Epithelial to Mesenchymal Transition and Cell Biology of Molecular Regulation in Endometrial Carcinogenesis





Abstract
1. Introduction
2. Clinical Feature of Endometrial Carcinoma
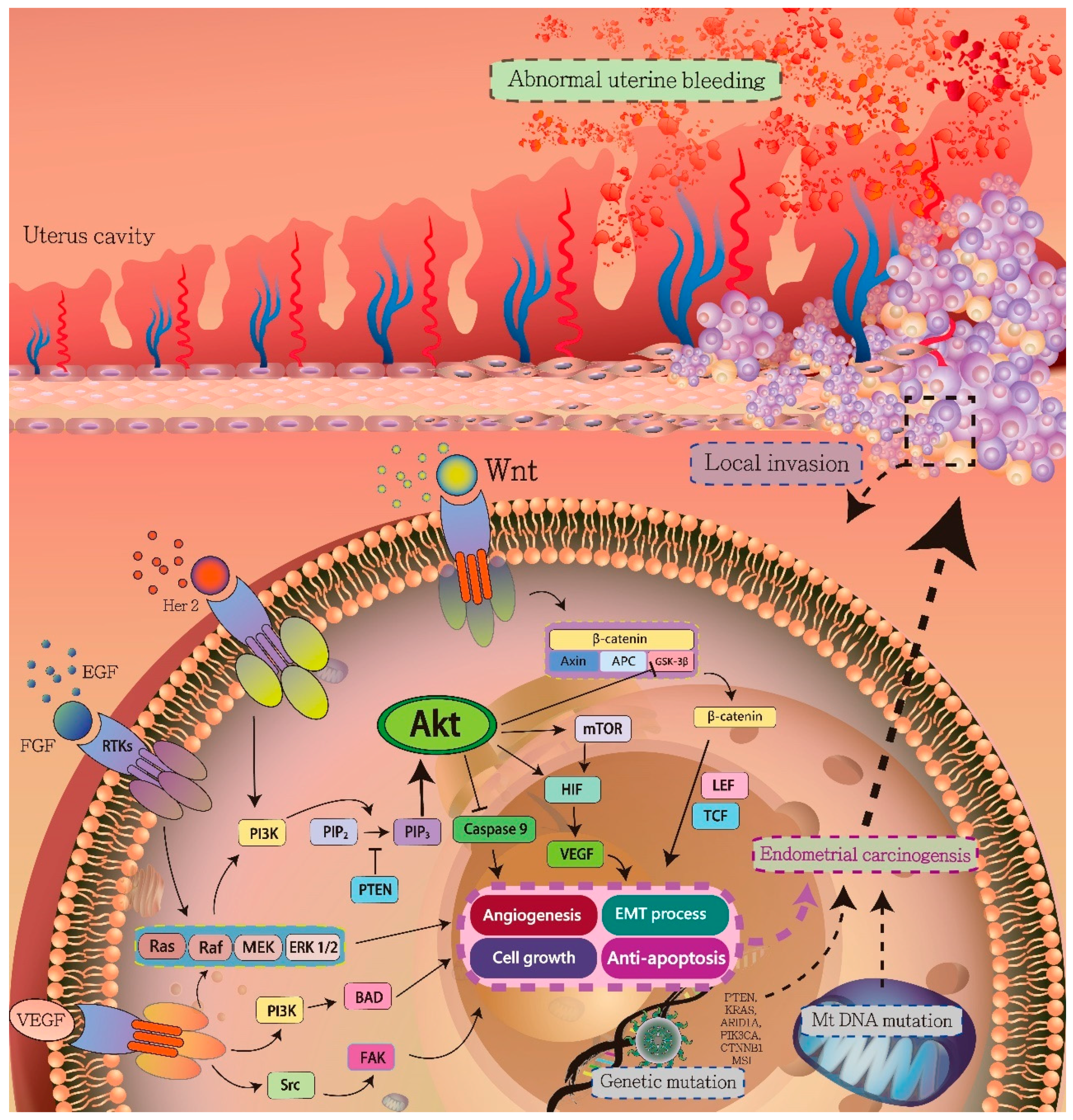
3. Signaling Pathways in Endometrial Carcinogenesis
3.1. HIF-1α/VEGF Axis
3.2. PI3K/AKT Pathway
3.3. Ras/Raf/MEK/ERK Signaling Pathway
3.4. Wnt/β-Catenin Signaling Pathway
3.5. Insulin/IGF-I Signaling Pathway
4. Epithelial-Mesenchymal Transition (EMT) Signaling Pathways in Endometrial Carcinoma
4.1. Classification of EMT
4.2. Effect of Tumor Microenvironment on TGF-β-Mediated EMT
4.3. Transcriptional Regulators in TGF-β and EMT
4.4. Estrogen Signaling in EMT
4.5. Progesterone Signaling in EMT
5. Conclusions
Funding
Conflicts of Interest
References
- Braun, M.M.; Overbeek-Wager, E.A.; Grumbo, R.J. Diagnosis and Management of Endometrial Cancer. Am. Fam. Phys. 2016, 93, 468–474. [Google Scholar]
- Sorosky, J.I. Endometrial cancer. Obstet. Gynecol. 2012, 120, 383–397. [Google Scholar] [CrossRef]
- Sanderson, P.A.; Critchley, H.O.; Williams, A.R.; Arends, M.J.; Saunders, P.T. New concepts for an old problem: The diagnosis of endometrial hyperplasia. Hum. Reprod. Update 2017, 23, 232–254. [Google Scholar] [CrossRef] [PubMed]
- Wik, E.; Raeder, M.B.; Krakstad, C.; Trovik, J.; Birkeland, E.; Hoivik, E.A.; Mjos, S.; Werner, H.M.; Mannelqvist, M.; Stefansson, I.M.; et al. Lack of estrogen receptor-alpha is associated with epithelial-mesenchymal transition and PI3K alterations in endometrial carcinoma. Clin. Cancer Res. 2013, 19, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar]
- Chang, Y.S.; Huang, H.D.; Yeh, K.T.; Chang, J.G. Identification of novel mutations in endometrial cancer patients by whole-exome sequencing. Int. J. Oncol. 2017, 50, 1778–1784. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W.J.; Hoivik, E.A.; Halle, M.K.; Taylor-Weiner, A.; Cherniack, A.D.; Berg, A.; Holst, F.; Zack, T.I.; Werner, H.M.; Staby, K.M.; et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat. Genet. 2016, 48, 848–855. [Google Scholar] [CrossRef]
- Lee, Y.C.; Lheureux, S.; Oza, A.M. Treatment strategies for endometrial cancer: Current practice and perspective. Curr. Opin. Obstet. Gynecol. 2017, 29, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Pisani, P.; Ferlay, J. Global cancer statistics. Cancer J. Clin. 1999, 49, 33–64. [Google Scholar] [CrossRef]
