Advanced Evolution of Pathogenesis Concepts in Cardiomyopathies

 ,
, 
 ,
, 
Abstract
1. Introduction
2. Clinical Features of Genetic Cardiomyopathy
3. DCM
4. HCM
5. RCM
6. ARVC (Arrhythmogenic Right Ventricular Cardiomyopathy)
7. LVNC
8. Genetic Mutations in Cardiomyopathy
9. Pathophysiology of Cardiomyopathy
9.1. Ras-Raf-MEK-ERK Pathway in Cardiac Myocyte Hypertrophy
9.2. G-Protein Signaling in Cardiomyopathy
9.3. Mechanotransduction Pathways
9.4. AKT/PI3K Signaling in Cardiomyopathy
10. Apoptosis Signaling in Cardiomyopathy and Heart Failure
11. Myocardial Fibrosis and Remodeling
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Augustine, D.X.; Howard, L. Left Ventricular Hypertrophy in Athletes: Differentiating Physiology From Pathology. Curr. Treat. Options Cardiovasc. Med. 2018, 20, 96. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Li, C.-J.; Hou, M.-F.; Chu, P.-Y. New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef]
- Fu, Y.; Eisen, H.J. Genetics of Dilated Cardiomyopathy. Curr. Cardiol. Rep. 2018, 20, 121. [Google Scholar] [CrossRef]
- Ehler, E. Actin-Associated Proteins and Cardiomyopathy-the ‘Unknown’ Beyond Troponin and Tropomyosin. Biophys. Rev. 2018, 10, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Kraft, T.; Montag, J. Altered Force Generation and Cell-to-Cell Contractile Imbalance in Hypertrophic Cardiomyopathy. Pflug. Arch 2019. [Google Scholar] [CrossRef] [PubMed]
- Vikhorev, P.G.; Vikhoreva, N.N. Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle. Int. J. Mol. Sci. 2018, 19, 2234. [Google Scholar] [CrossRef]
- Cooper, R.M.; Raphael, C.E.; Liebregts, M.; Anavekar, N.S.; Veselka, J. New Developments in Hypertrophic Cardiomyopathy. Can. J. Cardiol. 2017, 33, 1254–1265. [Google Scholar] [CrossRef]
- Dec, G.W.; Fuster, V. Idiopathic dilated cardiomyopathy. N. Engl. J. Med. 1994, 331, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.R.; Carniel, E.; Mestroni, L. Cardiomyopathy, familial dilated. Orphanet J. Rare Dis. 2006, 1, 27. [Google Scholar] [CrossRef]
- Wilson, K.; Lucchesi, P.A. Myofilament dysfunction as an emerging mechanism of volume overload heart failure. Pflug. Arch. 2014, 466, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Brayson, D.; Shanahan, C.M. Current insights into LMNA cardiomyopathies: Existing models and missing LINCs. Nucleus 2017, 8, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Morales, A. LMNA-Related Dilated Cardiomyopathy. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Peters, S.; Kumar, S.; Elliott, P.; Kalman, J.M.; Fatkin, D. Arrhythmic Genotypes in Familial Dilated Cardiomyopathy: Implications for Genetic Testing and Clinical Management. Heart Lung Circ. 2019, 28, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, V.; Joshi, M.; Suresh, S.; Sanchez, J.A.; Maulik, N.; Maulik, G. Diabetes, oxidative stress, molecular mechanism, and cardiovascular disease—An overview. Toxicol. Mech. Methods 2012, 22, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zabell, A.; Koh, W.; Tang, W.H. Lamin A/C Cardiomyopathies: Current Understanding and Novel Treatment Strategies. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 21. [Google Scholar] [CrossRef]
- Iskandrian, A.S.; Helfeld, H.; Lemlek, J.; Lee, J.; Iskandrian, B.; Heo, J. Differentiation between primary dilated cardiomyopathy and ischemic cardiomyopathy based on right ventricular performance. Am. Heart J. 1992, 123, 768–773. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.Y.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Cochran, T.R.; Briston, D.A.; Brown, S.R.; Sambatakos, P.J.; Miller, T.L.; Carrillo, A.A.; Corcia, L.; Sanchez, J.E.; Diamond, M.B.; et al. Pediatric cardiomyopathies: Causes, epidemiology, clinical course, preventive strategies and therapies. Future Cardiol. 2013, 9, 817–848. [Google Scholar] [CrossRef]
- Towbin, J.A.; Lowe, A.M.; Colan, S.D.; Sleeper, L.A.; Orav, E.J.; Clunie, S.; Messere, J.; Cox, G.F.; Lurie, P.R.; Hsu, D.; et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006, 296, 1867–1876. [Google Scholar] [CrossRef]
- Patel, M.D.; Mohan, J.; Schneider, C.; Bajpai, G.; Purevjav, E.; Canter, C.E.; Towbin, J.; Bredemeyer, A.; Lavine, K.J. Pediatric and adult dilated cardiomyopathy represent distinct pathological entities. JCI Insight 2017, 2, 94382. [Google Scholar] [CrossRef]
- Song, W.; Shou, W. Cardiac sodium channel Nav1.5 mutations and cardiac arrhythmia. Pediatr. Cardiol. 2012, 33, 943–949. [Google Scholar] [CrossRef]
- Messer, A.E.; Marston, S.B. Investigating the role of uncoupling of troponin I phosphorylation from changes in myofibrillar Ca2+-sensitivity in the pathogenesis of cardiomyopathy. Front. Physiol. 2014, 5, 315. [Google Scholar] [CrossRef]
- Bai, F.; Wang, L.; Kawai, M. A study of tropomyosin’s role in cardiac function and disease using thin-filament reconstituted myocardium. J. Muscle Res. Cell Motil. 2013, 34, 295–310. [Google Scholar] [CrossRef]
- Opie, L.H.; Commerford, P.J.; Gersh, B.J.; Pfeffer, M.A. Controversies in ventricular remodelling. Lancet 2006, 367, 356–367. [Google Scholar] [CrossRef]
- Fatkin, D. Familial dilated cardiomyopathy: Current challenges and future directions. Glob. Cardiol. Sci. Pract. 2012, 2012, 8. [Google Scholar] [CrossRef]
- Petretta, M.; Pirozzi, F.; Sasso, L.; Paglia, A.; Bonaduce, D. Review and metaanalysis of the frequency of familial dilated cardiomyopathy. Am. J. Cardiol. 2011, 108, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Ganesh Santhi, K.; Arnett Donna, K.; Assimes Themistocles, L.; Basson Craig, T.; Chakravarti, A.; Ellinor Patrick, T.; Engler Mary, B.; Goldmuntz, E.; Herrington David, M.; Hershberger Ray, E.; et al. Genetics and Genomics for the Prevention and Treatment of Cardiovascular Disease: Update. Circulation 2013, 128, 2813–2851. [Google Scholar] [CrossRef]
