Vascular Dysfunction in Alzheimer’s Disease: A Prelude to the Pathological Process or a Consequence of It?

 and
and 
Abstract
1. Introduction
2. Cerebrovascular Alterations in Alzheimer’s Disease
2.1. Neurovascular Coupling Deficits and Metabolic Dysfunction
2.2. Vascular Morphology and Angiogenesis
2.3. The Blood-Brain Barrier
2.3.1. The Neurovascular Unit
2.3.2. BBB Breakdown—A Leaky Barrier
2.3.3. Glucose Metabolism
2.4. Amyloid-Related Vascular Pathology and BBB Clearance Deficits in AD
2.4.1. A Link between Vasoactive Dysfunction and Aβ Pathology
2.4.2. Aβ-Endothelial Cell Interactions
2.4.3. Aβ-Vascular Smooth Muscle Cell and Aβ-Pericyte Interactions
2.4.4. The Role of Aβ in Aberrant Angiogenesis and BBB Dysfunction
2.5. Tau Pathology and Vascular Dysfunction
2.6. Vascular Cells as Inflammatory Mediators in the AD Brain
3. Systemic Vascular Health and Dementia Risk
3.1. Hypertension
3.2. Atherosclerosis
3.3. Hypercholesterolemia
4. Alzheimer’s Disease and Vascular Dementia
5. Implications for Therapy and Drug Design
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Vinters, H.V. Emerging concepts in Alzheimer’s disease. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 291–319. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Patterson, C. World Alzheimer Report 2018: The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International: London, UK, 2018. [Google Scholar]
- Förstl, H.; Kurz, A. Clinical features of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 288–290. [Google Scholar] [CrossRef]
- Calvo-Flores Guzmán, B.; Vinnakota, C.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. The GABAergic system as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2018, 146, 649–669. [Google Scholar] [CrossRef] [PubMed]
- Bachurin, S.O.; Bovina, E.V.; Ustyugov, A.A. Drugs in clinical trials for Alzheimer’s disease: The major trends. Med. Res. Rev. 2017, 37, 1186–1225. [Google Scholar] [CrossRef]
- Miyakawa, T. Vascular pathology in Alzheimer’s disease. Psychogeriatr. Off. J. Jpn. Psychogeriatr. Soc. 2010, 10, 39–44. [Google Scholar] [CrossRef]
- de la Torre, J. The vascular hypothesis of Alzheimer’s disease: A key to preclinical prediction of dementia using neuroimaging. J. Alzheimers Dis. 2018, 63, 35–52. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, J.C.; Mussivan, T. Can disturbed brain microcirculation cause Alzheimer’s disease? Neurol. Res. 1993, 15, 146–153. [Google Scholar] [CrossRef]
- Di Marco, L.Y.; Venneri, A.; Farkas, E.; Evans, P.C.; Marzo, A.; Frangi, A.F. Vascular dysfunction in the pathogenesis of Alzheimer’s disease—A review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol. Dis. 2015, 82, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Hays, C.C.; Zlatar, Z.Z.; Wierenga, C.E. The utility of cerebral blood flow as a biomarker of preclinical Alzheimer’s disease. Cell. Mol. Neurobiol. 2016, 36, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Binnewijzend, M.A.A.; Benedictus, M.R.; Kuijer, J.P.A.; van der Flier, W.M.; Teunissen, C.E.; Prins, N.D.; Wattjes, M.P.; van Berckel, B.N.M.; Scheltens, P.; Barkhof, F. Cerebral perfusion in the predementia stages of Alzheimer’s disease. Eur. Radiol. 2016, 26, 506–514. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Mosconi, L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 486–510. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Kalaria, R.N. Comparison between Alzheimer’s disease and vascular dementia: Implications for treatment. Neurol. Res. 2003, 25, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Erkinjuntti, T.; Román, G.; Gauthier, S.; Feldman, H.; Rockwood, K. Emerging therapies for vascular dementia and vascular cognitive impairment. Stroke 2004, 35, 1010–1017. [Google Scholar] [CrossRef]
- Custodio, N.; Montesinos, R.; Lira, D.; Herrera-Pérez, E.; Bardales, Y.; Valeriano-Lorenzo, L. Mixed dementia: A review of the evidence. Dement. Neuropsychol. 2017, 11, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Snowdon, D.A.; Greiner, L.H.; Mortimer, J.A.; Riley, K.P.; Greiner, P.A.; Markesbery, W.R. Brain infarction and the clinical expression of Alzheimer disease: The nun study. JAMA 1997, 277, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.; Esiri, M.M.; Jobst, K.A.; Morris, J.H.; King, E.M.F.; McDonald, B.; Joachim, C.; Litchfield, S.; Barnetson, L.; Smith, A.D. The effects of additional pathology on the cognitive deficit in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1997, 56, 165–170. [Google Scholar] [CrossRef]
- Tian, J.; Shi, J.; Bailey, K.; Mann, D.M.A. Relationships between arteriosclerosis, cerebral amyloid angiopathy and myelin loss from cerebral cortical white matter in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2004, 30, 46–56. [Google Scholar] [CrossRef]
- Kalback, W.; Esh, C.; Castaño, E.M.; Rahman, A.; Kokjohn, T.; Luehrs, D.C.; Sue, L.; Cisneros, R.; Gerber, F.; Richardson, C.; et al. Atherosclerosis, vascular amyloidosis and brain hypoperfusion in the pathogenesis of sporadic Alzheimer’s disease. Neurol. Res. 2004, 26, 525–539. [Google Scholar] [CrossRef]
- Robert, W.M.; Stanley, C.R., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genom. Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef]
- de Oliveira, F.F.; Chen, E.S.; Smith, M.C.; Bertolucci, P.H.F. Pharmacogenetics of angiotensin-converting enzyme inhibitors in patients with Alzheimer’s disease dementia. Curr. Alzheimer Res. 2018, 15, 386–398. [Google Scholar] [CrossRef]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 184. [Google Scholar] [CrossRef]
- Shamieh, S.E.; Costanian, C.; Kassir, R.; Visvkis-Siest, S.; Bissar-Tadmouri, N. APOE genotypes in Lebanon: Distribution and association with hypercholesterolemia and Alzheimer’s disease. Pers. Med. 2019, 16, 15–23. [Google Scholar] [CrossRef]
- Chartier-Hariln, M.-C.; Parfitt, M.; Legrain, S.; Pérez-Tur, J.; Brousseau, T.; Evans, A.; Berr, C.; Vldal, O.; Roques, P.; Gourlet, V.; et al. Apolipoprotein E, ε4 allele as a major risk factor for sporadic early and late-onset forms of Alzheimer’s disease: Analysis of the 19q13.2 chromosomal region. Hum. Mol. Genet. 1994, 3, 569–574. [Google Scholar] [CrossRef]
- de Oliveira, F.F.; Chen, E.S.; Cardoso Smith, M.A.; Ferreira Bertolucci, P.H. Effects of APOE gene haplotypes and measures of cardiovascular risk over cognitive and functional decline in one year in patients with Alzheimer’s disease dementia. Alzheimers Dement. J. Alzheimers Assoc. 2016, 12, P952. [Google Scholar] [CrossRef]
- Irie, F.; Fitzpatrick, A.L.; Lopez, O.L.; Kuller, L.H.; Peila, R.; Newman, A.B.; Launer, L.J. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE ε4: The Cardiovascular Health Study Cognition Study. Arch. Neurol. 2008, 65, 89–93. [Google Scholar] [CrossRef]
- Peila, R.; Rodriguez, B.L.; Launer, L.J. Type 2 diabetes, APOE gene and the risk for dementia and related pathologies: The Honolulu-Asia aging study. Diabetes 2002, 51, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Blair, C.K.; Folsom, A.R.; Knopman, D.S.; Bray, M.S.; Mosley, T.H.; Boerwinkle, E. APOE genotype and cognitive decline in a middle-aged cohort. Neurology 2005, 64, 268–276. [Google Scholar] [CrossRef]
- Bonte, F.J.; Ross, E.D.; Chehabi, H.H.; Devous, M.D., Sr. SPECT study of regional cerebral blood flow in Alzheimer disease. J. Comput. Assist. Tomogr. 1986, 10, 579–583. [Google Scholar] [CrossRef]
- Burns, A.; Philpot, M.P.; Costa, D.C.; Ell, P.J.; Levy, R. The investigation of Alzheimer’s disease with single photon emission tomography. J. Neurol. Neurosurg. Psychiatry 1989, 52, 248–253. [Google Scholar] [CrossRef]
- Hirsch, C.; Bartenstein, P.; Minoshima, S.; Mosch, D.; Willoch, F.; Buch, K.; Schad, D.; Schwaiger, M.; Kurz, A. Reduction of regional cerebral blood flow and cognitive impairment in patients with Alzheimer’s disease: Evaluation of an observer-independent analytic approach. Dement. Geriatr. Cogn. Disord. 1997, 8, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Mueller, S.T.; Walshe, T.M.; English, R.J.; Holman, B. Cerebral perfusion imaging in Alzheimer’s disease: Use of single photon emission computed tomography and iofetamine hydrochloride I 123. Arch. Neurol. 1987, 44, 165–168. [Google Scholar] [CrossRef]
- Eberling, J.L.; Jagust, W.J.; Reed, B.R.; Baker, M.G. Reduced temporal lobe blood flow in Alzheimer’s disease. Neurobiol. Aging 1992, 13, 483–491. [Google Scholar] [CrossRef]
- Johnson, N.A.; Jahng, G.-H.; Weiner, M.W.; Miller, B.L.; Chui, H.C.; Jagust, W.J.; Gorno-Tempini, M.L.; Schuff, N. Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: Initial experience. Radiology 2005, 234, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Lopez, O.L.; Carmichael, O.T.; Becker, J.T.; Kuller, L.H.; Gach, H.M. Mild cognitive impairment and Alzheimer disease: Patterns of altered cerebral blood flow at MR imaging. Radiology 2009, 250, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Bradley, K.; O’Sullivan, V.; Soper, N.; Nagy, Z.; King, E.F.; Smith, A.; Shepstone, B. Cerebral perfusion SPET correlated with Braak pathological stage in Alzheimer’s disease. Brain 2002, 125, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Brien, J.T.; Eagger, S.; Syed, G.M.; Sahakian, B.J.; Levy, R. A study of regional cerebral blood flow and cognitive performance in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1992, 55, 1182. [Google Scholar] [CrossRef][Green Version]
- Wilson, K.; Bowen, D.; Francis, P.; Tyrrell, P. Effect of central cholinergic stimulation on regional cerebral blood flow in Alzheimer’s disease. Br. J. Psychiatry 1991, 158, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Alsop, D.C.; Casement, M.; de Bazelaire, C.; Fong, T.; Press, D.Z. Hippocampal hyperperfusion in Alzheimer’s disease. NeuroImage 2008, 42, 1267–1274. [Google Scholar] [CrossRef]
- Hunter, R.; McLuskie, R.; Wyper, D.; Patterson, J.; Christie, J.E.; Brooks, D.N.; McCulloch, J.; Fink, G.; Goodwin, G.M. The pattern of function-related regional cerebral blood flow investigated by single photon emission tomography with 99mTc-HMPAO in patients with presenile Alzheimer’s disease and Korsakoff’s psychosis. Psychol. Med. 1989, 19, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Jobst, K.A.; Smith, A.D.; Barker, C.S.; Wear, A.; King, E.M.; Smith, A.; Anslow, P.A.; Molyneux, A.J.; Shepstone, B.J.; Soper, N. Association of atrophy of the medial temporal lobe with reduced blood flow in the posterior parietotemporal cortex in patients with a clinical and pathological diagnosis of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1992, 55, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Jagust, W.J.; Budinger, T.F.; Reed, B.R. The diagnosis of dementia with single photon emission computed tomography. Arch. Neurol. 1987, 44, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Schuff, N.; Matsumoto, S.; Kmiecik, J.; Studholme, C.; Du, A.; Ezekiel, F.; Miller, B.L.; Kramer, J.H.; Jagust, W.J.; Chui, H.C.; et al. Cerebral blood flow in ischemic vascular dementia and Alzheimer’s disease, measured by arterial spin-labeling magnetic resonance imaging. Alzheimers Dement. 2009, 5, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, P.; Sorg, C.; Förschler, A.; Grimmer, T.; Skokou, M.; Wohlschläger, A.; Perneczky, R.; Zimmer, C.; Kurz, A.; Preibisch, C. Perfusion abnormalities in mild cognitive impairment and mild dementia in Alzheimer’s disease measured by pulsed arterial spin labeling MRI. Eur. Arch. Psychiatry Clin. Neurosci. 2012, 262, 69–77. [Google Scholar] [CrossRef]
- Friedland, R.P.; Budinger, T.F.; Ganz, E.; Yano, Y.; Mathis, C.A.; Koss, B.; Ober, B.A.; Huesman, R.H.; Derenzo, S.E. Regional cerebral metabolic alterations in dementia of the Alzheimer type: Positron emission tomography with [18F] fluorodeoxyglucose. J. Comput. Assist. Tomogr. 1983, 7, 590–598. [Google Scholar] [CrossRef]
- Foster, N.L.; Chase, T.N.; Mansi, L.; Brooks, R.; Fedio, P.; Patronas, N.J.; Di Chiro, G. Cortical abnormalities in Alzheimer’s disease. Ann. Neurol. 1984, 16, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Chase, T.N.; Foster, N.L.; Mansi, L. Alzheimer’s disease and the parietal lobe. Lancet 1983, 322, 225. [Google Scholar] [CrossRef]
- Foster, N.L.; Chase, T.N.; Fedio, P.; Patronas, N.J.; Brooks, R.A.; Di Chiro, G. Alzheimer’s disease: Focal cortical changes shown by positron emission tomography. Neurology 1983, 33, 961–965. [Google Scholar] [CrossRef]
- Benson, D.F.; Kuhl, D.E.; Hawkins, R.A.; Phelps, M.E.; Cummings, J.L.; Tsai, S.Y. The fluorodeoxyglucose 18F scan in Alzheimer’s disease and multi-infarct dementia. Arch. Neurol. 1983, 40, 711–714. [Google Scholar] [CrossRef]
- Metter, E.J.; Riege, W.H.; Kameyama, M.; Kuhl, D.E.; Phelps, M.E. Cerebral metabolic relationships for selected brain regions in Alzheimer’s, Huntington’s and Parkinson’s diseases. J. Cereb. Blood Flow Metab. 1984, 4, 500–506. [Google Scholar] [CrossRef]
- Kuhl, D.E. Imaging local brain function with emission computed tomography. Radiology 1984, 150, 625–631. [Google Scholar] [CrossRef]
- Hirono, N.; Mori, E.; Yasuda, M.; Ishii, K.; Ikejiri, Y.; Imamura, T.; Shimomura, T.; Hashimoto, M.; Yamashita, H.; Sasaki, M. Lack of Association of Apolipoprotein E ε4 allele dose with cerebral glucose metabolism in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1998, 12, 362–367. [Google Scholar] [CrossRef]
- Silverman, D.H.; Small, G.W.; Chang, C.Y.; Lu, C.S.; de Aburto, M.A.K.; Chen, W.; Czernin, J.; Rapoport, S.I.; Pietrini, P.; Alexander, G.E.; et al. Positron emission tomography in evaluation of dementia: Regional brain metabolism and long-term outcome. JAMA 2001, 286, 2120–2127. [Google Scholar] [CrossRef]