- Amant, F.; Moerman, P.; Neven, P.; Timmerman, D.; Van Limbergen, E.; Vergote, I. Endometrial cancer. Lancet 2005, 366, 491–505. [Google Scholar] [CrossRef]
- Kimura, T.; Kamiura, S.; Yamamoto, T.; Seino-Noda, H.; Ohira, H.; Saji, F. Abnormal uterine bleeding and prognosis of endometrial cancer. Int. J. Gynaecol. Obstet. 2004, 85, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Seebacher, V.; Schmid, M.; Polterauer, S.; Hefler-Frischmuth, K.; Leipold, H.; Concin, N.; Reinthaller, A.; Hefler, L. The presence of postmenopausal bleeding as prognostic parameter in patients with endometrial cancer: A retrospective multi-center study. Bmc Cancer 2009, 9, 460. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Ding, D.-C.; Chu, T.-Y.; Hong, M.-K. Endocervical Cavity Anomaly Mimicking the Uterine Cavity and Delaying Diagnosis of Endometrial Adenocarcinoma: A Case Report. Reports 2018, 1, 5. [Google Scholar] [CrossRef]
- Gupta, D. Clinical Behavior and Treatment of Endometrial Cancer. Adv. Exp. Med. Biol. 2017, 943, 47–74. [Google Scholar]
- McAlpine, J.N.; Temkin, S.M.; Mackay, H.J. Endometrial cancer: Not your grandmother’s cancer. Cancer 2016, 122, 2787–2798. [Google Scholar] [CrossRef] [PubMed]
- MacKintosh, M.L.; Derbyshire, A.E.; McVey, R.J.; Bolton, J.; Nickkho-Amiry, M.; Higgins, C.L.; Kamieniorz, M.; Pemberton, P.W.; Kirmani, B.H.; Ahmed, B.; et al. The impact of obesity and bariatric surgery on circulating and tissue biomarkers of endometrial cancer risk. Int. J. Cancer 2019, 144, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, I.; Gentry-Maharaj, A.; Burnell, M.; Manchanda, R.; Singh, N.; Sharma, A.; Ryan, A.; Seif, M.W.; Amso, N.N.; Turner, G.; et al. Sensitivity of transvaginal ultrasound screening for endometrial cancer in postmenopausal women: A case-control study within the UKCTOCS cohort. Lancet. Oncol. 2011, 12, 38–48. [Google Scholar] [CrossRef]
- Steiner, E.; Juhasz-Bösz, I.; Emons, G.; Kölbl, H.; Kimmig, R.; Mallmann, P. Transvaginal Ultrasound for Endometrial Carcinoma Screening—Current Evidence-based Data. Geburtshilfe Frauenheilkd. 2012, 72, 1088–1091. [Google Scholar] [CrossRef]