- Ware, J.S.; Cook, S.A. Role of titin in cardiomyopathy: From DNA variants to patient stratification. Nat. Rev. Cardiol. 2018, 15, 241–252. [Google Scholar] [CrossRef]
- Linke, W.A. Sense and stretchability: The role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc. Res. 2008, 77, 637–648. [Google Scholar] [CrossRef]
- Gautel, M. The sarcomeric cytoskeleton: Who picks up the strain? Curr. Opin. Cell Biol. 2011, 23, 39–46. [Google Scholar] [CrossRef]
- Linke, W.A.; Hamdani, N. Gigantic business: Titin properties and function through thick and thin. Circ. Res. 2014, 114, 1052–1068. [Google Scholar] [CrossRef]
- Tskhovrebova, L.; Trinick, J. Roles of titin in the structure and elasticity of the sarcomere. J. Biomed. Biotechnol. 2010, 2010, 612482. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Lu, Q.W.; Harada, K.; Takahashi-Yanaga, F.; Minakami, R.; Ohta, M.; Sasaguri, T.; Ohtsuki, I. Ca2+-desensitizing effect of a deletion mutation Delta K210 in cardiac troponin T that causes familial dilated cardiomyopathy. Proc. Natl. Acad. Sci. USA 2002, 99, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Liew, A.C.; Vassiliou, V.S.; Cooper, R.; Raphael, C.E. Hypertrophic Cardiomyopathy—Past, Present and Future. J. Clin. Med. 2017, 6, 118. [Google Scholar] [CrossRef]
- Maron, B.J.; Ommen, S.R.; Semsarian, C.; Spirito, P.; Olivotto, I.; Maron, M.S. Hypertrophic cardiomyopathy: Present and future, with translation into contemporary cardiovascular medicine. J. Am. Coll. Cardiol. 2014, 64, 83–99. [Google Scholar] [CrossRef]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011, 124, 2761–2796. [Google Scholar] [CrossRef]
- Goyal, V.; Jassal, D.S.; Dhalla, N.S. Pathophysiology and prevention of sudden cardiac death. Can. J. Physiol. Pharmacol. 2016, 94, 237–244. [Google Scholar] [CrossRef]
- Yang, Q.; Sanbe, A.; Osinska, H.; Hewett, T.E.; Klevitsky, R.; Robbins, J. A mouse model of myosin binding protein C human familial hypertrophic cardiomyopathy. J. Clin. Investig. 1998, 102, 1292–1300. [Google Scholar] [CrossRef]
- Yang, Q.; Sanbe, A.; Osinska, H.; Hewett, T.E.; Klevitsky, R.; Robbins, J. In vivo modeling of myosin binding protein C familial hypertrophic cardiomyopathy. Circ. Res. 1999, 85, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Manabe, S.; Kasegawa, H.; Arai, H.; Takanashi, S. Management of systolic anterior motion of the mitral valve: A mechanism-based approach. Gen. Thorac. Cardiovasc. Surg. 2018, 66, 379–389. [Google Scholar] [CrossRef] [PubMed]
- MacIver, D.H.; Clark, A.L. Contractile Dysfunction in Sarcomeric Hypertrophic Cardiomyopathy. J. Card. Fail. 2016, 22, 731–737. [Google Scholar] [CrossRef]
- Lee, S.R.; Han, J. Mitochondrial Mutations in Cardiac Disorders. Adv. Exp. Med. Biol. 2017, 982, 81–111. [Google Scholar]
- Ren, X.; Hensley, N.; Brady, M.B.; Gao, W.D. The Genetic and Molecular Bases for Hypertrophic Cardiomyopathy: The Role for Calcium Sensitization. J. Cardiothorac. Vasc. Anesth. 2018, 32, 478–487. [Google Scholar] [CrossRef]
- Soetkamp, D.; Raedschelders, K.; Mastali, M.; Sobhani, K.; Bairey Merz, C.N.; Van Eyk, J. The continuing evolution of cardiac troponin I biomarker analysis: From protein to proteoform. Expert Rev. Proteomics 2017, 14, 973–986. [Google Scholar] [CrossRef]
- Zhuge, R.; Zhou, R.; Ni, X. Progress in Molecular Genetic Study of Mitochondrial Cardiomyopathy. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 2017, 39, 438–444. [Google Scholar]
- El-Hattab, A.W.; Dai, H.; Almannai, M.; Wang, J.; Faqeih, E.A.; Al Asmari, A.; Saleh, M.A.M.; Elamin, M.A.O.; Alfadhel, M.; Alkuraya, F.S.; et al. Molecular and clinical spectra of FBXL4 deficiency. Hum. Mutat. 2017, 38, 1649–1659. [Google Scholar] [CrossRef]
- Coppini, R.; Ferrantini, C.; Poggesi, C.; Mugelli, A.; Olivotto, I. Molecular targets and novel pharmacological options to prevent myocardial hypertrophic remodeling. Giornale Italiano di Cardiologia 2016, 17, 189–196. [Google Scholar]
- Landstrom, A.P.; Ackerman, M.J. Beyond the cardiac myofilament: Hypertrophic cardiomyopathy- associated mutations in genes that encode calcium-handling proteins. Curr. Mol. Med. 2012, 12, 507–518. [Google Scholar] [CrossRef]
- Kensler, R.W.; Shaffer, J.F.; Harris, S.P. Binding of the N-terminal fragment C0-C2 of cardiac MyBP-C to cardiac F-actin. J. Struct. Biol. 2011, 174, 44–51. [Google Scholar] [CrossRef]
- McNally, E.M.; Barefield, D.Y.; Puckelwartz, M.J. The genetic landscape of cardiomyopathy and its role in heart failure. Cell Metab. 2015, 21, 174–182. [Google Scholar] [CrossRef]
- Moore, J.R.; Leinwand, L.; Warshaw, D.M. Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ. Res. 2012, 111, 375–385. [Google Scholar] [CrossRef]
- Walsh, R.; Rutland, C.; Thomas, R.; Loughna, S. Cardiomyopathy: A systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology 2010, 115, 49–60. [Google Scholar] [CrossRef]
- Kittleson, M.D.; Meurs, K.M.; Munro, M.J.; Kittleson, J.A.; Liu, S.K.; Pion, P.D.; Towbin, J.A. Familial hypertrophic cardiomyopathy in maine coon cats: An animal model of human disease. Circulation 1999, 99, 3172–3180. [Google Scholar] [CrossRef]
- Marian, A.J.; Wu, Y.; Lim, D.S.; McCluggage, M.; Youker, K.; Yu, Q.T.; Brugada, R.; DeMayo, F.; Quinones, M.; Roberts, R. A transgenic rabbit model for human hypertrophic cardiomyopathy. J. Clin. Investig. 1999, 104, 1683–1692. [Google Scholar] [CrossRef]