- Fazekas, F.; Alavi, A.; Chawluk, J.B.; Zimmerman, R.A.; Hackney, D.; Bilaniuk, L.; Rosen, M.; Alves, W.M.; Hurtig, H.I.; Jamieson, D.G.; et al. Comparison of CT, MR and PET in Alzheimer’s dementia and normal aging. J. Nucl. Med. 1989, 30, 1607–1615. [Google Scholar]
- Hoffman, J.M.; Welsh-Bohmer, K.A.; Hanson, M.; Crain, B.; Hulette, C.; Earl, N.; Coleman, R.E. FDG PET imaging in patients with pathologically verified dementia. J. Nucl. Med. 2000, 41, 1920–1928. [Google Scholar]
- Holman, B.L.; Johnson, K.A.; Gerada, B.; Carvalho, P.A.; Satlin, A. The scintigraphic appearance of Alzheimer’s disease: A prospective study using technetium-99m-HMPAO SPECT. J. Nucl. Med. 1992, 33, 181–185. [Google Scholar] [PubMed]
- Jagust, W.; Thisted, R.; Devous, M.D., Sr.; Van Heertum, R.; Mayberg, H.; Jobst, K.; Smith, A.D.; Borys, N. SPECT perfusion imaging in the diagnosis of Alzheimer’s disease: A clinical-pathologic study. Neurology 2001, 56, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wolk, D.A.; Reddin, J.S.; Korczykowski, M.; Martinez, P.M.; Musiek, E.S.; Newberg, A.B.; Julin, P.; Arnold, S.E.; Greenberg, J.H.; et al. Voxel-level comparison of arterial spin-labeled perfusion MRI and FDG-PET in Alzheimer disease. Neurology 2011, 77, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Chen, Y.; Korczykowski, M.; Saboury, B.; Martinez, P.M.; Reddin, J.S.; Alavi, A.; Kimberg, D.Y.; Wolk, D.A.; Julin, P.; et al. Direct comparison of fluorodeoxyglucose positron emission tomography and arterial spin labeling magnetic resonance imaging in Alzheimer’s disease. Alzheimers Dement. 2012, 8, 51–59. [Google Scholar] [CrossRef]
- Gonzalez, R.G.; Fischman, A.J.; Guimaraes, A.R.; Carr, C.A.; Stern, C.E.; Halpern, E.F.; Growdon, J.H.; Rosen, B.R. Functional MR in the evaluation of dementia: Correlation of abnormal dynamic cerebral blood volume measurements with changes in cerebral metabolism on positron emission tomography with fludeoxyglucose F 18. Ajnr. Am. J. Neuroradiol. 1995, 16, 1763–1770. [Google Scholar] [PubMed]
- Yoshiura, T.; Mihara, F.; Kuwabara, Y.; Ogomori, K.; Kaneko, K.; Tanaka, A.; Sasaki, M.; Nakagawa, M.; Koga, H.; Yamanaka, T.; et al. MR Relative Cerebral Blood Flow Mapping of Alzheimer disease: Correlation with Tc-99m HMPAO SPECT. Acad. Radiol. 2002, 9, 1383–1387. [Google Scholar] [CrossRef]
- Rivera-Rivera, L.A.; Schubert, T.; Turski, P.; Johnson, K.M.; Berman, S.E.; Rowley, H.A.; Carlsson, C.M.; Johnson, S.C.; Wieben, O. Changes in intracranial venous blood flow and pulsatility in Alzheimer’s disease: A 4D flow MRI study. J. Cereb. Blood Flow Metab. 2016, 37, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Rivera, L.A.; Turski, P.; Johnson, K.M.; Hoffman, C.; Berman, S.E.; Kilgas, P.; Rowley, H.A.; Carlsson, C.M.; Johnson, S.C.; Wieben, O. 4D flow MRI for intracranial hemodynamics assessment in Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2015, 36, 1718–1730. [Google Scholar] [CrossRef]
- Okonkwo, O.C.; Xu, G.; Oh, J.M.; Dowling, N.M.; Carlsson, C.M.; Gallagher, C.L.; Birdsill, A.C.; Palotti, M.; Wharton, W.; Hermann, B.P.; et al. Cerebral blood flow is diminished in asymptomatic middle-aged adults with maternal history of Alzheimer’s disease. Cereb. Cortex 2014, 24, 978–988. [Google Scholar] [CrossRef]
- Ishii, K.; Sasaki, M.; Yamaji, S.; Sakamoto, S.; Kitagaki, H.; Mori, E. Demonstration of decreased posterior cingulate perfusion in mild Alzheimer’s disease by means of H215O positron emission tomography. Eur. J. Nucl. Med. 1997, 24, 670–673. [Google Scholar] [CrossRef]
- Callen, D.J.; Black, S.E.; Caldwell, C.B. Limbic system perfusion in Alzheimer’s disease measured by MRI-coregistered HMPAO SPET. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 899–906. [Google Scholar] [CrossRef]
- Minoshima, S.; Giordani, B.; Berent, S.; Frey, K.A.; Foster, N.L.; Kuhl, D.E. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann. Neurol. 1997, 42, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Thambisetty, M.; Beason-Held, L.; An, Y.; Kraut, M.A.; Resnick, S.M. APOE ε4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch. Neurol. 2010, 67, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, A.S.; Podraza, K.M.; Bangen, K.J.; Taylor, C.; Sherzai, A.; Sidhar, K.; Liu, T.T.; Dale, A.M.; Buxton, R.B. Cerebral perfusion and oxygenation differences in Alzheimer’s disease risk. Neurobiol. Aging 2009, 30, 1737–1748. [Google Scholar] [CrossRef]
- Kennedy, A.M.; Frackowiak, R.S.J.; Newman, S.K.; Bloomfield, P.M.; Seaward, J.; Roques, P.; Lewington, G.; Cunningham, V.J.; Rossor, M.N. Deficits in cerebral glucose metabolism demonstrated by positron emission tomography in individuals at risk of familial Alzheimer’s disease. Neurosci. Lett. 1995, 186, 17–20. [Google Scholar] [CrossRef]
- Jagust, W.J.; Bandy, D.; Chen, K.; Foster, N.L.; Landau, S.M.; Mathis, C.A.; Price, J.C.; Reiman, E.M.; Skovronsky, D.; Koeppe, R.A. The Alzheimer’s Disease Neuroimaging Initiative positron emission tomography core. Alzheimers Dement. 2010, 6, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Jones, K.; Holman, B.L.; Becker, J.A.; Spiers, P.A.; Satlin, A.; Albert, M.S. Preclinical prediction of Alzheimer’s disease using SPECT. Neurology 1998, 50, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Stomrud, E.; Forsberg, A.; Hagerstrom, D.; Ryding, E.; Blennow, K.; Zetterberg, H.; Minthon, L.; Hansson, O.; Londos, E. CSF biomarkers correlate with cerebral blood flow on SPECT in healthy elderly. Dement. Geriatr. Cogn. Disord. 2012, 33, 156–163. [Google Scholar] [CrossRef]
- Mattsson, N.; Tosun, D.; Insel, P.S.; Simonson, A.; Jack, C.R., Jr.; Beckett, L.A.; Donohue, M.; Jagust, W.; Schuff, N.; Weiner, M.W. Association of brain amyloid-beta with cerebral perfusion and structure in Alzheimer’s disease and mild cognitive impairment. Brain 2014, 137, 1550–1561. [Google Scholar] [CrossRef]
- Niwa, K.; Carlson, G.A.; Iadecola, C. Exogenous Aβ1–40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J. Cereb. Blood Flow Metab. 2000, 20, 1659–1668. [Google Scholar] [CrossRef]
- Maier, F.C.; Wehrl, H.F.; Schmid, A.M.; Mannheim, J.G.; Wiehr, S.; Lerdkrai, C.; Calaminus, C.; Stahlschmidt, A.; Ye, L.; Burnet, M.; et al. Longitudinal PET-MRI reveals β-amyloid deposition and rCBF dynamics and connects vascular amyloidosis to quantitative loss of perfusion. Nat. Med. 2014, 20, 1485. [Google Scholar] [CrossRef] [PubMed]
- Sojkova, J.; Beason-Held, L.; Zhou, Y.; An, Y.; Kraut, M.A.; Ye, W.; Ferrucci, L.; Mathis, C.A.; Klunk, W.E.; Wong, D.F.; et al. Longitudinal cerebral blood flow and amyloid deposition: An emerging pattern? J. Nucl. Med. 2008, 49, 1465–1471. [Google Scholar] [CrossRef]
- Gietl, A.F.; Warnock, G.; Riese, F.; Kälin, A.M.; Saake, A.; Gruber, E.; Leh, S.E.; Unschuld, P.G.; Kuhn, F.P.; Burger, C.; et al. Regional cerebral blood flow estimated by early PiB uptake is reduced in mild cognitive impairment and associated with age in an amyloid-dependent manner. Neurobiol. Aging 2015, 36, 1619–1628. [Google Scholar] [CrossRef]
- Oh, H.; Habeck, C.; Madison, C.; Jagust, W. Covarying alterations in Aβ deposition, glucose metabolism and gray matter volume in cognitively normal elderly. Hum. Brain Mapp. 2014, 35, 297–308. [Google Scholar] [CrossRef]
- Lowe, V.J.; Weigand, S.D.; Senjem, M.L.; Vemuri, P.; Jordan, L.; Kantarci, K.; Boeve, B.; Jack, C.R.; Knopman, D.; Petersen, R.C. Association of hypometabolism and amyloid levels in aging, normal subjects. Neurology 2014, 82, 1959–1967. [Google Scholar] [CrossRef]
- Knopman, D.S.; Jack, C.R., Jr.; Wiste, H.J.; Lundt, E.S.; Weigand, S.D.; Vemuri, P.; Lowe, V.J.; Kantarci, K.; Gunter, J.L.; Senjem, M.L.; et al. 18F-fluorodeoxyglucose positron emission tomography, aging and apolipoprotein E genotype in cognitively normal persons. Neurobiol. Aging 2014, 35, 2096–2106. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Jelic, V.; Basun, H.; Lannfelt, L.; Valind, S.; Winblad, B.; Nordberg, A. No difference in cerebral glucose metabolism in patients with Alzheimer disease and differing apolipoprotein E genotypes. Arch. Neurol. 1997, 54, 273–277. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Gelernter, J.; MacAvoy, M.G.; Avery, R.A.; Criden, M.; Okereke, O.; Varma, P.; Seibyl, J.P.; Hoffer, P.B. Absence of an Apolipoprotein E ε4 allele is associated with increased parietal regional cerebral blood flow asymmetry in Alzheimer disease. Arch. Neurol. 1998, 55, 1460–1466. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Higuchi, M.; Arai, H.; Nakagawa, T.; Higuchi, S.; Muramatsu, T.; Matsushita, S.; Kosaka, Y.; Itoh, M.; Sasaki, H. Regional cerebral glucose utilization is modulated by the dosage of apolipoprotein E type 4 allele and alpha1-antichymotrypsin type A allele in Alzheimer’s disease. Neuroreport 1997, 8, 2639–2643. [Google Scholar] [CrossRef]
- Kim, S.M.; Kim, M.J.; Rhee, H.Y.; Ryu, C.-W.; Kim, E.J.; Petersen, E.T.; Jahng, G.-H. Regional cerebral perfusion in patients with Alzheimer’s disease and mild cognitive impairment: Effect of APOE Epsilon4 allele. Neuroradiology 2013, 55, 25–34. [Google Scholar] [CrossRef]
- Sakamoto, S.; Matsuda, H.; Asada, T.; Ohnishi, T.; Nakano, S.; Kanetaka, H.; Takasaki, M. Apolipoprotein E genotype and early Alzheimer’s disease: A longitudinal SPECT study. J. Neuroimaging 2003, 13, 113–123. [Google Scholar] [CrossRef]
- Høgh, P.; Knudsen, G.M.; Kjær, K.H.; Jørgensen, O.S.; Paulson, O.B.; Waldemar, G. Single photon emission computed tomography and apolipoprotein E in Alzheimer’s disease: Impact of the ε4 allele on regional cerebral blood flow. J. Geriatr. Psychiatry Neurol. 2001, 14, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Mielke, R.; Zerres, K.; Uhlhaas, S.; Kessler, J.; Heiss, W.D. Apolipoprotein E polymorphism influences the cerebral metabolic pattern in Alzheimer’s disease. Neurosci. Lett. 1998, 254, 49–52. [Google Scholar] [CrossRef]
- Tanaka, S.; Kawamata, J.; Shimohama, S.; Akaki, H.; Akiguchi, I.; Kimura, J.; Ueda, K. Inferior temporal lobe atrophy and APOE genotypes in Alzheimer’s disease. X-ray computed tomography, magnetic resonance imaging and Xe-133 SPECT studies. Dement. Geriatr. Cogn. Disord. 1998, 9, 90–98. [Google Scholar] [CrossRef]
- Lehtovirta, M.; Kuikka, J.; Helisalmi, S.; Hartikainen, P.; Mannermaa, A.; Ryynänen, M.; Riekkinen, P.S.; Soininen, H. Longitudinal SPECT study in Alzheimer’s disease: Relation to apolipoprotein E polymorphism. J. Neurol. Neurosurg. Psychiatry 1998, 64, 742–746. [Google Scholar] [CrossRef]
- Lehtovirta, M.; Soininen, H.; Laakso, M.P.; Partanen, K.; Helisalmi, S.; Mannermaa, A.; Ryynänen, M.; Kuikka, J.; Hartikainen, P.; Riekkinen, P.J., Sr. SPECT and MRI analysis in Alzheimer’s disease: Relation to apolipoprotein E epsilon 4 allele. J. Neurol. Neurosurg. Psychiatry 1996, 60, 644–649. [Google Scholar] [CrossRef]
- Small, G.W.; Mazziotta, J.C.; Collins, M.T.; Baxter, L.R.; Phelps, M.E.; Mandelkern, M.A.; Kaplan, A.; La Rue, A.; Adamson, C.F.; Chang, L.; et al. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 1995, 273, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Caselli, R.J.; Yun, L.S.; Chen, K.; Bandy, D.; Minoshima, S.; Thibodeau, S.N.; Osborne, D. Preclinical evidence of Alzheimer’s Disease in Persons Homozygous for the ε4 allele for apolipoprotein E. N. Engl. J. Med. 1996, 334, 752–758. [Google Scholar] [CrossRef]
- Wierenga, C.E.; Clark, L.R.; Dev, S.I.; Shin, D.D.; Jurick, S.M.; Rissman, R.A.; Liu, T.T.; Bondi, M.W. Interaction of age and APOE genotype on cerebral blood flow at rest. J. Alzheimers Dis. 2013, 34, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Filippini, N.; MacIntosh, B.J.; Hough, M.G.; Goodwin, G.M.; Frisoni, G.B.; Smith, S.M.; Matthews, P.M.; Beckmann, C.F.; Mackay, C.E. Distinct patterns of brain activity in young carriers of the APOEε4 allele. Proc. Natl. Acad. Sci. USA 2009, 106, 7209–7214. [Google Scholar] [CrossRef]
- Filippini, N.; Ebmeier, K.P.; MacIntosh, B.J.; Trachtenberg, A.J.; Frisoni, G.B.; Wilcock, G.K.; Beckmann, C.F.; Smith, S.M.; Matthews, P.M.; Mackay, C.E. Differential effects of the APOE genotype on brain function across the lifespan. NeuroImage 2011, 54, 602–610. [Google Scholar] [CrossRef]
- Nizari, S.; Romero, I.A.; Hawkes, C.A. The role of perivascular innervation and neurally mediated vasoreactivity in the pathophysiology of Alzheimer’s disease. Clin. Sci. 2017, 131, 1207–1214. [Google Scholar] [CrossRef]
- Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012, 11, 1006–1012. [Google Scholar] [CrossRef]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Claassen, J.A.H.R.; Jansen, R.W.M.M. Cholinergically mediated augmentation of cerebral perfusion in Alzheimer’s disease and related cognitive disorders: The Cholinergic–Vascular Hypothesis. J. Gerontol. Ser. A 2006, 61, 267–271. [Google Scholar] [CrossRef]
- Van Beek, A.H.E.A.; Claassen, J.A.H.R. The cerebrovascular role of the cholinergic neural system in Alzheimer’s disease. Behav. Brain Res. 2011, 221, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E. Cholinergic modulation of the cortical microvascular bed. Prog. Brain Res. 2004, 145, 171–178. [Google Scholar] [PubMed]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Peppiatt, C.M.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nature 2006, 443, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Hempelmann, R.G.; Ziegler, A. Endothelium-dependent noradrenaline-induced relaxation of rat isolated cerebral arteries: Pharmacological characterization of receptor subtypes involved. Br. J. Pharmacol. 1993, 110, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Gannon, M.; Che, P.; Chen, Y.; Jiao, K.; Roberson, E.D.; Wang, Q. Noradrenergic dysfunction in Alzheimer’s disease. Front. Neurosci. 2015, 9, 220. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.T. Glutamatergic systems in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2003, 18, S15–S21. [Google Scholar] [CrossRef]
