; Tilemann, L.; Shapiro, S.; Takewa, Y.; Muller-Ehmsen, J.; Schwarz, M.; Garcia, M.J.; et al. Stimulating myocardial regeneration with periostin Peptide in large mammals improves function post-myocardial infarction but increases myocardial fibrosis. PLoS ONE 2013, 8, e59656. [Google Scholar] [CrossRef]
- Hermida, N.; Markl, A.; Hamelet, J.; Van Assche, T.; Vanderper, A.; Herijgers, P.; van Bilsen, M.; Hilfiker-Kleiner, D.; Noppe, G.; Beauloye, C.; et al. HMGCoA reductase inhibition reverses myocardial fibrosis and diastolic dysfunction through AMP-activated protein kinase activation in a mouse model of metabolic syndrome. Cardiovasc. Res. 2013, 99, 44–54. [Google Scholar] [CrossRef]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic effects of statins on the cardiovascular system. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, H.-F.; Hsieh, C.-C.; Wang, S.-C.; Chang, C.-Y.; Hung, C.-H.; Kuo, P.-L.; Liu, Y.-R.; Li, C.-Y.; Liu, P.-L. Simvastatin Attenuates Cardiac Fibrosis via Regulation of Cardiomyocyte-Derived Exosome Secretion. J. Clin. Med. 2019, 8, 794. https://doi.org/10.3390/jcm8060794
Kuo H-F, Hsieh C-C, Wang S-C, Chang C-Y, Hung C-H, Kuo P-L, Liu Y-R, Li C-Y, Liu P-L. Simvastatin Attenuates Cardiac Fibrosis via Regulation of Cardiomyocyte-Derived Exosome Secretion. Journal of Clinical Medicine. 2019; 8(6):794. https://doi.org/10.3390/jcm8060794
Chicago/Turabian StyleKuo, Hsuan-Fu, Chong-Chao Hsieh, Shu-Chi Wang, Chia-Yuan Chang, Chih-Hsin Hung, Po-Lin Kuo, Yu-Ru Liu, Chia-Yang Li, and Po-Len Liu. 2019. "Simvastatin Attenuates Cardiac Fibrosis via Regulation of Cardiomyocyte-Derived Exosome Secretion" Journal of Clinical Medicine 8, no. 6: 794. https://doi.org/10.3390/jcm8060794
APA StyleKuo, H.-F., Hsieh, C.-C., Wang, S.-C., Chang, C.-Y., Hung, C.-H., Kuo, P.-L., Liu, Y.-R., Li, C.-Y., & Liu, P.-L. (2019). Simvastatin Attenuates Cardiac Fibrosis via Regulation of Cardiomyocyte-Derived Exosome Secretion. Journal of Clinical Medicine, 8(6), 794. https://doi.org/10.3390/jcm8060794