- Niimura, H.; Bachinski, L.L.; Sangwatanaroj, S.; Watkins, H.; Chudley, A.E.; McKenna, W.; Kristinsson, A.; Roberts, R.; Sole, M.; Maron, B.J.; et al. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N. Engl. J. Med. 1998, 338, 1248–1257. [Google Scholar] [CrossRef]
- Bonne, G.; Carrier, L.; Bercovici, J.; Cruaud, C.; Richard, P.; Hainque, B.; Gautel, M.; Labeit, S.; James, M.; Beckmann, J.; et al. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat. Genet. 1995, 11, 438–440. [Google Scholar] [CrossRef]
- Van Dijk, S.J.; Dooijes, D.; dos Remedios, C.; Michels, M.; Lamers, J.M.; Winegrad, S.; Schlossarek, S.; Carrier, L.; ten Cate, F.J.; Stienen, G.J.; et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: Haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 2009, 119, 1473–1483. [Google Scholar] [CrossRef]
- Freiburg, A.; Gautel, M. A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur. J. Biochem. 1996, 235, 317–323. [Google Scholar] [CrossRef]
- Gilbert, R.; Kelly, M.G.; Mikawa, T.; Fischman, D.A. The carboxyl terminus of myosin binding protein C (MyBP-C, C-protein) specifies incorporation into the A-band of striated muscle. J. Cell Sci. 1996, 109 Pt 1, 101–111. [Google Scholar]
- Barefield, D.; Sadayappan, S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J. Mol. Cell. Cardiol. 2010, 48, 866–875. [Google Scholar] [CrossRef]
- Bezold, K.L.; Shaffer, J.F.; Khosa, J.K.; Hoye, E.R.; Harris, S.P. A gain-of-function mutation in the M-domain of cardiac myosin-binding protein-C increases binding to actin. J. Biol. Chem. 2013, 288, 21496–21505. [Google Scholar] [CrossRef]
- Kunst, G.; Kress, K.R.; Gruen, M.; Uttenweiler, D.; Gautel, M.; Fink, R.H. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ. Res. 2000, 86, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Sadayappan, S.; Gulick, J.; Osinska, H.; Martin, L.A.; Hahn, H.S.; Dorn, G.W., 2nd; Klevitsky, R.; Seidman, C.E.; Seidman, J.G.; Robbins, J. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ. Res. 2005, 97, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Sadayappan, S.; Osinska, H.; Klevitsky, R.; Lorenz, J.N.; Sargent, M.; Molkentin, J.D.; Seidman, C.E.; Seidman, J.G.; Robbins, J. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc. Natl. Acad. Sci. USA 2006, 103, 16918–16923. [Google Scholar] [CrossRef]
- Morner, S.; Richard, P.; Kazzam, E.; Hellman, U.; Hainque, B.; Schwartz, K.; Waldenstrom, A. Identification of the genotypes causing hypertrophic cardiomyopathy in northern Sweden. J. Mol. Cell. Cardiol. 2003, 35, 841–849. [Google Scholar] [CrossRef]
- Erdmann, J.; Daehmlow, S.; Wischke, S.; Senyuva, M.; Werner, U.; Raible, J.; Tanis, N.; Dyachenko, S.; Hummel, M.; Hetzer, R.; et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin. Genet. 2003, 64, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; Ommen, S.R.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Yield of genetic testing in hypertrophic cardiomyopathy. Mayo Clin. Proc. 2005, 80, 739–744. [Google Scholar] [CrossRef]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M.; et al. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef] [PubMed]
- Parvatiyar, M.S.; Landstrom, A.P.; Figueiredo-Freitas, C.; Potter, J.D.; Ackerman, M.J.; Pinto, J.R. A mutation in TNNC1-encoded cardiac troponin C, TNNC1-A31S, predisposes to hypertrophic cardiomyopathy and ventricular fibrillation. J. Biol. Chem. 2012, 287, 31845–31855. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Adekola, B.A.; Bos, J.M.; Ommen, S.R.; Ackerman, M.J. PLN-encoded phospholamban mutation in a large cohort of hypertrophic cardiomyopathy cases: Summary of the literature and implications for genetic testing. Am. Heart J. 2011, 161, 165–171. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Weisleder, N.; Batalden, K.B.; Bos, J.M.; Tester, D.J.; Ommen, S.R.; Wehrens, X.H.; Claycomb, W.C.; Ko, J.K.; Hwang, M.; et al. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J. Mol. Cell. Cardiol. 2007, 42, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Fujino, N.; Ino, H.; Hayashi, K.; Uchiyama, K.; Nagata, M.; Konno, T.; Katoh, H.; Sakamoto, Y.; Tsubokawa, T.; Ohsato, K.; et al. Abstract 915: A Novel Missense Mutation in Cardiac Ryanodine Receptor Gene as a Possible Cause of Hypertrophic Cardiomyopathy: Evidence From Familial Analysis. Circulation 2006, 114, II_165. [Google Scholar]
- Maron Martin, S.; Olivotto, I.; Zenovich Andrey, G.; Link Mark, S.; Pandian Natesa, G.; Kuvin Jeffery, T.; Nistri, S.; Cecchi, F.; Udelson James, E.; Maron Barry, J. Hypertrophic Cardiomyopathy Is Predominantly a Disease of Left Ventricular Outflow Tract Obstruction. Circulation 2006, 114, 2232–2239. [Google Scholar] [CrossRef]
- Dimitrow, P.P.; Rajtar-Salwa, R. Obstructive Form of Hypertrophic Cardiomyopathy-Left Ventricular Outflow Tract Gradient: Novel Methods of Provocation, Monitoring of Biomarkers, and Recent Advances in the Treatment. Biomed Res. Int. 2016, 2016, 1575130. [Google Scholar] [CrossRef] [PubMed]
- Seferovic, P.M.; Paulus, W.J. Clinical diabetic cardiomyopathy: A two-faced disease with restrictive and dilated phenotypes. Eur. Heart J. 2015, 36, 1718–1727. [Google Scholar] [CrossRef]
- Brown, K.N.; Diaz, R.R. Restrictive (Infiltrative) Cardiomyopathy. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2018. [Google Scholar]
- Kostareva, A.; Kiselev, A.; Gudkova, A.; Frishman, G.; Ruepp, A.; Frishman, D.; Smolina, N.; Tarnovskaya, S.; Nilsson, D.; Zlotina, A.; et al. Genetic Spectrum of Idiopathic Restrictive Cardiomyopathy Uncovered by Next-Generation Sequencing. PLoS ONE 2016, 11, e0163362. [Google Scholar] [CrossRef] [PubMed]