- Metea, M.R.; Newman, E.A. Glial cells dilate and constrict blood vessels: A mechanism of neurovascular coupling. J. Neurosci. 2006, 26, 2862–2870. [Google Scholar] [CrossRef]
- Govindpani, K.; Calvo-Flores Guzman, B.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. Towards a better understanding of GABAergic remodeling in Alzheimer’s disease. Int. J. Mol. Sci. 2017, 18, 1813. [Google Scholar] [CrossRef]
- Matthew, E.; Andreason, P.; Pettigrew, K.; Carson, R.E.; Herscovitch, P.; Cohen, R.; King, C.; Johanson, C.E.; Greenblatt, D.J.; Paul, S.M. Benzodiazepine receptors mediate regional blood flow changes in the living human brain. Proc. Natl. Acad. Sci. USA 1995, 92, 2775–2779. [Google Scholar] [CrossRef]
- Dzamba, D.; Harantova, L.; Butenko, O.; Anderova, M. Glial cells—The key elements of Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 894–911. [Google Scholar] [CrossRef]
- Fischer, V.W.; Siddiqi, A.; Yusufaly, Y. Altered angioarchitecture in selected areas of brains with Alzheimer’s disease. Acta Neuropathol. 1990, 79, 672–679. [Google Scholar] [CrossRef]
- Hassler, O. Vascular changes in senile brains. Acta Neuropathol. 1965, 5, 40–53. [Google Scholar] [CrossRef]
- Beskow, J.; Hassler, O.; Ottosson, J.O. Cerebral arterial deformities in relation to senile deterioration. Acta Psychiatr. Scand. 1971, 47, 111–119. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Kroon, S.N. Expression of leukocyte antigen CD34 by brain capillaries in Alzheimer’s disease and neurologically normal subjects. Acta Neuropathol. 1992, 84, 606–612. [Google Scholar] [CrossRef]
- Baloyannis, S.J.; Baloyannis, I.S. The vascular factor in Alzheimer’s disease: A study in Golgi technique and electron microscopy. J. Neurol. Sci. 2012, 322, 117–121. [Google Scholar] [CrossRef]
- de la Torre, J.C. Hemodynamic consequences of deformed microvessels in the brain in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1997, 826, 75–91. [Google Scholar] [CrossRef]
- Hunter, J.M.; Kwan, J.; Malek-Ahmadi, M.; Maarouf, C.L.; Kokjohn, T.A.; Belden, C.; Sabbagh, M.N.; Beach, T.G.; Roher, A.E. Morphological and pathological evolution of the brain microcirculation in aging and Alzheimer’s disease. PLoS ONE 2012, 7, e36893. [Google Scholar] [CrossRef]
- McGeer, P.L.; Zhu, S.G.; Dedhar, S. Immunostaining of human brain capillaries by antibodies to very late antigens. J. Neuroimmunol. 1990, 26, 213–218. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Hedera, P. Differential degeneration of the cerebral microvasculature in Alzheimer’s disease. Neuroreport 1995, 6, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Challa, V.R.; Thore, C.R.; Moody, D.M.; Anstrom, J.A.; Brown, W.R. Increase of white matter string vessels in Alzheimer’s disease. J. Alzheimers Dis. 2004, 6, 379–383; discussion 443–379. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.R. A review of string vessels or collapsed, empty basement membrane tubes. J. Alzheimers Dis. 2010, 21, 725–739. [Google Scholar] [CrossRef]
- Desai, B.S.; Schneider, J.A.; Li, J.-L.; Carvey, P.M.; Hendey, B. Evidence of angiogenic vessels in Alzheimer’s disease. J. Neural Transm. 2009, 116, 587–597. [Google Scholar] [CrossRef]
- Biron, K.E.; Dickstein, D.L.; Gopaul, R.; Jefferies, W.A. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer’s disease. PLoS ONE 2011, 6, e23789. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.P.; Ulmann-Schuler, A.; Staufenbiel, M.; Krucker, T. Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 3587–3592. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Yuasa, S.; Ueno, M.; Takakura, N.; Koseki, H.; Shirasawa, T. Abnormal blood vessel development in mice lacking presenilin-1. Mech. Dev. 2003, 120, 657–667. [Google Scholar] [CrossRef]
- Perlmutter, L.S.; Chui, H.C.; Saperia, D.; Athanikar, J. Microangiopathy and the colocalization of heparan sulfate proteoglycan with amyloid in senile plaques of Alzheimer’s disease. Brain Res. 1990, 508, 13–19. [Google Scholar] [CrossRef]
- Beckmann, N.; Schuler, A.; Mueggler, T.; Meyer, E.P.; Wiederhold, K.H.; Staufenbiel, M.; Krucker, T. Age-dependent cerebrovascular abnormalities and blood flow disturbances in APP23 mice modeling Alzheimer’s disease. J. Neurosci. 2003, 23, 8453–8459. [Google Scholar] [CrossRef] [PubMed]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef]
- Taichman, N.S.; Young, S.; Cruchley, A.T.; Taylor, P.; Paleolog, E. Human neutrophils secrete vascular endothelial growth factor. J. Leukoc. Biol. 1997, 62, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Gaudry, M.; Brégerie, O.; Andrieu, V.; El Benna, J.; Pocidalo, M.-A.; Hakim, J. Intracellular pool of vascular endothelial growth factor in human neutrophils. Blood 1997, 90, 4153–4161. [Google Scholar]
- Webb, N.J.A.; Myers, C.R.; Watson, C.J.; Bottomley, M.J.; Brenchley, P.E.C. Activated human neutrophils express vascular endothelial growth factor (VEGF). Cytokine 1998, 10, 254–257. [Google Scholar] [CrossRef] [PubMed]
- McCourt, M.; Wang, J.H.; Sookhai, S.; Redmond, H.P. Proinflammatory mediators stimulate neutrophil-directed angiogenesis. Arch. Surg. 1999, 134, 1325–1331. [Google Scholar] [CrossRef]
- Mayhan, W.G. VEGF increases permeability of the blood-brain barrier via a nitric oxide synthase/cGMP-dependent pathway. Am. J. Physiol. Cell Physiol. 1999, 276, C1148–C1153. [Google Scholar] [CrossRef]
- Provias, J.; Jeynes, B. Reduction in vascular endothelial growth factor expression in the superior temporal, hippocampal and brainstem regions in Alzheimer’s disease. Curr. Neurovasc. Res. 2014, 11, 202–209. [Google Scholar] [CrossRef]
- Huang, L.; Jia, J.; Liu, R. Decreased serum levels of the angiogenic factors VEGF and TGF-β1 in Alzheimer’s disease and amnestic mild cognitive impairment. Neurosci. Lett. 2013, 550, 60–63. [Google Scholar] [CrossRef]
- Mateo, I.; Llorca, J.; Infante, J.; Rodriguez-Rodriguez, E.; Fernandez-Viadero, C.; Pena, N.; Berciano, J.; Combarros, O. Low serum VEGF levels are associated with Alzheimer’s disease. Acta Neurol. Scand. 2007, 116, 56–58. [Google Scholar] [CrossRef]
- De Servi, B.; La Porta, C.A.M.; Bontempelli, M.; Comolli, R. Decrease of TGF-β1 plasma levels and increase of nitric oxide synthase activity in leukocytes as potential biomarkers of Alzheimer’s disease. Exp. Gerontol. 2002, 37, 813–821. [Google Scholar] [CrossRef]
- Tarkowski, E.; Issa, R.; Sjögren, M.; Wallin, A.; Blennow, K.; Tarkowski, A.; Kumar, P. Increased intrathecal levels of the angiogenic factors VEGF and TGF-β in Alzheimer’s disease and vascular dementia. Neurobiol. Aging 2002, 23, 237–243. [Google Scholar] [CrossRef]
- Paterson, R.W.; Bartlett, J.W.; Blennow, K.; Fox, N.C.; Alzheimer’s Disease Neuroimaging Initiative; Shaw, L.M.; Trojanowski, J.Q.; Zetterberg, H.; Schott, J.M. Cerebrospinal fluid markers including trefoil factor 3 are associated with neurodegeneration in amyloid-positive individuals. Transl. Psychiatry 2014, 4, e419. [Google Scholar] [CrossRef] [PubMed]
- Solerte, S.B.; Ferrari, E.; Cuzzoni, G.; Locatelli, E.; Giustina, A.; Zamboni, M.; Schifino, N.; Rondanelli, M.; Gazzaruso, C.; Fioravanti, M. Decreased release of the angiogenic peptide vascular endothelial growth factor in Alzheimer’s disease: Recovering effect with insulin and DHEA sulfate. Dement. Geriatr. Cogn. Disord. 2005, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.S.; Mathura, V.S.; Bachmeier, C.; Beaulieu-Abdelahad, D.; Laporte, V.; Weeks, O.; Mullan, M.; Paris, D. Alzheimer’s β-amyloid peptide blocks vascular endothelial growth factor mediated signaling via direct interaction with VEGFR-2. J. Neurochem. 2010, 112, 66–76. [Google Scholar] [CrossRef]
- Yang, S.-P.; Bae, D.-G.; Kang, H.J.; Gwag, B.J.; Gho, Y.S.; Chae, C.-B. Co-accumulation of vascular endothelial growth factor with β-amyloid in the brain of patients with Alzheimer’s disease. Neurobiol. Aging 2004, 25, 283–290. [Google Scholar] [CrossRef]
- Yang, S.P.; Kwon, B.O.; Gho, Y.S.; Chae, C.B. Specific interaction of VEGF165 with β-amyloid and its protective effect on β-amyloid-induced neurotoxicity. J. Neurochem. 2005, 93, 118–127. [Google Scholar] [CrossRef]
- Inai, T.; Mancuso, M.; Hashizume, H.; Baffert, F.; Haskell, A.; Baluk, P.; Hu-Lowe, D.D.; Shalinsky, D.R.; Thurston, G.; Yancopoulos, G.D.; et al. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels and appearance of basement membrane ghosts. Am. J. Pathol. 2004, 165, 35–52. [Google Scholar] [CrossRef]
- Jin, K.L.; Mao, X.O.; Greenberg, D.A. Vascular endothelial growth factor: Direct neuroprotective effect in in vitro ischemia. Proc. Natl. Acad. Sci. USA 2000, 97, 10242–10247. [Google Scholar] [CrossRef]
- Matsuzaki, H.; Tamatani, M.; Yamaguchi, A.; Namikawa, K.; Kiyama, H.; Vitek, M.P.; Mitsuda, N.; Tohyama, M. Vascular endothelial growth factor rescues hippocampal neurons from glutamate-induced toxicity: Signal transduction cascades. FASEB J. 2001, 15, 1218–1220. [Google Scholar] [CrossRef]
- Spuch, C.; Antequera, D.; Portero, A.; Orive, G.; Hernández, R.M.; Molina, J.A.; Bermejo-Pareja, F.; Pedraz, J.L.; Carro, E. The effect of encapsulated VEGF-secreting cells on brain amyloid load and behavioral impairment in a mouse model of Alzheimer’s disease. Biomaterials 2010, 31, 5608–5618. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xie, Z.-H.; Guo, Y.-J.; Zhao, C.-P.; Jiang, H.; Song, Y.; Zhu, Z.-Y.; Lai, C.; Xu, S.-L.; Bi, J.-Z. VEGF-induced angiogenesis ameliorates the memory impairment in APP transgenic mouse model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2011, 411, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Religa, P.; Cao, R.; Religa, D.; Xue, Y.; Bogdanovic, N.; Westaway, D.; Marti, H.H.; Winblad, B.; Cao, Y. VEGF significantly restores impaired memory behavior in Alzheimer’s mice by improvement of vascular survival. Sci. Rep. 2013, 3, 2053. [Google Scholar] [CrossRef] [PubMed]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Cohen, D.L.; Premkumar, D.R.D.; Nag, S.; LaManna, J.C.; Lust, W.D. Vascular endothelial growth factor in Alzheimer’s disease and experimental cerebral ischemia. Mol. Brain Res. 1998, 62, 101–105. [Google Scholar] [CrossRef]
- Zand, L.; Ryu, J.K.; McLarnon, J.G. Induction of angiogenesis in the beta-amyloid peptide-injected rat hippocampus. Neuroreport 2005, 16, 129–132. [Google Scholar] [CrossRef]
- Oosthuyse, B.; Moons, L.; Storkebaum, E.; Beck, H.; Nuyens, D.; Brusselmans, K.; Dorpe, J.V.; Hellings, P.; Gorselink, M.; Heymans, S.; et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 2001, 28, 131. [Google Scholar] [CrossRef]
- Schmid-Brunclik, N.; Bürgi-Taboada, C.; Antoniou, X.; Gassmann, M.; Ogunshola, O.O. Astrocyte responses to injury: VEGF simultaneously modulates cell death and proliferation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R864–R873. [Google Scholar] [CrossRef]
- Grammas, P.; Samany, P.G.; Thirumangalakudi, L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: Implications for disease pathogenesis. J. Alzheimers Dis. 2006, 9, 51–58. [Google Scholar] [CrossRef]
- Gariano, R.F.; Gardner, T.W. Retinal angiogenesis in development and disease. Nature 2005, 438, 960–966. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, H.; Chow, N.; Sallstrom, J.; Bell, R.D.; Deane, R.; Brooks, A.I.; Kanagala, S.; Rubio, A.; Sagare, A.; et al. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat. Med. 2005, 11, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Thirumangalakudi, L.; Samany, P.G.; Owoso, A.; Wiskar, B.; Grammas, P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Haas, T.L. Endothelial cell regulation of matrix metalloproteinases. Can. J. Physiol. Pharmacol. 2005, 83, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Sromek, S.M.; Grahovac, I.; Harik, S.I. Transferrin receptors of rat and human brain and cerebral microvessels and their status in Alzheimer’s disease. Brain Res. 1992, 585, 87–93. [Google Scholar] [CrossRef]
- Vagnucci, A.H.; Li, W.W. Alzheimer’s disease and angiogenesis. Lancet 2003, 361, 605–608. [Google Scholar] [CrossRef]
- Cameron, D.J.; Galvin, C.; Alkam, T.; Sidhu, H.; Ellison, J.; Luna, S.; Ethell, D.W. Alzheimer’s-related peptide amyloid-β plays a conserved role in angiogenesis. PLoS ONE 2012, 7, e39598. [Google Scholar] [CrossRef] [PubMed]
- Boscolo, E.; Folin, M.; Nico, B.; Grandi, C.; Mangieri, D.; Longo, V.; Scienza, R.; Zampieri, P.; Conconi, M.T.; Parnigotto, P.P.; et al. β amyloid angiogenic activity in vitro and in vivo. Int. J. Mol. Med. 2007, 19, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, W.A.; Price, K.A.; Biron, K.E.; Fenninger, F.; Pfeifer, C.G.; Dickstein, D.L. Adjusting the compass: New insights into the role of angiogenesis in Alzheimer’s disease. Alzheimers Res. Ther. 2013, 5, 64. [Google Scholar] [CrossRef]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The neurovascular unit—Concept review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef]
- Lecrux, C.; Hamel, E. The neurovascular unit in brain function and disease. Acta Physiol. 2011, 203, 47–59. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Cockerill, I.; Oliver, J.A.; Xu, H.; Fu, B.M.; Zhu, D. Blood-brain barrier integrity and clearance of amyloid-β from the BBB. Adv. Exp. Med. Biol. 2018, 1097, 26–278. [Google Scholar] [CrossRef]
- Lauer, D.; Reichenbach, A.; Birkenmeier, G. α2-macroglobulin-mediated degradation of amyloid β1–42: A mechanism to enhance amyloid β catabolism. Exp. Neurol. 2001, 167, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Bell, R.; Sagare, A.; Zlokovic, B. Clearance of amyloid-β peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Kanekiyo, T. Blood-brain barrier dysfunction and the pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1965. [Google Scholar] [CrossRef]
- Higuchi, Y.; Miyakawa, T.; Shimoji, A.; Katsuragi, S. Ultrastructural changes of blood vessels in the cerebral cortex in Alzheimer’s disease. Psychiatry Clin. Neurosci. 1987, 41, 283–290. [Google Scholar] [CrossRef]
- Claudio, L. Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer’s disease patients. Acta Neuropathol. 1996, 91, 6–14. [Google Scholar] [CrossRef]
- Vinters, H.V.; Secor, D.L.; Read, S.L.; Frazee, J.G.; Tomiyasu, U.; Stanley, T.M.; Ferreiro, J.A.; Akers, M.-A. Microvasculature in brain biopsy specimens from patients with Alzheimer’s disease: An immunohistochemical and ultrastructural study. Ultrastruct. Pathol. 1994, 18, 333–348. [Google Scholar] [CrossRef]
- Wisniewski, H.M.; Wegiel, J.; Wang, K.C.; Lach, B. Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer’s disease. Acta Neuropathol. 1992, 84, 117–127. [Google Scholar] [CrossRef]
- Stewart, P.A.; Hayakawa, K.; Akers, M.A.; Vinters, H.V. A morphometric study of the blood-brain barrier in Alzheimer’s disease. Lab. Investig. J. Tech. Methods Pathol. 1992, 67, 734–742. [Google Scholar]
- Mancardi, G.L.; Perdelli, F.; Rivano, C.; Leonardi, A.; Bugiani, O. Thickening of the basement membrane of cortical capillaries in Alzheimer’s disease. Acta Neuropathol. 1980, 49, 79–83. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood–brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef]
- Schultz, N.; Brännström, K.; Byman, E.; Moussaud, S.; Nielsen, H.M.; The Netherlands Brain Bank; Olofsson, A.; Wennström, M. Amyloid-beta 1-40 is associated with alterations in NG2+ pericyte population ex vivo and in vitro. Aging Cell 2018, 17, e12728. [Google Scholar] [CrossRef] [PubMed]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef]
- Takata, F.; Dohgu, S.; Matsumoto, J.; Takahashi, H.; Machida, T.; Wakigawa, T.; Harada, E.; Miyaji, H.; Koga, M.; Nishioku, T.; et al. Brain pericytes among cells constituting the blood-brain barrier are highly sensitive to tumor necrosis factor-α, releasing matrix metalloproteinase-9 and migrating in vitro. J. Neuroinflamm. 2011, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Kovac, A.; Erickson, M.A.; Banks, W.A. Brain microvascular pericytes are immunoactive in culture: Cytokine, chemokine, nitric oxide and LRP-1 expression in response to lipopolysaccharide. J. Neuroinflamm. 2011, 8, 139. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Sengillo, J.D.; Sullivan, J.S.; Henkel, J.S.; Appel, S.H.; Zlokovic, B.V. Blood–spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 111–120. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood–brain barrier. Nature 2010, 468, 557. [Google Scholar] [CrossRef]
- Miners, J.S.; Schulz, I.; Love, S. Differing associations between Aβ accumulation, hypoperfusion, blood–brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2017, 38, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Moore, P.; Weigel, P.H. Microvessels from Alzheimer’s disease brains kill neurons in vitro. Am. J. Pathol. 1999, 154, 337–342. [Google Scholar] [CrossRef]
- Grammas, P. A damaged microcirculation contributes to neuronal cell death in Alzheimer’s disease. Neurobiol. Aging 2000, 21, 199–205. [Google Scholar] [CrossRef]
- Yin, X.; Wright, J.; Wall, T.; Grammas, P. Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer’s disease. Am. J. Pathol. 2010, 176, 1600–1606. [Google Scholar] [CrossRef] [PubMed]
- Ciallella, J.R.; Figueiredo, H.; Smith-Swintosky, V.; McGillis, J.P. Thrombin induces surface and intracellular secretion of amyloid precursor protein from human endothelial cells. Thromb. Haemost. 1999, 81, 630–637. [Google Scholar]
- Peers, M.C.; Lenders, M.B.; Défossez, A.; Delacourte, A.; Mazzuca, M. Cortical angiopathy in Alzheimer’s disease: The formation of dystrophic perivascular neurites is related to the exudation of amyloid fibrils from the pathological vessels. Virchows Arch. A 1988, 414, 15–20. [Google Scholar] [CrossRef]
- Wisniewski, H.M.; Kozlowski, P.B. Evidence for blood-brain barrier changes in senile dementia of the Alzheimer type (SDAT). Ann. N. Y. Acad. Sci. 1982, 396, 119–129. [Google Scholar] [CrossRef]
- Slemmon, J.; Hughes, C.; Campbell, G.; Flood, D. Increased levels of hemoglobin-derived and other peptides in Alzheimer’s disease cerebellum. J. Neurosci. 1994, 14, 2225–2235. [Google Scholar] [CrossRef]
- Zipser, B.D.; Johanson, C.E.; Gonzalez, L.; Berzin, T.M.; Tavares, R.; Hulette, C.M.; Vitek, M.P.; Hovanesian, V.; Stopa, E.G. Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; McLarnon, J.G. A leaky blood–brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J. Cell. Mol. Med. 2009, 13, 2911–2925. [Google Scholar] [CrossRef]
- van de Haar, H.J.; Burgmans, S.; Jansen, J.F.A.; van Osch, M.J.P.; van Buchem, M.A.; Muller, M.; Hofman, P.A.M.; Verhey, F.R.J.; Backes, W.H. Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef]
- van de Haar, H.J.; Jansen, J.F.A.; van Osch, M.J.P.; van Buchem, M.A.; Muller, M.; Wong, S.M.; Hofman, P.A.M.; Burgmans, S.; Verhey, F.R.J.; Backes, W.H. Neurovascular unit impairment in early Alzheimer’s disease measured with magnetic resonance imaging. Neurobiol. Aging 2016, 45, 190–196. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Bauer, H.-C.; Krizbai, I.A.; Bauer, H.; Traweger, A. “You Shall Not Pass”—Tight junctions of the blood brain barrier. Front. Neurosci. 2014, 8, 392. [Google Scholar] [CrossRef]
- Marco, S.; Skaper, S.D. Amyloid β-peptide1–42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci. Lett. 2006, 401, 219–224. [Google Scholar] [CrossRef]
- Kook, S.-Y.; Hong, H.S.; Moon, M.; Ha, C.M.; Chang, S.; Mook-Jung, I. Aβ1–42-RAGE interaction disrupts tight junctions of the blood–brain barrier via Ca2+-calcineurin signaling. J. Neurosci. 2012, 32, 8845–8854. [Google Scholar] [CrossRef]
- Krizbai, I.A.; Bauer, H.; Bresgen, N.; Eckl, P.M.; Farkas, A.; Szatmári, E.; Traweger, A.; Wejksza, K.; Bauer, H.-C. Effect of oxidative stress on the junctional proteins of cultured cerebral endothelial cells. Cell. Mol. Neurobiol. 2005, 25, 129–139. [Google Scholar] [CrossRef]
- Fischer, S.; Wobben, M.; Marti, H.H.; Renz, D.; Schaper, W. Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc. Res. 2002, 63, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Okayama, N.; Gute, D.; Krsmanovic, A.; Battarbee, H.; Alexander, J.S. Hypoxia/aglycemia increases endothelial permeability: Role of second messengers and cytoskeleton. Am. J. Physiol. Cell Physiol. 1999, 277, C1066–C1074. [Google Scholar] [CrossRef] [PubMed]
- Navarro, V.; Sanchez-Mejias, E.; Jimenez, S.; Muñoz-Castro, C.; Sanchez-Varo, R.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Microglia in Alzheimer’s disease: Activated, dysfunctional or degenerative. Front. Aging Neurosci. 2018, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Li, L.; Sun, X.-H. Monocytes and Alzheimer’s disease. Neurosci. Bull. 2011, 27, 115–122. [Google Scholar] [CrossRef]
- Fiala, M.; Liu, Q.N.; Sayre, J.; Pop, V.; Brahmandam, V.; Graves, M.C.; Vinters, H.V. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood–brain barrier. Eur. J. Clin. Investig. 2002, 32, 360–371. [Google Scholar] [CrossRef]
- Boven, L.A.; Middel, J.; Verhoef, J.; De Groot, C.J.A.; Nottet, H.S.L.M. Monocyte infiltration is highly associated with loss of the tight junction protein zonula occludens in HIV-1-associated dementia. Neuropathol. Appl. Neurobiol. 2000, 26, 356–360. [Google Scholar] [CrossRef]
- Dallasta, L.M.; Pisarov, L.A.; Esplen, J.E.; Werley, J.V.; Moses, A.V.; Nelson, J.A.; Achim, C.L. Blood-brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am. J. Pathol. 1999, 155, 1915–1927. [Google Scholar] [CrossRef]
- Khoury, J.E.; Luster, A.D. Mechanisms of microglia accumulation in Alzheimer’s disease: Therapeutic implications. Trends Pharmacol. Sci. 2008, 29, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Pietronigro, E.; Bianca, V.D.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease–like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880. [Google Scholar] [CrossRef]
- Salloway, S.; Gur, T.; Berzin, T.; Zipser, B.; Correia, S.; Hovanesian, V.; Fallon, J.; Kuo-Leblanc, V.; Glass, D.; Hulette, C.; et al. Effect of APOE genotype on microvascular basement membrane in Alzheimer’s disease. J. Neurol. Sci. 2002, 203–204, 183–187. [Google Scholar] [CrossRef]
- Nishitsuji, K.; Hosono, T.; Nakamura, T.; Bu, G.; Michikawa, M. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J. Biol. Chem. 2011, 286, 17536–17542. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.R.; Rege, S.V.; Ma, Q.; Zhao, Z.; Miller, C.A.; Winkler, E.A.; Zlokovic, B.V. Accelerated pericyte degeneration and blood–brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2015, 36, 216–227. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Fullerton, S.M.; Shirman, G.A.; Strittmatter, W.J.; Matthew, W.D. Impairment of the blood–nerve and blood–brain barriers in apolipoprotein E knockout mice. Exp. Neurol. 2001, 169, 13–22. [Google Scholar] [CrossRef]
- Halliday, M.R.; Pomara, N.; Sagare, A.P.; Mack, W.J.; Frangione, B.; Zlokovic, B.V. Relationship between cyclophilin A levels and matrix metalloproteinase 9 activity in cerebrospinal fluid of cognitively normal apolipoprotein E4 carriers and blood-brain barrier breakdown. JAMA Neurol. 2013, 70, 1198–1200. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, M.M.; Van Nostrand, W.E.; Otte-Höller, I.; Wesseling, P.; De Waal, R.M.W. Amyloid-β-induced degeneration of human brain pericytes is dependent on the apolipoprotein E genotype. Ann. N. Y. Acad. Sci. 2000, 903, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Harik, S.I. Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J. Neurochem. 1989, 53, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Harik, S.I. Changes in the glucose transporter of brain capillaries. Can. J. Physiol. Pharmacol. 1992, 70, S113–S117. [Google Scholar] [CrossRef]
- Simpson, I.A.; Chundu, K.R.; Davies-Hill, T.; Honer, W.G.; Davies, P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann. Neurol. 1994, 35, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D.; Chung, H.C.; Shah, G.N. GLUT-1 Expression in the cerebra of patients with Alzheimer’s disease. Neurobiol. Aging 1997, 18, 469–474. [Google Scholar] [CrossRef]
- Horwood, N.; Davies, D.C. Immunolabelling of hippocampal microvessel glucose transporter protein is reduced in Alzheimer’s disease. Virchows Arch. 1994, 425, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Vogelsang, P.; Giil, L.M.; Lund, A.; Vedeler, C.A.; Parkar, A.P.; Nordrehaug, J.E.; Kristoffersen, E.K. Reduced glucose transporter-1 in brain derived circulating endothelial cells in mild Alzheimer’s disease patients. Brain Res. 2018, 1678, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Jagust, W.J.; Seab, J.P.; Huesman, R.H.; Valk, P.E.; Mathis, C.A.; Reed, B.R.; Coxson, P.G.; Budinger, T.F. Diminished glucose transport in Alzheimer’s disease: Dynamic PET studies. J. Cereb. Blood Flow Metab. 1991, 11, 323–330. [Google Scholar] [CrossRef]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521. [Google Scholar] [CrossRef]
- Nishida, Y.; Winkler, E.; Sagare, A.; De Vivo, D.; Zlokovic, B. Decreased glucose transporter 1 expression at the blood-brain barrier exacerbates Alzheimer disease-like phenotypes in mouse models. J. Neurol. Sci. 2017, 381, 768. [Google Scholar] [CrossRef]
- Zheng, P.-P.; Romme, E.; van der Spek, P.J.; Dirven, C.M.F.; Willemsen, R.; Kros, J.M. Glut1/SLC2A1 is crucial for the development of the blood-brain barrier in vivo. Ann. Neurol. 2010, 68, 835–844. [Google Scholar] [CrossRef]
- Seidner, G.; Alvarez, M.G.; Yeh, J.-I.; O’Driscoll, K.R.; Klepper, J.; Stump, T.S.; Wang, D.; Spinner, N.B.; Birnbaum, M.J.; De Vivo, D.C. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat. Genet. 1998, 18, 188–191. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C. Alzheimer’s disease and the amyloid cascade hypothesis: A critical review. Int. J. Alzheimers Dis. 2012, 2012, 369808. [Google Scholar] [CrossRef]
- Thomas, T.; Thomas, G.; McLendon, C.; Sutton, T.; Mullan, M. β-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature 1996, 380, 168–171. [Google Scholar] [CrossRef]
- Crawford, F.; Suo, Z.; Fang, C.; Mullan, M. Characteristics of the in vitro vasoactivity of β-amyloid peptides. Exp. Neurol. 1998, 150, 159–168. [Google Scholar] [CrossRef]
- Paris, D.; Town, T.; Mori, T.; Parker, T.A.; Humphrey, J.; Mullan, M. Soluble β-amyloid peptides mediate vasoactivity via activation of a pro-inflammatory pathway. Neurobiol. Aging 2000, 21, 183–197. [Google Scholar] [CrossRef]
- Paris, D.; Humphrey, J.; Quadros, A.; Patel, N.; Crescentini, R.; Crawford, F.; Mullan, M. Vasoactive effects of Aβ in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer’s disease: Role of inflammation. Neurol. Res. 2003, 25, 642–651. [Google Scholar] [CrossRef]
- Hald, E.S.; Timm, C.D.; Alford, P.W. Amyloid beta influences vascular smooth muscle contractility and mechanoadaptation. J. Biomech. Eng. 2016, 138, 111007. [Google Scholar] [CrossRef] [PubMed]
- Niwa, K.; Porter, V.A.; Kazama, K.; Cornfield, D.; Carlson, G.A.; Iadecola, C. A beta-peptides enhance vasoconstriction in cerebral circulation. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H2417–H2424. [Google Scholar] [CrossRef]