- Parvatiyar, M.S.; Pinto, J.R.; Dweck, D.; Potter, J.D. Cardiac troponin mutations and restrictive cardiomyopathy. J. Biomed. Biotechnol. 2010, 2010, 350706. [Google Scholar] [CrossRef]
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of contraction in striated muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [CrossRef]
- Gordon, A.M.; Regnier, M.; Homsher, E. Skeletal and cardiac muscle contractile activation: Tropomyosin “rocks and rolls”. News Physiol. Sci. 2001, 16, 49–55. [Google Scholar] [CrossRef]
- Kobayashi, T.; Solaro, R.J. Increased Ca2+ affinity of cardiac thin filaments reconstituted with cardiomyopathy-related mutant cardiac troponin I. J. Biol. Chem. 2006, 281, 13471–13477. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.V.; Liang, J.; Potter, J.D. Mutations in human cardiac troponin I that are associated with restrictive cardiomyopathy affect basal ATPase activity and the calcium sensitivity of force development. J. Biol. Chem. 2005, 280, 30909–30915. [Google Scholar] [CrossRef]
- Kaski, J.P.; Syrris, P.; Burch, M.; Tome-Esteban, M.T.; Fenton, M.; Christiansen, M.; Andersen, P.S.; Sebire, N.; Ashworth, M.; Deanfield, J.E.; et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 2008, 94, 1478–1484. [Google Scholar] [CrossRef]
- Sweet, M.; Taylor, M.R.; Mestroni, L. Diagnosis, prevalence, and screening of familial dilated cardiomyopathy. Expert Opin. Orphan Drugs 2015, 3, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Husser, D.; Ueberham, L.; Jacob, J.; Heuer, D.; Riedel-Heller, S.; Walker, J.; Hindricks, G.; Bollmann, A. Prevalence of clinically apparent hypertrophic cardiomyopathy in Germany-An analysis of over 5 million patients. PLoS ONE 2018, 13, e0196612. [Google Scholar] [CrossRef] [PubMed]
- Pereira, N.L.; Grogan, M.; Dec, G.W. Spectrum of Restrictive and Infiltrative Cardiomyopathies: Part 1 of a 2-Part Series. J. Am. Coll. Cardiol. 2018, 71, 1130–1148. [Google Scholar] [CrossRef]
- Pereira, N.L.; Grogan, M.; Dec, G.W. Spectrum of Restrictive and Infiltrative Cardiomyopathies: Part 2 of a 2-Part Series. J. Am. Coll. Cardiol. 2018, 71, 1149–1166. [Google Scholar] [CrossRef]
- Yadav, S.; Sitbon, Y.H.; Kazmierczak, K.; Szczesna-Cordary, D. Hereditary heart disease: Pathophysiology, clinical presentation, and animal models of HCM, RCM, and DCM associated with mutations in cardiac myosin light chains. Pflug. Arch. 2019. [Google Scholar] [CrossRef]
- Rammos, A.; Meladinis, V.; Vovas, G.; Patsouras, D. Restrictive Cardiomyopathies: The Importance of Noninvasive Cardiac Imaging Modalities in Diagnosis and Treatment-A Systematic Review. Radiol. Res. Pract. 2017, 2017, 2874902. [Google Scholar] [CrossRef]
- Mogensen, J.; Kubo, T.; Duque, M.; Uribe, W.; Shaw, A.; Murphy, R.; Gimeno, J.R.; Elliott, P.; McKenna, W.J. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J. Clin. Investig. 2003, 111, 209–216. [Google Scholar] [CrossRef]
- Gemayel, C.; Pelliccia, A.; Thompson, P.D. Arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2001, 38, 1773–1781. [Google Scholar] [CrossRef]
- Ohno, S. The genetic background of arrhythmogenic right ventricular cardiomyopathy. J. Arrhythmia 2016, 32, 398–403. [Google Scholar] [CrossRef]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [CrossRef]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Marcus, F.I.; Edson, S.; Towbin, J.A. Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy: A Practical Guide for Physicians. J. Am. Coll. Cardiol. 2013, 61, 1945–1948. [Google Scholar] [CrossRef]
- James, C.A.; Bhonsale, A.; Tichnell, C.; Murray, B.; Russell, S.D.; Tandri, H.; Tedford, R.J.; Judge, D.P.; Calkins, H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J. Am. Coll. Cardiol. 2013, 62, 1290–1297. [Google Scholar] [CrossRef]
- Dalal, D.; James, C.; Devanagondi, R.; Tichnell, C.; Tucker, A.; Prakasa, K.; Spevak, P.J.; Bluemke, D.A.; Abraham, T.; Russell, S.D.; et al. Penetrance of mutations in plakophilin-2 among families with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Am. Coll. Cardiol. 2006, 48, 1416–1424. [Google Scholar] [CrossRef]
- Kirchhof, P.; Fabritz, L.; Zwiener, M.; Witt, H.; Schafers, M.; Zellerhoff, S.; Paul, M.; Athai, T.; Hiller, K.H.; Baba, H.A.; et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation 2006, 114, 1799–1806. [Google Scholar] [CrossRef]
- Que, D.; Yang, P.; Song, X.; Liu, L. Traditional vs. genetic pathogenesis of arrhythmogenic right ventricular cardiomyopathy. Europace 2015, 17, 1770–1776. [Google Scholar] [CrossRef]
- Roux-Buisson, N.; Gandjbakhch, E.; Donal, E.; Probst, V.; Deharo, J.C.; Chevalier, P.; Klug, D.; Mansencal, N.; Delacretaz, E.; Cosnay, P.; et al. Prevalence and significance of rare RYR2 variants in arrhythmogenic right ventricular cardiomyopathy/dysplasia: Results of a systematic screening. Heart Rhythm 2014, 11, 1999–2009. [Google Scholar] [CrossRef]
- Coonar, A.S.; Protonotarios, N.; Tsatsopoulou, A.; Needham, E.W.; Houlston, R.S.; Cliff, S.; Otter, M.I.; Murday, V.A.; Mattu, R.K.; McKenna, W.J. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation 1998, 97, 2049–2058. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Danieli, G.A.; Buja, G.; Daliento, L.; Fasoli, G.; Scognamiglio, R.; Corrado, D.; Thiene, G. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Hum. Mol. Genet. 1994, 3, 959–962. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef]
- Hulot, J.S.; Jouven, X.; Empana, J.P.; Frank, R.; Fontaine, G. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 2004, 110, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Dalal, D.; Nasir, K.; Bomma, C.; Prakasa, K.; Tandri, H.; Piccini, J.; Roguin, A.; Tichnell, C.; James, C.; Russell, S.D.; et al. Arrhythmogenic right ventricular dysplasia: A United States experience. Circulation 2005, 112, 3823–3832. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef]