- Candela, P.; Saint-Pol, J.; Kuntz, M.; Boucau, M.-C.; Lamartiniere, Y.; Gosselet, F.; Fenart, L. In vitro discrimination of the role of LRP1 at the BBB cellular level: Focus on brain capillary endothelial cells and brain pericytes. Brain Res. 2015, 1594, 15–26. [Google Scholar] [CrossRef]
- Iadecola, C.; Zhang, F.; Niwa, K.; Eckman, C.; Turner, S.K.; Fischer, E.; Younkin, S.; Borchelt, D.R.; Hsiao, K.K.; Carlson, G.A. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat. Neurosci. 1999, 2, 157. [Google Scholar] [CrossRef]
- Niwa, K.; Kazama, K.; Younkin, L.; Younkin, S.G.; Carlson, G.A.; Iadecola, C. Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H315–H323. [Google Scholar] [CrossRef]
- Niwa, K.; Younkin, L.; Ebeling, C.; Turner, S.K.; Westaway, D.; Younkin, S.; Ashe, K.H.; Carlson, G.A.; Iadecola, C. Aβ1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc. Natl. Acad. Sci. USA 2000, 97, 9735–9740. [Google Scholar] [CrossRef]
- Zlokovic, B.V.; Yamada, S.; Holtzman, D.; Ghiso, J.; Frangione, B. Clearance of amyloid β-peptide from brain: Transport or metabolism? Nat. Med. 2000, 6, 718–719. [Google Scholar] [CrossRef]
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood–brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid β-peptide interaction mediates differential brain efflux of aβ isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, L.B.; Dohgu, S.; Hwang, M.C.; Farr, S.A.; Murphy, M.P.; Fleegal-DeMotta, M.A.; Lynch, J.L.; Robinson, S.M.; Niehoff, M.L.; Johnson, S.N.; et al. Testing the neurovascular hypothesis of Alzheimer’s disease: LRP-1 antisense reduces blood-brain barrier clearance, increases brain levels of amyloid-beta protein and impairs cognition. J. Alzheimers Dis. 2009, 17, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Sagare, A.P.; Ma, Q.; Halliday, M.R.; Kong, P.; Kisler, K.; Winkler, E.A.; Ramanathan, A.; Kanekiyo, T.; Bu, G.; et al. Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nat. Neurosci. 2015, 18, 978–987. [Google Scholar] [CrossRef]
- Quinn, K.A.; Grimsley, P.G.; Dai, Y.-P.; Tapner, M.; Chesterman, C.N.; Owensby, D.A. Soluble low density lipoprotein receptor-related protein (LRP) circulates in human plasma. J. Biol. Chem. 1997, 272, 23946–23951. [Google Scholar] [CrossRef] [PubMed]
- Sagare, A.; Deane, R.; Bell, R.D.; Johnson, B.; Hamm, K.; Pendu, R.; Marky, A.; Lenting, P.J.; Wu, Z.; Zarcone, T.; et al. Clearance of amyloid-β by circulating lipoprotein receptors. Nat. Med. 2007, 13, 1029. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Mackic, J.B.; Stins, M.; McComb, J.G.; Calero, M.; Ghiso, J.; Kim, K.S.; Yan, S.D.; Stern, D.; Schmidt, A.M.; Frangione, B.; et al. Human blood-brain barrier receptors for Alzheimer’s amyloid-beta 1-40. Asymmetrical binding, endocytosis and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J. Clin. Investig. 1998, 102, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907. [Google Scholar] [CrossRef]
- Donahue, J.E.; Flaherty, S.L.; Johanson, C.E.; Duncan, J.A.; Silverberg, G.D.; Miller, M.C.; Tavares, R.; Yang, W.; Wu, Q.; Sabo, E.; et al. RAGE, LRP-1 and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 2006, 112, 405–415. [Google Scholar] [CrossRef]
- Urmoneit, B.; Prikulis, I.; Wihl, G.; D’Urso, D.; Frank, R.; Heeren, J.; Beisiegel, U.; Prior, R. Cerebrovascular smooth muscle cells internalize Alzheimer amyloid beta protein via a lipoprotein pathway: Implications for cerebral amyloid angiopathy. Lab. Investig. J. Tech. Methods Pathol. 1997, 77, 157–166. [Google Scholar]
- Kanekiyo, T.; Liu, C.-C.; Shinohara, M.; Li, J.; Bu, G. LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer’s amyloid-β. J. Neurosci. 2012, 32, 16458–16465. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, M.; Robitaille, Y.; Boileau, G.; DesGroseillers, L.; Marcinkiewicz, M. Declining expression of neprilysin in Alzheimer disease vasculature: Possible involvement in cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 2002, 61, 849–856. [Google Scholar] [CrossRef]
- Ellis, R.J.; Olichney, J.M.; Thal, L.J.; Mirra, S.S.; Morris, J.C.; Beekly, D.; Heyman, A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: The CERAD experience, Part XV. Neurology 1996, 46, 1592–1596. [Google Scholar] [CrossRef]
- Olichney, J.M.; Hansen, L.A.; Hofstetter, C.R.; Grundman, M.; Katzman, R.; Thal, L.J. Cerebral infarction in Alzheimer’s disease is associated with severe amyloid angiopathy and hypertension. Arch. Neurol. 1995, 52, 702–708. [Google Scholar] [CrossRef]
- Ferreiro, J.A.; Ansbacher, L.E.; Vinters, H.V. Stroke related to cerebral amyloid angiopathy: The significance of systemic vascular disease. J. Neurol. 1989, 236, 267–272. [Google Scholar] [CrossRef]
- Weller, R.O.; Subash, M.; Preston, S.D.; Mazanti, I.; Carare, R.O. Perivascular drainage of amyloid-β peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 2008, 18, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Mok, S.S.; Turner, B.J.; Beyreuther, K.; Masters, C.L.; Barrow, C.J.; Small, D.H. Toxicity of substrate-bound amyloid peptides on vascular smooth muscle cells is enhanced by homocysteine. Eur. J. Biochem. 2002, 269, 3014–3022. [Google Scholar] [CrossRef]
- Kalaria, R.N. Cerebrovascular degeneration is related to amyloid-beta protein deposition in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1997, 826, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Weller, R.O.; Massey, A.; Newman, T.A.; Hutchings, M.; Kuo, Y.M.; Roher, A.E. Cerebral amyloid angiopathy: Amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am. J. Pathol. 1998, 153, 725–733. [Google Scholar] [CrossRef]
- Davis, J.; Xu, F.; Deane, R.; Romanov, G.; Previti, M.L.; Zeigler, K.; Zlokovic, B.V.; Van Nostrand, W.E. Early-onset and robust cerebral microvascular accumulation of amyloid β-protein in transgenic mice expressing low levels of a vasculotropic dutch/iowa mutant form of amyloid β-protein precursor. J. Biol. Chem. 2004, 279, 20296–20306. [Google Scholar] [CrossRef]
- Chow, N.; Bell, R.D.; Deane, R.; Streb, J.W.; Chen, J.; Brooks, A.; Van Nostrand, W.; Miano, J.M.; Zlokovic, B.V. Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer’s phenotype. Proc. Natl. Acad. Sci. USA 2007, 104, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Deane, R.; Chow, N.; Long, X.; Sagare, A.; Singh, I.; Streb, J.W.; Guo, H.; Rubio, A.; Van Nostrand, W.; et al. SRF and myocardin regulate LRP-mediated amyloid-β clearance in brain vascular cells. Nat. Cell Biol. 2008, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmus, M.M.; Otte-Höller, I.; van Triel, J.J.; Veerhuis, R.; Maat-Schieman, M.L.C.; Bu, G.; de Waal, R.M.W.; Verbeek, M.M. Lipoprotein receptor-related protein-1 mediates amyloid-β-mediated cell death of cerebrovascular cells. Am. J. Pathol. 2007, 171, 1989–1999. [Google Scholar] [CrossRef]
- Mazur-Kolecka, B.; Frackowiak, J.; Wiśniewski, H.M. Apolipoproteins E3 and E4 induce and transthyretin prevents accumulation of the Alzheimer’s β-amyloid peptide in cultured vascular smooth muscle cells. Brain Res. 1995, 698, 217–222. [Google Scholar] [CrossRef]
- Miners, J.S.; Van Helmond, Z.; Chalmers, K.; Wilcock, G.; Love, S.; Kehoe, P.G. Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 2006, 65, 1012–1021. [Google Scholar] [CrossRef]
- Miners, J.S.; Kehoe, P.; Love, S. Neprilysin protects against cerebral amyloid angiopathy and Aβ-induced degeneration of cerebrovascular smooth muscle cells. Brain Pathol. 2011, 21, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Ruzali, W.A.; Kehoe, P.G.; Love, S. Influence of LRP-1 and apolipoprotein E on amyloid-β uptake and toxicity to cerebrovascular smooth muscle cells. J. Alzheimers Dis. 2013, 33, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. ApoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Investig. 2008, 118, 4002–4013. [Google Scholar] [CrossRef]
- Yang, D.S.; Small, D.H.; Seydel, U.; Smith, J.D.; Hallmayer, J.; Gandy, S.E.; Martins, R.N. Apolipoprotein E promotes the binding and uptake of β-amyloid into Chinese hamster ovary cells in an isoform-specific manner. Neuroscience 1999, 90, 1217–1226. [Google Scholar] [CrossRef]
- Tokuda, T.; Calero, M.; Matsubara, E.; Vidal, R.; Kumar, A.; Permanne, B.; Zlokovic, B.; Smith, J.D.; Ladu, M.J.; Rostagno, A.; et al. Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer’s amyloid β peptides. Biochem. J. 2000, 348, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.; Kouiavskaia, D.; Migliorini, M.; Robinson, S.; Saenko, E.L.; Gorlatova, N.; Li, D.; Lawrence, D.; Hyman, B.T.; Weisgraber, K.H.; et al. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J. Lipid Res. 2005, 46, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Dhanasekaran, P.; Baldwin, F.; Weisgraber, K.H.; Lund-Katz, S.; Phillips, M.C. Lipid binding-induced conformational change in human apolipoprotein E: Evidence for two lipid-bound states on spherical particles. J. Biol. Chem. 2001, 276, 40949–40954. [Google Scholar] [CrossRef] [PubMed]
- Tamamizu-Kato, S.; Cohen, J.K.; Drake, C.B.; Kosaraju, M.G.; Drury, J.; Narayanaswami, V. Interaction with amyloid β peptide compromises the lipid binding function of apolipoprotein E. Biochemistry 2008, 47, 5225–5234. [Google Scholar] [CrossRef]
- Olichney, J.M.; Hansen, L.A.; Hofstetter, C.R.; Lee, J.-H.; Katzman, R.; Thal, L.J. Association between severe cerebral amyloid angiopathy and cerebrovascular lesions in Alzheimer disease is not a spurious one attributable to apolipoprotein E4. Arch. Neurol. 2000, 57, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Ghebremedhin, E.; Rub, U.; Yamaguchi, H.; Del Tredici, K.; Braak, H. Two types of sporadic cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 2002, 61, 282–293. [Google Scholar] [CrossRef]
- Thal, D.R.; Papassotiropoulos, A.; Saido, T.C.; Griffin, W.S.T.; Mrak, R.E.; Kölsch, H.; Tredici, K.D.; Attems, J.; Ghebremedhin, E. Capillary cerebral amyloid angiopathy identifies a distinct APOE ε4-associated subtype of sporadic Alzheimer’s disease. Acta Neuropathol. 2010, 120, 169–183. [Google Scholar] [CrossRef]
- Hultman, K.; Strickland, S.; Norris, E.H. The APOE ε4/ε4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 2013, 33, 1251–1258. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Paul, J.; Norris, E.H.; Bronstein, R.; Ahn, H.J.; Zamolodchikov, D.; Bhuvanendran, S.; Fenz, K.M.; Strickland, S. Fibrinogen and β-amyloid association alters thrombosis and fibrinolysis: A possible contributing factor to Alzheimer’s disease. Neuron 2010, 66, 695–709. [Google Scholar] [CrossRef]
- Ahn, H.J.; Zamolodchikov, D.; Cortes-Canteli, M.; Norris, E.H.; Glickman, J.F.; Strickland, S. Alzheimer’s disease peptide β-amyloid interacts with fibrinogen and induces its oligomerization. Proc. Natl. Acad. Sci. USA 2010, 107, 21812–21817. [Google Scholar] [CrossRef]
- Zamolodchikov, D.; Strickland, S. Aβ delays fibrin clot lysis by altering fibrin structure and attenuating plasminogen binding to fibrin. Blood 2012, 119, 3342–3351. [Google Scholar] [CrossRef]
- van Oijen, M.; Witteman, J.C.; Hofman, A.; Koudstaal, P.J.; Breteler, M.M. Fibrinogen is associated with an increased risk of Alzheimer disease and vascular dementia. Stroke 2005, 36, 2637–2641. [Google Scholar] [CrossRef] [PubMed]
- Davis-Salinas, J.; Saporito-Irwin, S.M.; Cotman, C.W.; Van Nostrand, W.E. Amyloid β-protein induces its own production in cultured degenerating cerebrovascular smooth muscle cells. J. Neurochem. 1995, 65, 931–934. [Google Scholar] [CrossRef]
- Subasinghe, S.; Unabia, S.; Barrow, C.J.; Mok, S.S.; Aguilar, M.-I.; Small, D.H. Cholesterol is necessary both for the toxic effect of Aβ peptides on vascular smooth muscle cells and for Aβ binding to vascular smooth muscle cell membranes. J. Neurochem. 2003, 84, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Davis-Salinas, J.; Van Nostrand, W.E. Amyloid β-protein aggregation nullifies its pathologic properties in cultured cerebrovascular smooth muscle cells. J. Biol. Chem. 1995, 270, 20887–20890. [Google Scholar] [CrossRef]
- Delacourte, A.; Defossez, A.; Persuy, P.; Peers, M.C. Observation of morphological relationships between angiopathic blood vessels and degenerative neurites in Alzheimer’s disease. Virchows Arch. A 1987, 411, 199–204. [Google Scholar] [CrossRef]
- Miyakawa, T.; Katsuragi, S.; Watanabe, K.; Shimoji, A.; Ikeuchi, Y. Ultrastructural studies of amyloid fibrils and senile plaques in human brain. Acta Neuropathol 1986, 70, 202–208. [Google Scholar] [CrossRef]
- Miyakawa, T.; Shimoji, A.; Kuramoto, R.; Higuchi, Y. The relationship between senile plaques and cerebral blood vessels in Alzheimer’s disease and senile dementia. Morphological mechanism of senile plaque production. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1982, 40, 121–129. [Google Scholar] [CrossRef]
- Xu, F.; Fu, Z.; Dass, S.; Kotarba, A.E.; Davis, J.; Smith, S.O.; Van Nostrand, W.E. Cerebral vascular amyloid seeds drive amyloid β-protein fibril assembly with a distinct anti-parallel structure. Nat. Commun. 2016, 7, 13527. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann. Neurol. 2011, 70, 532–540. [Google Scholar] [CrossRef]
- Ujiie, M.; Dickstein, D.L.; Carlow, D.A.; Jefferies, W.A. Blood–brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation 2003, 10, 463–470. [Google Scholar] [CrossRef]
- Dickstein, D.L.; Biron, K.E.; Ujiie, M.; Pfeifer, C.G.; Jeffries, A.R.; Jefferies, W.A. Aβ peptide immunization restores blood-brain barrier integrity in Alzheimer disease. FASEB J. 2006, 20, 426–433. [Google Scholar] [CrossRef]
- Biron, K.E.; Dickstein, D.L.; Gopaul, R.; Fenninger, F.; Jefferies, W.A. Cessation of neoangiogenesis in Alzheimer’s disease follows amyloid-beta immunization. Sci. Rep. 2013, 3, 1354. [Google Scholar] [CrossRef]