- Towbin, J.A.; Lorts, A.; Jefferies, J.L. Left ventricular non-compaction cardiomyopathy. Lancet 2015, 386, 813–825. [Google Scholar] [CrossRef]
- Henderson, D.J.; Anderson, R.H. The development and structure of the ventricles in the human heart. Pediatr. Cardiol. 2009, 30, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Ichida, F.; Tsubata, S.; Bowles, K.R.; Haneda, N.; Uese, K.; Miyawaki, T.; Dreyer, W.J.; Messina, J.; Li, H.; Bowles, N.E.; et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 2001, 103, 1256–1263. [Google Scholar] [CrossRef]
- Ichida, F.; Hamamichi, Y.; Miyawaki, T.; Ono, Y.; Kamiya, T.; Akagi, T.; Hamada, H.; Hirose, O.; Isobe, T.; Yamada, K.; et al. Clinical features of isolated noncompaction of the ventricular myocardium: Long-term clinical course, hemodynamic properties, and genetic background. J. Am. Coll. Cardiol. 1999, 34, 233–240. [Google Scholar] [CrossRef]
- Towbin, J.A. Left Ventricular Noncompaction: A New Form of Heart Failure. Heart Fail. Clin. 2010, 6, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Szydłowski, L.; Łoskot, M.; Markiewicz-Łoskot, G.; Hollek, A.; Moric-Janiszewska, E.; Węglarz, L. Isolated ventricular non-compaction: Clinical study and genetic review. EP Europace 2006, 8, 1064–1067. [Google Scholar]
- Cosson, L.; Toutain, A.; Simard, G.; Kulik, W.; Matyas, G.; Guichet, A.; Blasco, H.; Maakaroun-Vermesse, Z.; Vaillant, M.C.; Le Caignec, C.; et al. Barth syndrome in a female patient. Mol. Genet. Metab. 2012, 106, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Hoedemaekers, Y.M.; Caliskan, K.; Michels, M.; Frohn-Mulder, I.; van der Smagt, J.J.; Phefferkorn, J.E.; Wessels, M.W.; ten Cate, F.J.; Sijbrands, E.J.; Dooijes, D.; et al. The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Probst, S.; Oechslin, E.; Schuler, P.; Greutmann, M.; Boye, P.; Knirsch, W.; Berger, F.; Thierfelder, L.; Jenni, R.; Klaassen, S. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ. Cardiovasc. Genet. 2011, 4, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Digilio, M.C.; Bernardini, L.; Gagliardi, M.G.; Versacci, P.; Baban, A.; Capolino, R.; Dentici, M.L.; Roberti, M.C.; Angioni, A.; Novelli, A.; et al. Syndromic non-compaction of the left ventricle: Associated chromosomal anomalies. Clin. Genet. 2013, 84, 362–367. [Google Scholar] [CrossRef]
- Tang, S.; Batra, A.; Zhang, Y.; Ebenroth, E.S.; Huang, T. Left ventricular noncompaction is associated with mutations in the mitochondrial genome. Mitochondrion 2010, 10, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, M.; Li, B.Y.; Chen, H.; Xu, X.; Song, L.S.; Guatimosim, S.; Zhu, W.; Yong, W.; Zhang, W.; Bu, G.; et al. FKBP12 is a critical regulator of the heart rhythm and the cardiac voltage-gated sodium current in mice. Circ. Res. 2011, 108, 1042–1052. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, W.; Sun, X.; Yoshimoto, M.; Chen, Z.; Zhu, W.; Liu, J.; Shen, Y.; Yong, W.; Li, D.; et al. Fkbp1a controls ventricular myocardium trabeculation and compaction by regulating endocardial Notch1 activity. Development 2013, 140, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Donahoe, P.K. The immunophilin FKBP12: A molecular guardian of the TGF-beta family type I receptors. Front. Biosci. 2004, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.N.; Henderson, D.J.; Doudney, K.; Gaston-Massuet, C.; Phillips, H.M.; Paternotte, C.; Arkell, R.; Stanier, P.; Copp, A.J. Disruption of scribble (Scrb1) causes severe neural tube defects in the circletail mouse. Hum. Mol. Genet. 2003, 12, 87–98. [Google Scholar] [CrossRef]
- Phillips, H.M.; Hildreth, V.; Peat, J.D.; Murdoch, J.N.; Kobayashi, K.; Chaudhry, B.; Henderson, D.J. Non-cell-autonomous roles for the planar cell polarity gene Vangl2 in development of the coronary circulation. Circ. Res. 2008, 102, 615–623. [Google Scholar] [CrossRef]
- Phillips, H.M.; Murdoch, J.N.; Chaudhry, B.; Copp, A.J.; Henderson, D.J. Vangl2 acts via RhoA signaling to regulate polarized cell movements during development of the proximal outflow tract. Circ. Res. 2005, 96, 292–299. [Google Scholar] [CrossRef]
- Phillips, H.M.; Rhee, H.J.; Murdoch, J.N.; Hildreth, V.; Peat, J.D.; Anderson, R.H.; Copp, A.J.; Chaudhry, B.; Henderson, D.J. Disruption of planar cell polarity signaling results in congenital heart defects and cardiomyopathy attributable to early cardiomyocyte disorganization. Circ. Res. 2007, 101, 137–145. [Google Scholar] [CrossRef]
- Sinha, T.; Wang, B.; Evans, S.; Wynshaw-Boris, A.; Wang, J. Disheveled mediated planar cell polarity signaling is required in the second heart field lineage for outflow tract morphogenesis. Dev. Biol. 2012, 370, 135–144. [Google Scholar] [CrossRef]
- England, J.; Loughna, S.; Rutland, C.S. Multiple Species Comparison of Cardiac Troponin T and Dystrophin: Unravelling the DNA behind Dilated Cardiomyopathy. J. Cardiovasc. Dev. Dis. 2017, 4, 8. [Google Scholar] [CrossRef]
- Long, P.A.; Evans, J.M.; Olson, T.M. Diagnostic Yield of Whole Exome Sequencing in Pediatric Dilated Cardiomyopathy. J. Cardiovasc. Dev. Dis. 2017, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T.; Fitzgerald, K.K. Dystrophic Cardiomyopathy: Complex Pathobiological Processes to Generate Clinical Phenotype. J. Cardiovasc. Dev. Dis. 2017, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Riabowol, K.T.; Vosatka, R.J.; Ziff, E.B.; Lamb, N.J.; Feramisco, J.R. Microinjection of fos-specific antibodies blocks DNA synthesis in fibroblast cells. Mol. Cell. Biol. 1988, 8, 1670–1676. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.J.; Chien, K.R. Signaling Pathways for Cardiac Hypertrophy and Failure. N. Engl. J. Med. 1999, 341, 1276–1283. [Google Scholar] [CrossRef]