- Menendez-Gonzalez, M.; Perez-Pinera, P.; Martinez-Rivera, M.; Lopez Muniz, A.; Vega, J.A. Immunotherapy for Alzheimer’s disease: Rational basis in ongoing clinical trials. Curr. Pharm. Des. 2011, 17, 508–520. [Google Scholar] [CrossRef]
- Paris, D.; Townsend, K.; Quadros, A.; Humphrey, J.; Sun, J.; Brem, S.; Wotoczek-Obadia, M.; DelleDonne, A.; Patel, N.; Obregon, D.F.; et al. Inhibition of angiogenesis by Aβ peptides. Angiogenesis 2004, 7, 75–85. [Google Scholar] [CrossRef]
- Solito, R.; Corti, F.; Fossati, S.; Mezhericher, E.; Donnini, S.; Ghiso, J.; Giachetti, A.; Rostagno, A.; Ziche, M. Dutch and arctic mutant peptides of β amyloid1–40 differentially affect the FGF-2 pathway in brain endothelium. Exp. Cell Res. 2009, 315, 385–395. [Google Scholar] [CrossRef]
- Donnini, S.; Cantara, S.; Morbidelli, L.; Giachetti, A.; Ziche, M. FGF-2 overexpression opposes the beta amyloid toxic injuries to the vascular endothelium. Cell Death Differ. 2006, 13, 1088. [Google Scholar] [CrossRef][Green Version]
- Gonzalez-Velasquez, F.J.; Moss, M.A. Soluble aggregates of the amyloid-β protein activate endothelial monolayers for adhesion and subsequent transmigration of monocyte cells. J. Neurochem. 2008, 104, 500–513. [Google Scholar] [CrossRef]
- Blanc, E.M.; Toborek, M.; Mark, R.J.; Hennig, B.; Mattson, M.P. Amyloid β-peptide induces cell monolayer albumin permeability, impairs glucose transport and induces apoptosis in vascular endothelial cells. J. Neurochem. 1997, 68, 1870–1881. [Google Scholar] [CrossRef]
- Fossati, S.; Cam, J.; Meyerson, J.; Mezhericher, E.; Romero, I.A.; Couraud, P.O.; Weksler, B.B.; Ghiso, J.; Rostagno, A. Differential activation of mitochondrial apoptotic pathways by vasculotropic amyloid-β variants in cells composing the cerebral vessel walls. FASEB J. 2009, 24, 229–241. [Google Scholar] [CrossRef]
- Gorevic, P.; Ghersi-Egea, J.-F.; Strazielle, N.; Dehouck, M.-P.; Frangione, B.; Ghiso, J.; Patlak, C.; Fenstermacher, J. In vitro evidence that β-amyloid peptide 1–40 diffuses across the blood–brain barrier and affects its permeability. J. Neuropathol. Exp. Neurol. 2000, 59, 29–38. [Google Scholar] [CrossRef]
- Uchihara, T.; Akiyama, H.; Kondo, H.; Ikeda, K. Activated microglial cells are colocalized with perivascular deposits of amyloid-β protein in Alzheimer’s disease brain. Stroke 1997, 28, 1948–1950. [Google Scholar] [CrossRef]
- Giri, R.; Shen, Y.; Stins, M.; Du Yan, S.; Schmidt, A.M.; Stern, D.; Kim, K.-S.; Zlokovic, B.; Kalra, V.K. β-Amyloid-induced migration of monocytes across human brain endothelial cells involves RAGE and PECAM-1. Am. J. Physiol. Cell Physiol. 2000, 279, C1772–C1781. [Google Scholar] [CrossRef]
- Castillo-Carranza, D.L.; Nilson, A.N.; Van Skike, C.E.; Jahrling, J.B.; Patel, K.; Garach, P.; Gerson, J.E.; Sengupta, U.; Abisambra, J.; Nelson, P.; et al. cerebral microvascular accumulation of tau oligomers in Alzheimer’s disease and related tauopathies. Aging Dis. 2017, 8, 257–266. [Google Scholar] [CrossRef]
- Williams, S.; Chalmers, K.; Wilcock, G.K.; Love, S. Relationship of neurofibrillary pathology to cerebral amyloid angiopathy in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2005, 31, 414–421. [Google Scholar] [CrossRef]
- Thaker, U.; McDonagh, A.M.; Iwatsubo, T.; Lendon, C.L.; Pickering-Brown, S.M.; Mann, D.M.A. Tau load is associated with apolipoprotein E genotype and the amount of amyloid β protein, Aβ40, in sporadic and familial Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2003, 29, 35–44. [Google Scholar] [CrossRef]
- Shafiei, S.S.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. Tau oligomers: Cytotoxicity, propagation and mitochondrial damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.E.; Robbins, A.B.; Hu, M.; Cao, X.; Betensky, R.A.; Clark, T.; Das, S.; Hyman, B.T. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1289–E1298. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease—A brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed]
- Gasic-Milenkovic, J.; Dukic-Stefanovic, S.; Deuther-Conrad, W.; Gartner, U.; Munch, G. Beta-amyloid peptide potentiates inflammatory responses induced by lipopolysaccharide, interferon-γ and ‘advanced glycation endproducts’ in a murine microglia cell line. Eur. J. Neurosci. 2003, 17, 813–821. [Google Scholar] [CrossRef]
- Manocha, G.D.; Floden, A.M.; Rausch, K.; Kulas, J.A.; McGregor, B.A.; Rojanathammanee, L.; Puig, K.R.; Puig, K.L.; Karki, S.; Nichols, M.R.; et al. APP regulates microglial phenotype in a mouse model of Alzheimer’s disease. J. Neurosci. 2016, 36, 8471–8486. [Google Scholar] [CrossRef]
- Currais, A.; Quehenberger, O.; Armando, A.M.; Daugherty, D.; Maher, P.; Schubert, D. Amyloid proteotoxicity initiates an inflammatory response blocked by cannabinoids. NPJ Aging Mech. Dis. 2016, 2, 16012. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, A.; Zaheer, S.; Thangavel, R.; Wu, Y.; Sahu, S.K.; Yang, B. Glia maturation factor modulates β-amyloid-induced glial activation, inflammatory cytokine/chemokine production and neuronal damage. Brain Res. 2008, 1208, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Ovase, R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 837–842. [Google Scholar] [CrossRef]
- Tripathy, D.; Thirumangalakudi, L.; Grammas, P. Expression of macrophage inflammatory protein 1-α is elevated in Alzheimer’s vessels and is regulated by oxidative stress. J. Alzheimers Dis. 2007, 11, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Ovase, R. Cerebrovascular transforming growth factor-β contributes to inflammation in the Alzheimer’s disease brain. Am. J. Pathol. 2002, 160, 1583–1587. [Google Scholar] [CrossRef]
- Goumans, M.-J.; Liu, Z.; ten Dijke, P. TGF-β signaling in vascular biology and dysfunction. Cell Res. 2008, 19, 116. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Masliah, E.; Mallory, M.; McConlogue, L.; Johnson-Wood, K.; Lin, C.; Mucke, L. Amyloidogenic role of cytokine TGF-β1 in transgenic mice and in Alzheimer’s disease. Nature 1997, 389, 603–606. [Google Scholar] [CrossRef]
- Frautschy, S.A.; Yang, F.; Calderón, L.; Cole, G.M. Rodent models of Alzheimer’s disease: Rat aβ infusion approaches to amyloid deposits. Neurobiol. Aging 1996, 17, 311–321. [Google Scholar] [CrossRef]
- Lesné, S.; Docagne, F.; Gabriel, C.l.; Liot, G.; Lahiri, D.K.; Buée, L.; Plawinski, L.; Delacourte, A.; MacKenzie, E.T.; Buisson, A.; et al. Transforming growth factor-β1 potentiates amyloid-β generation in astrocytes and in transgenic mice. J. Biol. Chem. 2003, 278, 18408–18418. [Google Scholar] [CrossRef]
- Gray, C.W.; Patel, A.J. Regulation of β-amyloid precursor protein isoform mRNAs by transforming growth factor-β1 and interleukin-1β in astrocytes. Mol. Brain Res. 1993, 19, 251–256. [Google Scholar] [CrossRef]
- Amara, F.M.; Junaid, A.; Clough, R.R.; Liang, B. TGF-β1, regulation of Alzheimer amyloid precursor protein mRNA expression in a normal human astrocyte cell line: mRNA stabilization. Mol. Brain Res. 1999, 71, 42–49. [Google Scholar] [CrossRef]
- Ignotz, R.A.; Massague, J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986, 261, 4337–4345. [Google Scholar]
- Smyth, L.C.D.; Rustenhoven, J.; Park, T.I.H.; Schweder, P.; Jansson, D.; Heppner, P.A.; O’Carroll, S.J.; Mee, E.W.; Faull, R.L.M.; Curtis, M.; et al. Unique and shared inflammatory profiles of human brain endothelia and pericytes. J. Neuroinflamm. 2018, 15, 138. [Google Scholar] [CrossRef]
- Forloni, G.; Demicheli, F.; Giorgi, S.; Bendotti, C.; Angeretti, N. Expression of amyloid precursor protein mRNAs in endothelial, neuronal and glial cells: Modulation by interleukin-1. Mol. Brain Res. 1992, 16, 128–134. [Google Scholar] [CrossRef]
- Goldgaber, D.; Harris, H.W.; Hla, T.; Maciag, T.; Donnelly, R.J.; Jacobsen, J.S.; Vitek, M.P.; Gajdusek, D.C. Interleukin 1 regulates synthesis of amyloid β-protein precursor mRNA in human endothelial cells. Proc. Natl. Acad. Sci. USA 1989, 86, 7606–7610. [Google Scholar] [CrossRef] [PubMed]
- Dobbie, M.S.; Hurst, R.D.; Klein, N.J.; Surtees, R.A.H. Upregulation of intercellular adhesion molecule-1 expression on human endothelial cells by tumour necrosis factor-α in an in vitro model of the blood–brain barrier. Brain Res. 1999, 830, 330–336. [Google Scholar] [CrossRef]
- De Cesaris, P.; Starace, D.; Starace, G.; Filippini, A.; Stefanini, M.; Ziparo, E. Activation of Jun N-terminal kinase/stress-activated protein kinase pathway by tumor necrosis factor α leads to intercellular adhesion molecule-1 expression. J. Biol. Chem. 1999, 274, 28978–28982. [Google Scholar] [CrossRef]
- Curtis, T.M.; Rotundo, R.F.; Vincent, P.A.; McKeown-Longo, P.J.; Saba, T.M. TNF-alpha-induced matrix Fn disruption and decreased endothelial integrity are independent of Fn proteolysis. Am. J. Physiol. 1998, 275, L126–L138. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Anne Pereira, H.; Kumar, P.; Grammas, P. Expression of CAP37, a novel inflammatory mediator, in Alzheimer’s disease. Neurobiol. Aging 1996, 17, 753–759. [Google Scholar] [CrossRef]
- Smith, C.W.; Marlin, S.D.; Rothlein, R.; Toman, C.; Anderson, D.C. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J. Clin. Investig. 1989, 83, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- Long, E.O. ICAM-1: Getting a grip on leukocyte adhesion. J. Immunol. 2011, 186, 5021–5023. [Google Scholar] [CrossRef]
- Kallmann, B.A.; Hummel, V.; Lindenlaub, T.; Ruprecht, K.; Toyka, K.V.; Rieckmann, P. Cytokine-induced modulation of cellular adhesion to human cerebral endothelial cells is mediated by soluble vascular cell adhesion molecule-1. Brain 2000, 123, 687–697. [Google Scholar] [CrossRef]
- Modur, V.; Zimmerman, G.A.; Prescott, S.M.; McIntyre, T.M. Endothelial cell inflammatory responses to tumor necrosis factor α: Ceramide-dependent and -independent mitogen-activated protein kinase cascades. J. Biol. Chem. 1996, 271, 13094–13102. [Google Scholar] [CrossRef]
- Miho, N.; Ishida, T.; Kuwaba, N.; Ishida, M.; Shimote-Abe, K.; Tabuchi, K.; Oshima, T.; Yoshizumi, M.; Chayama, K. Role of the JNK pathway in thrombin-induced ICAM-1 expression in endothelial cells. Cardiovasc. Res. 2005, 68, 289–298. [Google Scholar] [CrossRef]
- Alabanza, L.M.; Bynoe, M.S. Thrombin induces an inflammatory phenotype in a human brain endothelial cell line. J. Neuroimmunol. 2012, 245, 48–55. [Google Scholar] [CrossRef][Green Version]
- Tripathy, D.; Sanchez, A.; Yin, X.; Luo, J.; Martinez, J.; Grammas, P. Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia. Front. Aging Neurosci. 2013, 5, 19. [Google Scholar] [CrossRef]
- Crawford, A.; Angelosanto, J.M.; Nadwodny, K.L.; Blackburn, S.D.; Wherry, E.J. A Role for the chemokine RANTES in cegulating CD8 T cell responses during chronic viral infection. PLoS Pathog. 2011, 7, e1002098. [Google Scholar] [CrossRef]
- Tripathy, D.; Thirumangalakudi, L.; Grammas, P. RANTES upregulation in the Alzheimer’s disease brain: A possible neuroprotective role. Neurobiol. Aging 2010, 31, 8–16. [Google Scholar] [CrossRef]
- Suo, Z.; Tan, J.; Placzek, A.; Crawford, F.; Fang, C.; Mullan, M. Alzheimer’s β-amyloid peptides induce inflammatory cascade in human vascular cells: The roles of cytokines and CD40. Brain Res. 1998, 807, 110–117. [Google Scholar] [CrossRef]
- Vukic, V.; Callaghan, D.; Walker, D.; Lue, L.-F.; Liu, Q.Y.; Couraud, P.-O.; Romero, I.A.; Weksler, B.; Stanimirovic, D.B.; Zhang, W. Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol. Dis. 2009, 34, 95–106. [Google Scholar] [CrossRef]
- Callaghan, D.; Vukic, V.; Jones, A.; Walker, D.; Lue, L.; Woulfe, J.; Beach, T.; Sue, L.; Stanimirovic, D.; Zhang, W. Expression and regulation of inflammatory genes in human cerebrovascular endothelial cells induced by Aβ peptides. Alzheimers Dis. New Adv. Bologna Italy 2007, 83–87. [Google Scholar]
- Yamada, M.; Itoh, Y.; Shintaku, M.; Kawamura, J.; Jensson, O.; Thorsteinsson, L.; Suematsu, N.; Matsushita, M.; Otomo, E. Immune reactions associated with cerebral amyloid angiopathy. Stroke 1996, 27, 1155–1162. [Google Scholar] [CrossRef]
- Koeppen, M.; Eckle, T.; Eltzschig, H.K. The hypoxia-inflammation link and potential drug targets. Curr. Opin. Anaesthesiol. 2011, 24, 363–369. [Google Scholar] [CrossRef]
- Volk, T.; Hensel, M.; Schuster, H.; Kox, W.J. Secretion of MCP-1 and IL-6 by cytokine stimulated production of reactive oxygen species in endothelial cells. Mol. Cell. Biochem. 2000, 206, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Jansson, D.; Rustenhoven, J.; Feng, S.; Hurley, D.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; Faull, R.L.M.; Dragunow, M. A role for human brain pericytes in neuroinflammation. J. Neuroinflamm. 2014, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Guijarro-Muñoz, I.; Compte, M.; Álvarez-Cienfuegos, A.; Álvarez-Vallina, L.; Sanz, L. Lipopolysaccharide activates toll-like receptor 4 (TLR4)-mediated NF-κB signaling pathway and proinflammatory response in human pericytes. J. Biol. Chem. 2014, 289, 2457–2468. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.; Eckart, A.; Haidari, S.; Tirniceriu, A.; Lorenz, M.; von Brühl, M.-L.; Gärtner, F.; Khandoga, A.G.; Legate, K.R.; Pless, R.; et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and & ‘instruct’ them with pattern-recognition and motility programs. Nat. Immunol. 2012, 14, 41. [Google Scholar] [CrossRef]
- Pieper, C.; Pieloch, P.; Galla, H.-J. Pericytes support neutrophil transmigration via interleukin-8 across a porcine co-culture model of the blood–brain barrier. Brain Res. 2013, 1524, 1–11. [Google Scholar] [CrossRef]
- Proebstl, D.; Voisin, M.-B.; Woodfin, A.; Whiteford, J.; D’Acquisto, F.; Jones, G.E.; Rowe, D.; Nourshargh, S. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J. Exp. Med. 2012, 209, 1219. [Google Scholar] [CrossRef] [PubMed]