- Lázár-Molnár, E.; Hegyesi, H.; Tóth, S.; Falus, A. Autocrine And Paracrine Regulation By Cytokines And Growth Factors In Melanoma. Cytokine 2000, 12, 547–554. [Google Scholar] [CrossRef]
- Brand, T.; Schneider, M.D. The TGF beta superfamily in myocardium: Ligands, receptors, transduction, and function. J. Mol. Cell. Cardiol. 1995, 27, 5–18. [Google Scholar] [CrossRef]
- Hefti, M.A.; Harder, B.A.; Eppenberger, H.M.; Schaub, M.C. Signaling pathways in cardiac myocyte hypertrophy. J. Mol. Cell. Cardiol. 1997, 29, 2873–2892. [Google Scholar] [CrossRef] [PubMed]
- Gelb, B.D.; Cavé, H.; Dillon, M.W.; Gripp, K.W.; Lee, J.A.; Mason-Suares, H.; Rauen, K.A.; Williams, B.; Zenker, M.; Vincent, L.M.; et al. ClinGen’s RASopathy Expert Panel consensus methods for variant interpretation. Genet. Med. 2018, 20, 1334–1345. [Google Scholar] [CrossRef]
- Dhandapany, P.S.; Razzaque, M.A.; Muthusami, U.; Kunnoth, S.; Edwards, J.J.; Mulero-Navarro, S.; Riess, I.; Pardo, S.; Sheng, J.; Rani, D.S.; et al. RAF1 mutations in childhood-onset dilated cardiomyopathy. Nat. Genet. 2014, 46, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Gelb, B.D.; Tartaglia, M. RAS signaling pathway mutations and hypertrophic cardiomyopathy: Getting into and out of the thick of it. J. Clin. Investig. 2011, 121, 844–847. [Google Scholar] [CrossRef]
- Pandit, B.; Sarkozy, A.; Pennacchio, L.A.; Carta, C.; Oishi, K.; Martinelli, S.; Pogna, E.A.; Schackwitz, W.; Ustaszewska, A.; Landstrom, A.; et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat. Genet. 2007, 39, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef]
- Rockman, H.A.; Koch, W.J.; Lefkowitz, R.J. Seven-transmembrane-spanning receptors and heart function. Nature 2002, 415, 206–212. [Google Scholar] [CrossRef]
- Hamm, H.E. The many faces of G protein signaling. J. Biol. Chem. 1998, 273, 669–672. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Weeks, K.L.; Pretorius, L.; McMullen, J.R. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 2010, 128, 191–227. [Google Scholar] [CrossRef]
- Strniskova, M.; Barancik, M.; Neckar, J.; Ravingerova, T. Mitogen-activated protein kinases in the acute diabetic myocardium. Mol. Cell. Biochem. 2003, 249, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ichikawa, T.; Li, J.; Si, Q.; Yang, H.; Chen, X.; Goldblatt, C.S.; Meyer, C.J.; Li, X.; Cai, L.; et al. Diabetic Downregulation of Nrf2 Activity via ERK Contributes to Oxidative Stress–Induced Insulin Resistance in Cardiac Cells In Vitro and In Vivo. Diabetes 2011, 60, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Obermann, W.M.; Gautel, M.; Steiner, F.; van der Ven, P.F.; Weber, K.; Furst, D.O. The structure of the sarcomeric M band: Localization of defined domains of myomesin, M-protein, and the 250-kD carboxy-terminal region of titin by immunoelectron microscopy. J. Cell Biol. 1996, 134, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Gautel, M. Cytoskeletal protein kinases: Titin and its relations in mechanosensing. Pflug. Arch. 2011, 462, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Puchner, E.M.; Alexandrovich, A.; Kho, A.L.; Hensen, U.; Schafer, L.V.; Brandmeier, B.; Grater, F.; Grubmuller, H.; Gaub, H.E.; Gautel, M. Mechanoenzymatics of titin kinase. Proc. Natl. Acad. Sci. USA 2008, 105, 13385–13390. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Xiang, F.; Yakovenko, A.; Vihola, A.; Hackman, P.; Rostkova, E.; Kristensen, J.; Brandmeier, B.; Franzen, G.; Hedberg, B.; et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science 2005, 308, 1599–1603. [Google Scholar] [CrossRef]
- Bogomolovas, J.; Gasch, A.; Simkovic, F.; Rigden, D.J.; Labeit, S.; Mayans, O. Titin kinase is an inactive pseudokinase scaffold that supports MuRF1 recruitment to the sarcomeric M-line. Open Biol. 2014, 4, 140041. [Google Scholar] [CrossRef]
- Arber, S.; Hunter, J.J.; Ross, J., Jr.; Hongo, M.; Sansig, G.; Borg, J.; Perriard, J.C.; Chien, K.R.; Caroni, P. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell 1997, 88, 393–403. [Google Scholar] [CrossRef]
- Knoll, R.; Hoshijima, M.; Hoffman, H.M.; Person, V.; Lorenzen-Schmidt, I.; Bang, M.L.; Hayashi, T.; Shiga, N.; Yasukawa, H.; Schaper, W.; et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 2002, 111, 943–955. [Google Scholar] [CrossRef]
- Knoll, R.; Kostin, S.; Klede, S.; Savvatis, K.; Klinge, L.; Stehle, I.; Gunkel, S.; Kotter, S.; Babicz, K.; Sohns, M.; et al. A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circ. Res. 2010, 106, 695–704. [Google Scholar] [CrossRef]
- Witt, C.C.; Witt, S.H.; Lerche, S.; Labeit, D.; Back, W.; Labeit, S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J. 2008, 27, 350–360. [Google Scholar] [CrossRef]
- Chauveau, C.; Bonnemann, C.G.; Julien, C.; Kho, A.L.; Marks, H.; Talim, B.; Maury, P.; Arne-Bes, M.C.; Uro-Coste, E.; Alexandrovich, A.; et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum. Mol. Genet. 2014, 23, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.; Pawałowska, M.; Lubin, J.; Markowska, J. Signalling pathways in endometrial cancer. Contemp. Oncol. 2014, 18, 143–148. [Google Scholar]
- Yang, C.-Y.; Chen, C.-S.; Yiang, G.-T.; Cheng, Y.-L.; Yong, S.-B.; Wu, M.-Y.; Li, C.-J. New Insights into the Immune Molecular Regulation of the Pathogenesis of Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2018, 19, 588. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, T.; Matsui, T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 2011, 17, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, A.; Cornu, M.; Cybulski, N.; Polak, P.; Betz, C.; Trapani, F.; Terracciano, L.; Heim, M.H.; Ruegg, M.A.; Hall, M.N. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012, 15, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, I.; Sato, K.; Izumiya, Y.; Schiekofer, S.; Ito, M.; Liao, R.; Colucci, W.S.; Walsh, K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Investig. 2005, 115, 2108–2118. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, I.; Walsh, K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006, 20, 3347–3365. [Google Scholar] [CrossRef]
- Michael, A.; Haq, S.; Chen, X.; Hsich, E.; Cui, L.; Walters, B.; Shao, Z.; Bhattacharya, K.; Kilter, H.; Huggins, G.; et al. Glycogen synthase kinase-3beta regulates growth, calcium homeostasis, and diastolic function in the heart. J. Biol. Chem. 2004, 279, 21383–21393. [Google Scholar] [CrossRef] [PubMed]
- Lal, H.; Ahmad, F.; Woodgett, J.; Force, T. The GSK-3 family as therapeutic target for myocardial diseases. Circ. Res. 2015, 116, 138–149. [Google Scholar] [CrossRef]
- Cheng, H.; Woodgett, J.; Maamari, M.; Force, T. Targeting GSK-3 family members in the heart: A very sharp double-edged sword. J. Mol. Cell. Cardiol. 2011, 51, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, B.L.; Konhilas, J.P.; Luczak, E.D.; Leinwand, L.A. Soy diet worsens heart disease in mice. J. Clin. Investig. 2006, 116, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Luckey, S.W.; Walker, L.A.; Smyth, T.; Mansoori, J.; Messmer-Kratzsch, A.; Rosenzweig, A.; Olson, E.N.; Leinwand, L.A. The role of Akt/GSK-3beta signaling in familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2009, 46, 739–747. [Google Scholar] [CrossRef]
- Narula, J.; Kolodgie, F.D.; Virmani, R. Apoptosis and cardiomyopathy. Curr. Opin. Cardiol. 2000, 15, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.K.; Kwon, H.M.; Byun, K.H.; Kim, D.; Choi, E.Y.; Kang, T.S.; Kang, S.M.; Chun, K.J.; Jang, Y.; Kim, H.S.; et al. Apoptosis in dilated cardiomyopathy. Korean J. Intern. Med. 2000, 15, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Schaper, J.; Lorenz-Meyer, S.; Suzuki, K. The role of apoptosis in dilated cardiomyopathy. Herz 1999, 24, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Communal, C.; Singh, K.; Pimentel, D.R.; Colucci, W.S. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation 1998, 98, 1329–1334. [Google Scholar] [CrossRef]
- Geng, Y.-J.; Ishikawa, Y.; Vatner Dorothy, E.; Wagner Thomas, E.; Bishop Sanford, P.; Vatner Stephen, F.; Homcy Charles, J. Apoptosis of Cardiac Myocytes in Gsα Transgenic Mice. Circ. Res. 1999, 84, 34–42. [Google Scholar] [CrossRef]
- Iwai-Kanai, E.; Hasegawa, K.; Araki, M.; Kakita, T.; Morimoto, T.; Sasayama, S. α- and β-Adrenergic Pathways Differentially Regulate Cell Type–Specific Apoptosis in Rat Cardiac Myocytes. Circulation 1999, 100, 305–311. [Google Scholar] [CrossRef]
- Teiger, E.; Than, V.D.; Richard, L.; Wisnewsky, C.; Tea, B.S.; Gaboury, L.; Tremblay, J.; Schwartz, K.; Hamet, P. Apoptosis in pressure overload-induced heart hypertrophy in the rat. J. Clin. Investig. 1996, 97, 2891–2897. [Google Scholar] [CrossRef] [PubMed]
- Narula, N.; Zaragoza, M.V.; Sengupta, P.P.; Li, P.; Haider, N.; Verjans, J.; Waymire, K.; Vannan, M.; Wallace, D.C. Adenine nucleotide translocase 1 deficiency results in dilated cardiomyopathy with defects in myocardial mechanics, histopathological alterations, and activation of apoptosis. JACC Cardiovasc. Imaging 2011, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Machackova, J.; Barta, J.; Dhalla, N.S. Myofibrillar remodeling in cardiac hypertrophy, heart failure and cardiomyopathies. Can. J. Cardiol. 2006, 22, 953–968. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Gürtl, B.; Kratky, D.; Guelly, C.; Zhang, L.; Gorkiewicz, G.; Das, S.K.; Tamilarasan, K.P.; Hoefler, G. Apoptosis and fibrosis are early features of heart failure in an animal model of metabolic cardiomyopathy. Int. J. Exp. Pathol. 2009, 90, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Narula, J.; Haider, N.; Virmani, R.; DiSalvo, T.G.; Kolodgie, F.D.; Hajjar, R.J.; Schmidt, U.; Semigran, M.J.; Dec, G.W.; Khaw, B.A. Apoptosis in myocytes in end-stage heart failure. N. Engl. J. Med. 1996, 335, 1182–1189. [Google Scholar] [CrossRef]
- Olivetti, G.; Abbi, R.; Quaini, F.; Kajstura, J.; Cheng, W.; Nitahara, J.A.; Quaini, E.; Di Loreto, C.; Beltrami, C.A.; Krajewski, S.; et al. Apoptosis in the failing human heart. N. Engl. J. Med. 1997, 336, 1131–1141. [Google Scholar] [CrossRef]
- Krown, K.A.; Page, M.T.; Nguyen, C.; Zechner, D.; Gutierrez, V.; Comstock, K.L.; Glembotski, C.C.; Quintana, P.J.; Sabbadini, R.A. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J. Clin. Investig. 1996, 98, 2854–2865. [Google Scholar] [CrossRef]
- Torre-Amione, G.; Kapadia, S.; Lee, J.; Durand, J.B.; Bies, R.D.; Young, J.B.; Mann, D.L. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation 1996, 93, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Akyurek, O.; Akyurek, N.; Sayin, T.; Dincer, I.; Berkalp, B.; Akyol, G.; Ozenci, M.; Oral, D. Association between the severity of heart failure and the susceptibility of myocytes to apoptosis in patients with idiopathic dilated cardiomyopathy. Int. J. Cardiol. 2001, 80, 29–36. [Google Scholar] [CrossRef]
- Gonzalez, A.; Fortuno, M.A.; Querejeta, R.; Ravassa, S.; Lopez, B.; Lopez, N.; Diez, J. Cardiomyocyte apoptosis in hypertensive cardiomyopathy. Cardiovasc. Res. 2003, 59, 549–562. [Google Scholar] [CrossRef]