- Ayres-Sander, C.E.; Lauridsen, H.; Maier, C.L.; Sava, P.; Pober, J.S.; Gonzalez, A.L. Transendothelial migration enables subsequent transmigration of neutrophils through underlying pericytes. PLoS ONE 2013, 8, e60025. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, J.; Takata, F.; Machida, T.; Takahashi, H.; Soejima, Y.; Funakoshi, M.; Futagami, K.; Yamauchi, A.; Dohgu, S.; Kataoka, Y. Tumor necrosis factor-α-stimulated brain pericytes possess a unique cytokine and chemokine release profile and enhance microglial activation. Neurosci. Lett. 2014, 578, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Balabanov, R.; Beaumont, T.; Dore-Duffy, P. Role of central nervous system microvascular pericytes in activation of antigen-primed splenic T-lymphocytes. J. Neurosci. Res. 1999, 55, 578–587. [Google Scholar] [CrossRef]
- Verbeek, M.M.; Westphal, J.R.; Ruiter, D.J.; de Waal, R.M. T lymphocyte adhesion to human brain pericytes is mediated via very late antigen-4/vascular cell adhesion molecule-1 interactions. J. Immunol. 1995, 154, 5876. [Google Scholar] [PubMed]
- Pieper, C.; Marek, J.J.; Unterberg, M.; Schwerdtle, T.; Galla, H.-J. Brain capillary pericytes contribute to the immune defense in response to cytokines or LPS in vitro. Brain Res. 2014, 1550, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rozzini, L.; Chilovi, B.V.; Bertoletti, E.; Conti, M.; Del Rio, I.; Trabucchi, M.; Padovani, A. Angiotensin converting enzyme (ACE) inhibitors modulate the rate of progression of amnestic mild cognitive impairment. Int. J. Geriatr. Psychiatry 2006, 21, 550–555. [Google Scholar] [CrossRef]
- Launer, L.J. Demonstrating the case that AD is a vascular disease: Epidemiologic evidence. Ageing Res. Rev. 2002, 1, 61–77. [Google Scholar] [CrossRef]
- Skoog, I.; Andreasson, L.A.; Landahl, S.; Lernfelt, B. A population-based study on blood pressure and brain atrophy in 85-year-olds. Hypertension 1998, 32, 404–409. [Google Scholar] [CrossRef]
- Saavedra, J. Evidence to consider angiotensin ii receptor blockers for the treatment of early Alzheimer’s disease. Cell. Mol. Neurobiol. 2016, 36, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, P.G.; Davies, N.M.; Martin, R.M.; Ben-Shlomo, Y. Associations of angiotensin targeting antihypertensive drugs with mortality and hospitalization in primary care patients with dementia. J. Alzheimers Dis. 2013, 33, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Stampfer, M.J. Cardiovascular disease and Alzheimer’s disease: Common links. J. Intern. Med. 2006, 260, 211–223. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Reitz, C.; Honig, L.S.; Tang, M.X.; Shea, S.; Mayeux, R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology 2005, 65, 545. [Google Scholar] [CrossRef]
- Whitmer, R.A.; Sidney, S.; Selby, J.; Johnston, S.C.; Yaffe, K. Midlife cardiovascular risk factors and risk of dementia in late life. Neurology 2005, 64, 277. [Google Scholar] [CrossRef]
- Purnell, C.; Gao, S.; Callahan, C.M.; Hendrie, H.C. Cardiovascular risk factors and incident Alzheimer disease: A systematic review of the literature. Alzheimer Dis. Assoc. Disord. 2009, 23, 1–10. [Google Scholar] [CrossRef]
- Mills, K.; Bundy, J.; Kelly, T.; Reed, J.; Kearney, P.; Reynolds, K.; Chen, J.; He, J. Global disparities of hypertension prevalence and control: A systematic analysis of population-based studies from 90 countries. Circulation 2016, 134, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Gabin, J.M.; Tambs, K.; Saltvedt, I.; Sund, E.; Holmen, J. Association between blood pressure and Alzheimer disease measured up to 27 years prior to diagnosis: The HUNT Study. Alzheimers Res. Ther. 2017, 31, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Shi, J.; Zhang, Y.; Wang, B.; Liu, W.; Chen, Z.; Tong, Q. Central angiotensin II stimulation promotes β amyloid production in Sprague Dawley rats. PLoS ONE 2011, 6, e16037. [Google Scholar] [CrossRef] [PubMed]
- Estato, V.; Nascimento, A.; Antunes, B.; Gomes, F.; Coelho, L.; Rangel, R.; Garzoni, L.; Daliry, A.; Bousquet, P.; Tibiriçá, E. Cerebral microvascular dysfunction and inflammation are improved by centrally acting antihypertensive drugs in metabolic syndrome. Metab. Syndr. Relat. Disord. 2017, 15, 26–35. [Google Scholar] [CrossRef]
- Rosenbaum, D.; Mattina, A.; Koch, E.; Rossant, F.; Gallo, A.; Kachenoura, N.; Paques, M.; Redheuil, A.; Girerd, X. Effects of age, blood pressure and antihypertensive treatments on retinal arterioles remodeling assessed by adaptive optics. J. Hypertens. 2016, 34, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, I.; Brown, L.; Mack, W.J.; Chui, H. Impact of angiotensin receptor blockers on Alzheimer disease neuropathology in a large brain autopsy series. Arch. Neurol. 2012, 69, 1632–1638. [Google Scholar] [CrossRef]
- Bosch, J.; O’Donnell, M.; Swaminathan, B.; Lonn, E.M.; Sharma, M.; Dagenais, G.; Diaz, R.; Khunti, K.; Lewis, B.S.; Avezum, A.; et al. Effects of blood pressure and lipid lowering on cognition: Results from the HOPE-3 study. Neurology 2019, 92, e1435–e1446. [Google Scholar] [CrossRef] [PubMed]
- O’Caoimh, R.; Healy, L.; Gao, Y.; Svendrovski, A.; Kerins, D.M.; Eustace, J.; Kehoe, P.G.; Guyatt, G.; Molloy, D.W. Effects of centrally acting angiotensin converting enzyme inhibitors on functional decline in patients with Alzheimer’s disease. J. Alzheimers Dis. 2014, 40, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Fazal, K.; Perera, G.; Khondoker, M.; Howard, R.; Stewart, R. Associations of centrally acting ACE inhibitors with cognitive decline and survival in Alzheimer’s disease. BJPsych Open 2017, 3, 158–164. [Google Scholar] [CrossRef]
- Yarchoan, M.; Xie, S.X.; Kling, M.A.; Toledo, J.B.; Wolk, D.A.; Lee, E.B.; Van Deerlin, V.; Lee, V.M.Y.; Trojanowski, J.Q.; Arnold, S.E. Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain 2012, 135, 3749–3756. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Capuano, A.W.; Leurgans, S.E.; Bennett, D.A.; Schneider, J.A. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in older persons: A cross-sectional study. Lancet Neurol. 2016, 15, 934–943. [Google Scholar] [CrossRef]
- Dearborn, J.; Zhang, Y.; Qiao, Y.; Suri, M.F.; Liu, L.; Gottesman, R.; Rawlings, A.; Mosley, T.; Alonso, A.; Knopman, D.; et al. Intracranial atherosclerosis and dementia: The Atherosclerosis Risk in Communities (ARIC) Study. Neurology 2017, 88, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Dolan, H.; Crain, B.; Troncoso, J.; Resnick, S.M.; Zonderman, A.B.; Obrien, R.J. Atherosclerosis, dementia and Alzheimer disease in the Baltimore Longitudinal Study of Aging cohort. Ann. Neurol. 2010, 68, 231. [Google Scholar]
- Khan, T.A.; Shah, T.; Prieto, D.; Zhang, W.; Price, J.; Fowkes, G.R.; Cooper, J.; Talmud, P.J.; Humphries, S.E.; Sundstrom, J.; et al. Apolipoprotein E genotype, cardiovascular biomarkers and risk of stroke: Systematic review and meta-analysis of 14 015 stroke cases and pooled analysis of primary biomarker data from up to 60 883 individuals. Int. J. Epidemiol. 2013, 42, 475–492. [Google Scholar] [CrossRef]
- Davignon, J. Apolipoprotein E and atherosclerosis: Beyond lipid effect. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 267–269. [Google Scholar] [CrossRef]
- Curtiss, L. ApoE in atherosclerosis: A protein with multiple hats. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1852–1853. [Google Scholar] [CrossRef]
- Riedel, B.C.; Thompson, P.M.; Brinton, R.D. Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2016, 160, 134. [Google Scholar] [CrossRef] [PubMed]
- Elosua, R.; Ordovas, J.M.; Cupples, L.A.; Fox, C.S.; Polak, J.F.; Wolf, P.A.; D’Agostino, R.A., Sr.; O’Donnell, C.J. Association of APOE genotype with carotid atherosclerosis in men and women: The Framingham Heart Study. J. Lipid Res. 2004, 45, 1868. [Google Scholar] [CrossRef] [PubMed]
- Kwakowsky, A.; Koszegi, Z.; Cheong, R.Y.; Abraham, I.M. Neuroprotective effects of non-classical estrogen-like signaling activators: From mechanism to potential implications. CNS Neurol. Disord. Drug Targets 2013, 12, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Kwakowsky, A.; Milne, M.R.; Waldvogel, H.J.; Faull, R.L. Effect of estradiol on neurotrophin receptors in basal forebrain cholinergic neurons: Relevance for Alzheimer’s disease. Int. J. Mol. Sci. 2016, 17, 2122. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hendrie, H.C.; Hall, K.S.; Hui, S. The relationships between age, sex and the incidence of dementia and Alzheimer disease: A meta-analysis. Arch. Gen. Psychiatry 1998, 55, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Vemuri, P.; Rocca, W.A. Clinical epidemiology of Alzheimer’s disease: Assessing sex and gender differences. Clin. Epidemiol. 2014, 6, 37–48. [Google Scholar] [CrossRef]
- Altmann, A.; Tian, L.; Henderson, V.W.; Greicius, M.D. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann. Neurol. 2014, 75, 563–573. [Google Scholar] [CrossRef]
- Ylilauri, M.P.; Voutilainen, S.; Lönnroos, E.; Mursu, J.; Virtanen, H.E.; Koskinen, T.T.; Salonen, J.T.; Tuomainen, T.-P.; Virtanen, J.K. Association of dietary cholesterol and egg intakes with the risk of incident dementia or Alzheimer disease: The Kuopio Ischaemic Heart Disease Risk Factor Study. Am. J. Clin. Nutr. 2017, 105, 476–484. [Google Scholar] [CrossRef]
- Reitz, C.; Tang, M.-X.; Luchsinger, J.; Mayeux, R. Relation of plasma lipids to Alzheimer disease and vascular dementia. Arch. Neurol. 2004, 61, 705–714. [Google Scholar] [CrossRef]
- Ramanan, V.K.; Przybelski, S.A.; Graff-Radford, J.; Castillo, A.M.; Lowe, V.J.; Mielke, M.M.; Roberts, R.O.; Reid, R.I.; Knopman, D.S.; Jack, C.R.; et al. Statins and brain health: Alzheimer’s Disease and cerebrovascular disease biomarkers in older adults. J. Alzheimers Dis. 2018, 65, 1345–1352. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Sasaki, K.; Hata, J.; Hirakawa, Y.; Fujimi, K.; Ninomiya, T.; Suzuki, S.O.; Kanba, S.; Kiyohara, Y.; Iwaki, T. Association of Alzheimer disease pathology with abnormal lipid metabolism. Neurology 2011, 77, 1068. [Google Scholar] [CrossRef]
- O’Brien, J.T.; Thomas, A. Vascular dementia. Lancet 2015, 386, 1698–1706. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Ballard, C. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis. Assoc. Disord. 1999, 13, S115–S123. [Google Scholar] [CrossRef]
- Jellinger, K.A.; Attems, J. Prevalence and pathogenic role of cerebrovascular lesions in Alzheimer disease. J. Neurol. Sci. 2005, 229–230, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Chi, N.F.; Chien, L.N.; Ku, H.L.; Hu, C.J.; Chiou, H.Y. Alzheimer disease and risk of stroke: A population-based cohort study. Neurology 2013, 80, 705–711. [Google Scholar] [CrossRef]
- Roman, G.C.; Tatemichi, T.K.; Erkinjuntti, T.; Cummings, J.L.; Masdeu, J.C.; Garcia, J.H.; Amaducci, L.; Orgogozo, J.M.; Brun, A.; Hofman, A.; et al. Vascular dementia: Diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology 1993, 43, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Attems, J. Neuropathological evaluation of mixed dementia. J. Neurol. Sci. 2007, 257, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Graham, N.L.; Emery, T.; Hodges, J.R. Distinctive cognitive profiles in Alzheimer’s disease and subcortical vascular dementia. J. Neurol. Neurosurg. Psychiatry 2004, 75, 61–71. [Google Scholar]
- Bowler, J.V.; Eliasziw, M.; Steenhuis, R.; Munoz, D.G.; Fry, R.; Merskey, H.; Hachinski, V.C. Comparative evolution of alzheimer disease, vascular dementia and mixed dementia. Arch. Neurol. 1997, 54, 697–703. [Google Scholar] [CrossRef]
- Erkinjuntti, T. Clinical deficits of Alzheimer’s disease with cerebrovascular disease and probable VaD. Int. J. Clin. Pract. Suppl. 2001, 120, 14–23. [Google Scholar]
- Kalaria, R. Similarities between Alzheimer’s disease and vascular dementia. J. Neurol. Sci. 2002, 203, 29–34. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Struble, R.G.; Clark, A.W.; Price, D.L. Alzheimer disease: Plaques, tangles and the basal forebrain. Ann. Neurol. 1982, 12, 494. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.M. Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer’s disease. J. Comp. Neurol. 2013, 521, 4124–4144. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Adem, A.; Sabbagh, M. New acetylcholinesterase inhibitors for Alzheimer’s disease. Int. J. Alzheimers Dis. 2012, 2012, 8. [Google Scholar] [CrossRef]
- Nakano, S.; Asada, T.; Matsuda, H.; Uno, M.; Takasaki, M. Donepezil hydrochloride preserves regional cerebral blood flow in patients with Alzheimer’s disease. J. Nucl. Med. 2001, 42, 1441–1445. [Google Scholar] [PubMed]
- Nobili, F.; Koulibaly, M.; Vitali, P.; Migneco, O.; Mariani, G.; Ebmeier, K.; Pupi, A.; Robert, P.H.; Rodriguez, G.; Darcourt, J. Brain perfusion follow-up in Alzheimer’s patients during treatment with acetylcholinesterase inhibitors. J. Nucl. Med. 2002, 43, 983–990. [Google Scholar]
- Lojkowska, W.; Ryglewicz, D.; Jedrzejczak, T.; Minc, S.; Jakubowska, T.; Jarosz, H.; Bochynska, A. The effect of cholinesterase inhibitors on the regional blood flow in patients with Alzheimer’s disease and vascular dementia. J. Neurol. Sci. 2003, 216, 119–126. [Google Scholar] [CrossRef]
- Shimizu, S.; Kanetaka, H.; Hirose, D.; Sakurai, H.; Hanyu, H. Differential effects of acetylcholinesterase inhibitors on clinical responses and cerebral blood flow changes in patients with Alzheimer’s disease: A 12-month, randomized and open-label trial. Dement. Geriatr. Cogn. Disord. Extra 2015, 5, 135–146. [Google Scholar] [CrossRef]