- Agrotis, A.; Kalinina, N.; Bobik, A. Transforming growth factor-beta, cell signaling and cardiovascular disorders. Curr. Vasc. Pharmacol. 2005, 3, 55–61. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef]
- Khan, R.; Sheppard, R. Fibrosis in heart disease: Understanding the role of transforming growth factor-beta in cardiomyopathy, valvular disease and arrhythmia. Immunology 2006, 118, 10–24. [Google Scholar] [CrossRef]
- Sanderson, J.E.; Lai, K.B.; Shum, I.O.; Wei, S.; Chow, L.T. Transforming growth factor-beta(1) expression in dilated cardiomyopathy. Heart 2001, 86, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Borger, M.A.; Williams, W.G.; Weisel, R.D.; Mickle, D.A.G.; Wigle, E.D.; Li, R.-K. Regional overexpression of insulin-like growth factor-I and transforming growth factor-β1 in the myocardium of patients with hypertrophic obstructive cardiomyopathy. J. Thorac. Cardiovasc. Surg. 2002, 123, 89–95. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef]
- Lijnen, P.J.; Petrov, V.V.; Fagard, R.H. Induction of cardiac fibrosis by transforming growth factor-beta(1). Mol. Genet. Metab. 2000, 71, 418–435. [Google Scholar] [CrossRef]
- Courcelles, M.; Fremin, C.; Voisin, L.; Lemieux, S.; Meloche, S.; Thibault, P. Phosphoproteome dynamics reveal novel ERK1/2 MAP kinase substrates with broad spectrum of functions. Mol. Syst. Biol. 2013, 9, 669. [Google Scholar] [CrossRef] [PubMed]
- Olivetti, G.; Melissari, M.; Balbi, T.; Quaini, F.; Cigola, E.; Sonnenblick, E.H.; Anversa, P. Myocyte cellular hypertrophy is responsible for ventricular remodelling in the hypertrophied heart of middle aged individuals in the absence of cardiac failure. Cardiovasc. Res. 1994, 28, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Anversa, P.; Kajstura, J.; Olivetti, G. Myocyte death in heart failure. Curr. Opin. Cardiol. 1996, 11, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Joyce, J.; Margulies, K.B.; Tsuda, T. Enhanced Bioactive Myocardial Transforming Growth Factor-β in Advanced Human Heart Failure. Circ. J. 2014, 78, 2711–2718. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
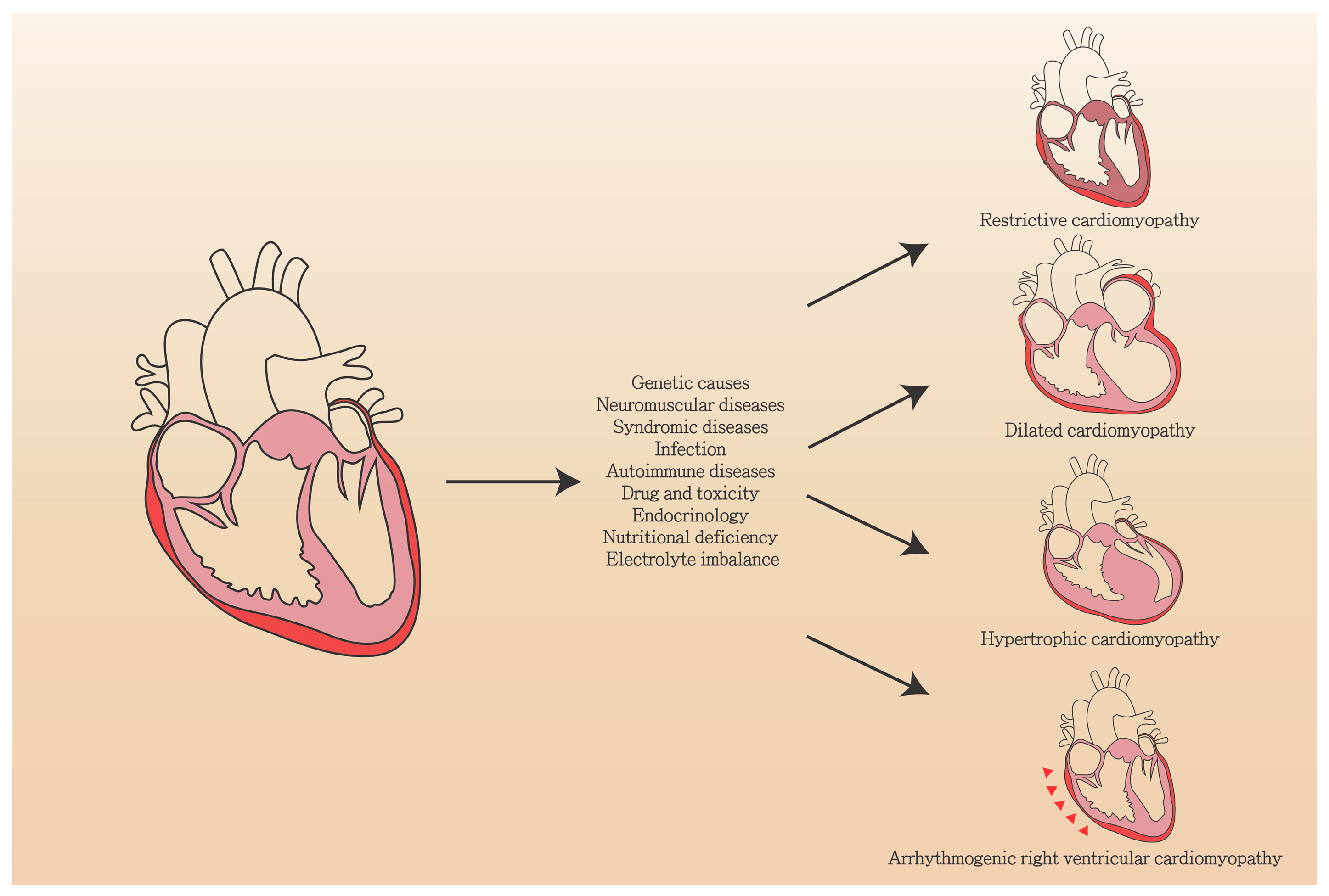
| Etiology | Features |
|---|---|
| Gene mutation | LMNA, MYH7, TNNT2, RBM20, TTN, BAG3, SCN5A, FLNC, TPM1, PLN, TNNC1, TNNI3, EYA4, NEBL, NEXN, ANKRD1, CSRP3, DES, SGCD, ILK, PDLIM3, ACTC1, ABCC9, CRYAB, ACTN2, TCAP, LDB3, VCL, LAMA4, MYH6, MYBPC3, MYPN, CTF1, DEM, DNAJC19, DSC2, DSP, EMD, FHL2, FKTN, FOXD4, LAMP2, PSEN1, PSEN2, SDHA, SYNE1, SYNE2, TAZ, TCAP, TMPO, TPM1, DMD |
| Neuromuscular diseases | Duchenne muscular dystrophy, Becker muscular dystrophy (Mutation in dystrophin gene) |
| Syndromic diseases | Mitochondrial dysfunction, Tafazzin |
| Infection | Virus (parvovirus B19, HPV6, HIV), bacteria, Fungus, parasite |
| Autoimmune diseases | Polymyositis/dermatomyositis, Churg-Strauss syndrome, Wegener’s granulomatosis, Systemic lupus erythematosus, Sarcoidosis, Giant cell myocarditis |
| Drug and toxicity | Ethanol, Cocaine, Amphetamines, Iron overload, Antineoplastic drugs (paclitaxel, hypomethylating agents, monoclonal antibodies, tyrosine kinase inhibitors), Psychiatric drugs (Clozapine, Olanzapine, Chlorpromazine, Risperidone, Lithium, Methylphenidate, Tricyclic antidepressants, Phenothiazines) |
| Endocrinology | Hypothyroidism, Hyperthyroidism, Cushing’s disease, Addison disease, Pheochromocytoma, Stress, Diabetes mellitus |
| Nutritional deficiency | Thiamine, Zinc, Copper, Selenium |
| Electrolyte imbalance | Hypocalcemia, Hypophosphatemia |
| Cardiomyopathy | Gene Mutation |
|---|---|
| Dilated cardiomyopathy | LMNA, MYH7, TNNT2, RBM20, TTN, BAG3, SCN5A, FLNC, TPM1, PLN, TNNC1, TNNI3, EYA4, NEBL, NEXN, ANKRD1, CSRP3, DES, SGCD, ILK, PDLIM3, ACTC1, ABCC9, CRYAB, ACTN2, TCAP, LDB3, VCL, LAMA4, MYH6, MYBPC3, MYPN, CTF1, DEM, DNAJC19, DSC2, DSP, EMD, FHL2, FKTN, FOXD4, LAMP2, PSEN1, PSEN2, SDHA, SYNE1, SYNE2, TAZ, TCAP, TMPO |
| Hypertrophic cardiomyopathy | TTN, MYH7, MYH6, MYL2, MYL3, MYBPC3, MYLK2, TNNT2, TNNI3, TPM1, ACTC, TNNC1, LDB3, CSRP3, TCAP, VCL, ACTN2, MYOZ2, NEXN, JPH2, PLN, ANKRD1, CAV3, COX15, CRYAB, GLA, LAMP2, PRKAG2 |
| Restrictive cardiomyopathy | TNNI3, TNNT2, TPN1, MYH7, DES, MYBPC3, LMNA, FLNC, LAMP2 |
| Arrhythmogenic right ventricular cardiomyopathy | DSP, PKP2, DSG2, DSC2, JUP, TMEM43, CTNNA3, DES, LMNA, PLN, RYR2, TGFB3, TTN, SCN5A, ARVC3, ARVC6, |
| Left ventricular non-compaction cardiomyopathy | LDB3, DTNA, TAZ, LMNA, NKX2-5, MYH7, ACTC, TNNT2, TNN13, MYBPC3, SCN5A, SNTA1, PRDM16, TPM1, NSD1, RPS6KA3, PMP22, CASQ2, MYH6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.-J.; Chen, C.-S.; Yiang, G.-T.; Tsai, A.P.-Y.; Liao, W.-T.; Wu, M.-Y. Advanced Evolution of Pathogenesis Concepts in Cardiomyopathies. J. Clin. Med. 2019, 8, 520. https://doi.org/10.3390/jcm8040520
Li C-J, Chen C-S, Yiang G-T, Tsai AP-Y, Liao W-T, Wu M-Y. Advanced Evolution of Pathogenesis Concepts in Cardiomyopathies. Journal of Clinical Medicine. 2019; 8(4):520. https://doi.org/10.3390/jcm8040520
Chicago/Turabian StyleLi, Chia-Jung, Chien-Sheng Chen, Giou-Teng Yiang, Andy Po-Yi Tsai, Wan-Ting Liao, and Meng-Yu Wu. 2019. "Advanced Evolution of Pathogenesis Concepts in Cardiomyopathies" Journal of Clinical Medicine 8, no. 4: 520. https://doi.org/10.3390/jcm8040520
APA StyleLi, C.-J., Chen, C.-S., Yiang, G.-T., Tsai, A. P.-Y., Liao, W.-T., & Wu, M.-Y. (2019). Advanced Evolution of Pathogenesis Concepts in Cardiomyopathies. Journal of Clinical Medicine, 8(4), 520. https://doi.org/10.3390/jcm8040520