- Ebmeier, K.P.; Hunter, R.; Curran, S.M.; Dougal, N.J.; Murray, C.L.; Wyper, D.J.; Patterson, J.; Hanson, M.T.; Siegfried, K.; Goodwin, G.M. Effects of a single dose of the acetylcholinesterase inhibitor velnacrine on recognition memory and regional cerebral blood flow in Alzheimer’s disease. Psychopharmacology 1992, 108, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, G.D.; Messier, A.A.; Miller, M.C.; Machan, J.T.; Majmudar, S.S.; Stopa, E.G.; Donahue, J.E.; Johanson, C.E. Amyloid efflux transporter expression at the blood-brain barrier declines in normal aging. J. Neuropathol. Exp. Neurol. 2010, 69, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Sato, N.; Kurinami, H.; Takeuchi, D.; Takeda, S.; Shimamura, M.; Yamashita, T.; Uchiyama, Y.; Rakugi, H.; Morishita, R. Reduction of brain β-amyloid (Aβ) by fluvastatin, a hydroxymethylglutaryl-CoA reductase inhibitor, through increase in degradation of amyloid precursor protein C-terminal fragments (APP-CTFs) and Abeta clearance. J. Biol. Chem. 2010, 285, 22091–22102. [Google Scholar] [CrossRef]
- Moon, J.H.; Kang, S.B.; Park, J.S.; Lee, B.W.; Kang, E.S.; Ahn, C.W.; Lee, H.C.; Cha, B.S. Up-regulation of hepatic low-density lipoprotein receptor-related protein 1: A possible novel mechanism of antiatherogenic activity of hydroxymethylglutaryl-coenzyme A reductase inhibitor Atorvastatin and hepatic LRP1 expression. Metab. Clin. Exp. 2011, 60, 930–940. [Google Scholar] [CrossRef]
- Shah, N.P.; Swiger, K.J.; Martin, S.S. Impact on cognitive function—Are all statins the same? Curr. Atheroscler. Rep. 2014, 17, 466. [Google Scholar] [CrossRef] [PubMed]
- Schultz, B.G.; Patten, D.K.; Berlau, D.J. The role of statins in both cognitive impairment and protection against dementia: A tale of two mechanisms. Transl. Neurodegener. 2018, 7, 5. [Google Scholar] [CrossRef]
- Sagare, A.P.; Deane, R.; Zlokovic, B.V. Low-density lipoprotein receptor-related protein 1: A physiological Aβ homeostatic mechanism with multiple therapeutic opportunities. Pharmacol. Ther. 2012, 136, 94–105. [Google Scholar] [CrossRef]
- Deane, R.J. Is RAGE still a therapeutic target for Alzheimer’s disease? Future Med. Chem. 2012, 4, 915–925. [Google Scholar] [CrossRef]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.-J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, D.; Wang, Y.; Huang, H.; Zhao, Y.; Zhou, H. Meta-analysis of expression and function of neprilysin in Alzheimer’s disease. Neurosci. Lett. 2017, 657, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Zhuravin, I.A.; Nalivaeva, N.N.; Kozlova, D.I.; Kochkina, E.G.; Fedorova, Y.B.; Gavrilova, S.I. The activity of blood serum cholinesterases and neprilysin as potential biomarkers of mild-cognitive impairment and Alzheimer’s disease. Zhurnal Nevrologii i psikhiatrii Imeni SS Korsakova 2015, 115, 110–117. [Google Scholar] [CrossRef]
- Maruyama, M.; Higuchi, M.; Takaki, Y.; Matsuba, Y.; Tanji, H.; Nemoto, M.; Tomita, N.; Matsui, T.; Iwata, N.; Mizukami, H.; et al. Cerebrospinal fluid neprilysin is reduced in prodromal Alzheimer’s disease. Ann. Neurol. 2005, 57, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M. Cerebral amyloid angiopathy and gene polymorphisms. J. Neurol. Sci. 2004, 226, 41–44. [Google Scholar] [CrossRef]
- Belyaev, N.D.; Nalivaeva, N.N.; Makova, N.Z.; Turner, A.J. Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: Implications for Alzheimer disease. EMBO Rep. 2009, 10, 94–100. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.-J.; Li, T.; Li, J.; Tang, Y.; Le, W. Valproic acid reduces neuritic plaque formation and improves learning deficits in APPSwe/PS1A246E transgenic mice via preventing the prenatal hypoxia-induced down-regulation of neprilysin. CNS Neurosci. Ther. 2014, 20, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, K.E.; Farr, S.A.; Banks, W.A.; Crider, A.M.; Morley, J.E.; Witt, K.A. Somatostatin receptor subtype-4 agonist NNC 26-9100 mitigates the effect of soluble Aβ42 oligomers via a metalloproteinase-dependent mechanism. Brain Res. 2013, 1520, 145–156. [Google Scholar] [CrossRef][Green Version]
- Nalivaeva, N.N.; Turner, A.J. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Barrera-Ocampo, A.; Lopera, F. Amyloid-beta immunotherapy: The hope for Alzheimer disease? Colomb. Med. 2016, 47, 203–212. [Google Scholar] [PubMed]
- van Dyck, C.H. Anti-amyloid-β monoclonal antibodies for Alzheimer’s disease: Pitfalls and promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, J.; Williams, L.; Stella, H.; Leitermann, K.; Mikulskis, A.; O’Gorman, J.; Sevigny, J. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimers Dement. Transl. Res. Clin. Interv. 2016, 2, 169–176. [Google Scholar] [CrossRef]
- Salloway, S.; Honigberg, L.A.; Cho, W.; Ward, M.; Friesenhahn, M.; Brunstein, F.; Quartino, A.; Clayton, D.; Mortensen, D.; Bittner, T.; et al. Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti-amyloid-beta antibody double-blind, placebo-controlled, randomized phase II study in mild-to-moderate Alzheimer’s disease (BLAZE). Alzheimers Res. Ther. 2018, 10, 96. [Google Scholar] [CrossRef]
- Cummings, J.L.; Cohen, S.; van Dyck, C.H.; Brody, M.; Curtis, C.; Cho, W.; Ward, M.; Friesenhahn, M.; Rabe, C.; Brunstein, F.; et al. ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 2018, 90, e1889–e1897. [Google Scholar] [CrossRef] [PubMed]
- Tariot, P.N.; Lopera, F.; Langbaum, J.B.; Thomas, R.G.; Hendrix, S.; Schneider, L.S.; Rios-Romenets, S.; Giraldo, M.; Acosta, N.; Tobon, C.; et al. The Alzheimer’s Prevention Initiative Autosomal-Dominant Alzheimer’s Disease Trial: A study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer’s disease, including a placebo-treated noncarrier cohort. Alzheimers Dement. Transl. Res. Clin. Interv. 2018, 4, 150–160. [Google Scholar] [CrossRef]
- Farlow, M.; Arnold, S.E.; van Dyck, C.H.; Aisen, P.S.; Snider, B.J.; Porsteinsson, A.P.; Friedrich, S.; Dean, R.A.; Gonzales, C.; Sethuraman, G.; et al. Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement. 2012, 8, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 Trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.; Moller, C.; Tegerstedt, K.; Lord, A.; Laudon, H.; Sjodahl, J.; Soderberg, L.; Spens, E.; Sahlin, C.; Waara, E.R.; et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-beta protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J. Alzheimers Dis. 2015, 43, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Lannfelt, L.; Möller, C.; Basun, H.; Osswald, G.; Sehlin, D.; Satlin, A.; Logovinsky, V.; Gellerfors, P. Perspectives on future Alzheimer therapies: Amyloid-β protofibrils - a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Bohrmann, B.; Baumann, K.; Benz, J.; Gerber, F.; Huber, W.; Knoflach, F.; Messer, J.; Oroszlan, K.; Rauchenberger, R.; Richter, W.F.; et al. Gantenerumab: A novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J. Alzheimers Dis. 2012, 28, 49–69. [Google Scholar] [CrossRef]
- Lasser, R.; Ostrowitzki, S.; Scheltens, P.; Boada, M.; Dubois, B.; Dorflinger, E.; Balas, B.; Nikolcheva, T.; Volz, D.; Ashford, E.; et al. Efficacy and safety of gantenerumab in prodromal Alzheimer’s disease: Results from Scarlet Road—A global, multicenter trial. Alzheimers Dement. J. Alzheimers Assoc. 2015, 11, P331–P332. [Google Scholar] [CrossRef]
- The Lancet Neurology. Solanezumab: Too late in mild Alzheimer’s disease? Lancet Neurol. 2017, 16, 97. [Google Scholar] [CrossRef]
- Sarazin, M.; Dorothee, G.; de Souza, L.C.; Aucouturier, P. Immunotherapy in Alzheimer’s disease: Do we have all the pieces of the puzzle? Biol. Psychiatry 2013, 74, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Manassero, G.; Guglielmotto, M.; Zamfir, R.; Borghi, R.; Colombo, L.; Salmona, M.; Perry, G.; Odetti, P.; Arancio, O.; Tamagno, E.; et al. Beta-amyloid 1-42 monomers but not oligomers, produce PHF-like conformation of Tau protein. Aging Cell 2016, 15, 914–923. [Google Scholar] [CrossRef]
- Clementi, M.E.; Marini, S.; Coletta, M.; Orsini, F.; Giardina, B.; Misiti, F. Aβ(31–35) and Aβ(25–35) fragments of amyloid beta-protein induce cellular death through apoptotic signals: Role of the redox state of methionine-35. FEBS Lett. 2005, 579, 2913–2918. [Google Scholar] [CrossRef]
- Barritt, J.D.; Viles, J.H. Truncated amyloid-β(11–40/42) from Alzheimer disease binds Cu2+ with a femtomolar affinity and influences fiber assembly. J. Biol. Chem. 2015, 290, 27791–27802. [Google Scholar] [CrossRef] [PubMed]
- Lemere, C.A. Immunotherapy for Alzheimer’s disease: Hoops and hurdles. Mol. Neurodegener. 2013, 8, 36. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Wang, Y.-R.; Xiang, Y.; Zhou, H.-D.; Giunta, B.; Mañucat-Tan, N.B.; Tan, J.; Zhou, X.-F.; Wang, Y.-J. Clearance of amyloid-beta in Alzheimer’s disease: Shifting the action site from center to periphery. Mol. Neurobiol. 2015, 51, 1–7. [Google Scholar] [CrossRef]
- Grammas, P.; Martinez, J.; Sanchez, A.; Yin, X.; Riley, J.; Gay, D.; Desobry, K.; Tripathy, D.; Luo, J.; Evola, M.; et al. A new paradigm for the treatment of Alzheimer’s disease: Targeting vascular activation. J. Alzheimers Dis. 2014, 40, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Ardura-Fabregat, A.; Boddeke, E.W.G.M.; Boza-Serrano, A.; Brioschi, S.; Castro-Gomez, S.; Ceyzériat, K.; Dansokho, C.; Dierkes, T.; Gelders, G.; Heneka, M.T.; et al. Targeting neuroinflammation to treat Alzheimer’s disease. CNS Drugs 2017, 31, 1057–1082. [Google Scholar] [CrossRef]
- Jaturapatporn, D.; Isaac, M.G.; McCleery, J.; Tabet, N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523. [Google Scholar] [CrossRef]
- Hudry, E.; Dashkoff, J.; Roe, A.D.; Takeda, S.; Koffie, R.M.; Hashimoto, T.; Scheel, M.; Spires-Jones, T.; Arbel-Ornath, M.; Betensky, R.; et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci. Transl. Med. 2013, 5, 212ra161. [Google Scholar] [CrossRef]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.D.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.L.; Goebel, W.D.; James, M.J. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 2012, 335, 1503–1506. [Google Scholar] [CrossRef]
- Cummings, J.L.; Zhong, K.; Kinney, J.W.; Heaney, C.; Moll-Tudla, J.; Joshi, A.; Pontecorvo, M.; Devous, M.; Tang, A.; Bena, J. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene in moderate Alzheimer’s disease. Alzheimers Res. Ther. 2016, 8, 4. [Google Scholar] [CrossRef]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.-L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Gilman, S.; Fox, N.C.; Blennow, K.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Doody, R.; van Dyck, C.H.; et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 2009, 73, 2061–2070. [Google Scholar] [CrossRef]
- Paganini-Hill, A.; Henderson, V.W. Estrogen deficiency and risk of Alzheimer’s disease in women. Am. J. Epidemiol. 1994, 140, 256–261. [Google Scholar] [CrossRef]
- Rocca, W.A.; Grossardt, B.R.; Shuster, L.T. Oophorectomy, menopause, estrogen treatment and cognitive aging: Clinical evidence for a window of opportunity. Brain Res. 2011, 1379, 188–198. [Google Scholar] [CrossRef]
- Milne, M.R.; Haug, C.A.; Ábrahám, I.M.; Kwakowsky, A. Estradiol modulation of neurotrophin receptor expression in female mouse basal forebrain cholinergic neurons in vivo. Endocrinology 2014, 156, 613–626. [Google Scholar] [CrossRef]
- Kunzler, J.; Youmans, K.L.; Yu, C.; Ladu, M.J.; Tai, L.M. APOE modulates the effect of estrogen therapy on Aβ accumulation EFAD-Tg mice. Neurosci. Lett. 2014, 560, 131–136. [Google Scholar] [CrossRef]
- Yaffe, K.; Haan, M.; Byers, A.; Tangen, C.; Kuller, L. Estrogen use, APOE and cognitive decline: Evidence of gene-environment interaction. Neurology 2000, 54, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Kaufer, D.; Gandy, S. APOE ε4 and bapineuzumab Infusing pharmacogenomics into Alzheimer disease therapeutics. Neurology 2009, 73, 2052–2053. [Google Scholar] [CrossRef] [PubMed]


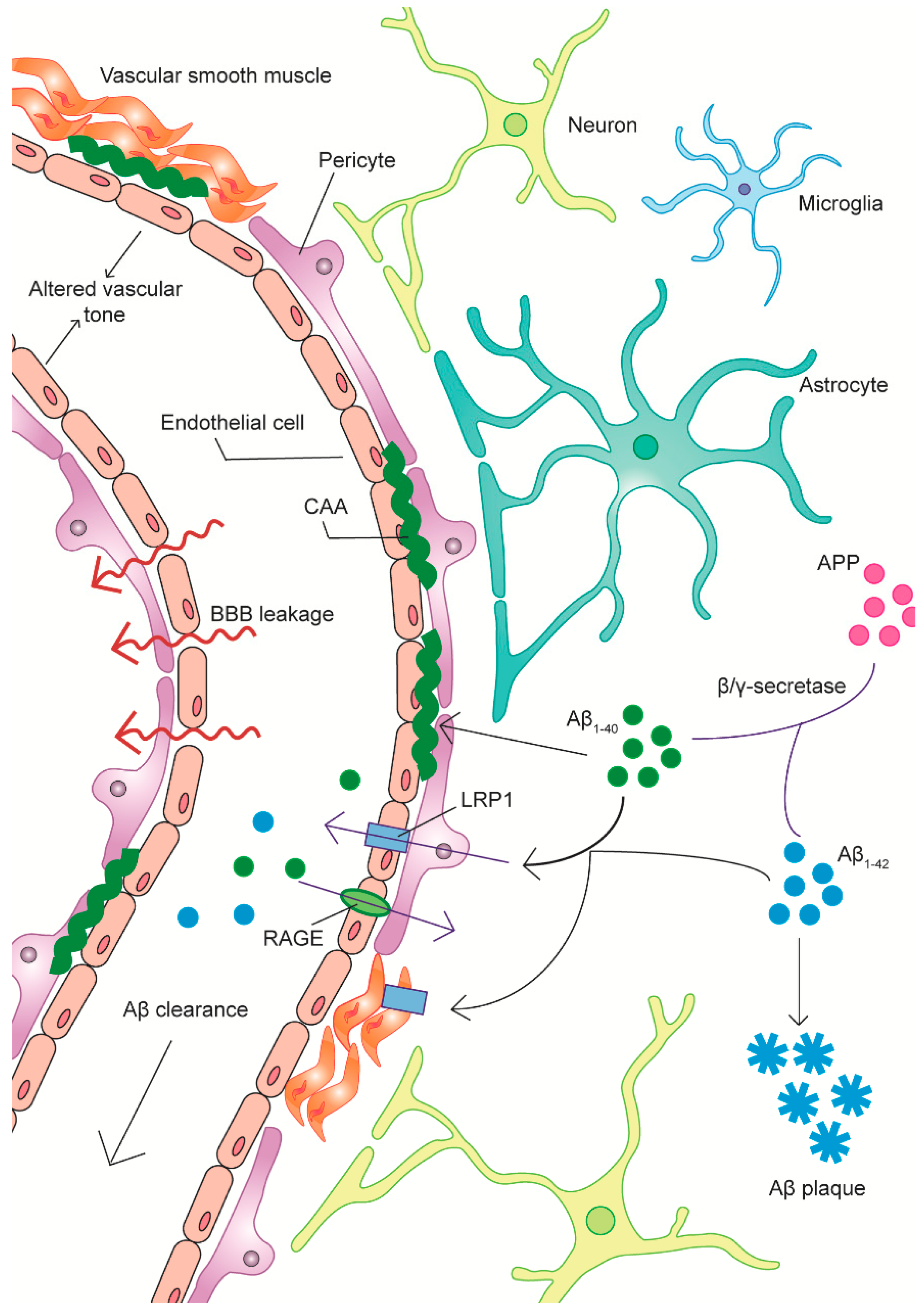{kind=link}
| AD Pathology | Therapeutic Strategies | Potential Targets and Mechanisms |
|---|---|---|
| Aβ aggregation/clearance deficits | Targeting amyloid-β (Aβ) degrading enzymes |
|
| Targeting vascular Aβ transport processes |
| |
| Reducing total brain amyloid load |
| |
| Aberrant angiogenesis | Pro-/anti-angiogenic treatments |
|
| Vascular-mediated inflammation | Anti-inflammatory agents |
|
| Altered vascular tone and cerebral blood flow (CBF) | Ameliorating neurotransmitter dysfunction |
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Govindpani, K.; McNamara, L.G.; Smith, N.R.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. Vascular Dysfunction in Alzheimer’s Disease: A Prelude to the Pathological Process or a Consequence of It? J. Clin. Med. 2019, 8, 651. https://doi.org/10.3390/jcm8050651
Govindpani K, McNamara LG, Smith NR, Vinnakota C, Waldvogel HJ, Faull RL, Kwakowsky A. Vascular Dysfunction in Alzheimer’s Disease: A Prelude to the Pathological Process or a Consequence of It? Journal of Clinical Medicine. 2019; 8(5):651. https://doi.org/10.3390/jcm8050651
Chicago/Turabian StyleGovindpani, Karan, Laura G McNamara, Nicholas R Smith, Chitra Vinnakota, Henry J Waldvogel, Richard LM Faull, and Andrea Kwakowsky. 2019. "Vascular Dysfunction in Alzheimer’s Disease: A Prelude to the Pathological Process or a Consequence of It?" Journal of Clinical Medicine 8, no. 5: 651. https://doi.org/10.3390/jcm8050651
APA StyleGovindpani, K., McNamara, L. G., Smith, N. R., Vinnakota, C., Waldvogel, H. J., Faull, R. L., & Kwakowsky, A. (2019). Vascular Dysfunction in Alzheimer’s Disease: A Prelude to the Pathological Process or a Consequence of It? Journal of Clinical Medicine, 8(5), 651. https://doi.org/10.3390/jcm8050651