- Bagaria, M.; Shields, E.; Bakkum-Gamez, J.N. Novel approaches to early detection of endometrial cancer. Curr. Opin. Obstet. Gynecol. 2017, 29, 40–46. [Google Scholar] [CrossRef]
- Connor, J.P.; Andrews, J.I.; Anderson, B.; Buller, R.E. Computed tomography in endometrial carcinoma. Obstet. Gynecol. 2000, 95, 692–696. [Google Scholar]
- Kitson, S.J.; Lindsay, J.; Sivalingam, V.N.; Lunt, M.; Ryan, N.A.J.; Edmondson, R.J.; Rutter, M.K.; Crosbie, E.J. The unrecognized burden of cardiovascular risk factors in women newly diagnosed with endometrial cancer: A prospective case control study. Gynecol. Oncol. 2018, 148, 154–160. [Google Scholar] [CrossRef]
- Winterhoff, B.; Konecny, G.E. Targeting fibroblast growth factor pathways in endometrial cancer. Curr. Probl. Cancer 2017, 41, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Creutzberg, C.; Amant, F.; Bosse, T.; Gonzalez-Martin, A.; Ledermann, J.; Marth, C.; Nout, R.; Querleu, D.; Mirza, M.R.; et al. ESMO-ESGO-ESTRO Consensus Conference on Endometrial Cancer: Diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, 16–41. [Google Scholar] [CrossRef] [PubMed]
- Grigsby, P.W.; Perez, C.A.; Kuten, A.; Simpson, J.R.; Garcia, D.M.; Camel, H.M.; Kao, M.S.; Galakatos, A.E. Clinical stage I endometrial cancer: Prognostic factors for local control and distant metastasis and implications of the new FIGO surgical staging system. Int. J. Radiat. Oncol. Biol. Phys. 1992, 22, 905–911. [Google Scholar] [CrossRef]
- Morrow, C.P.; Bundy, B.N.; Kurman, R.J.; Creasman, W.T.; Heller, P.; Homesley, H.D.; Graham, J.E. Relationship between surgical-pathological risk factors and outcome in clinical stage I and II carcinoma of the endometrium: A Gynecologic Oncology Group study. Gynecol. Oncol. 1991, 40, 55–65. [Google Scholar] [CrossRef]
- Lu, Z.; Chen, J. Introduction of WHO classification of tumours of female reproductive organs, fourth edition. Chin. J. Pathol. 2014, 43, 649–650. [Google Scholar]
- Cuevas, D.; Valls, J.; Gatius, S.; Roman-Canal, B.; Estaran, E.; Dorca, E.; Santacana, M.; Vaquero, M.; Eritja, N.; Velasco, A.; et al. Targeted sequencing with a customized panel to assess histological typing in endometrial carcinoma. Virchows Arch. Int. J. Pathol. 2019. [Google Scholar] [CrossRef]
- Huang, H.N.; Lin, M.C.; Tseng, L.H.; Chiang, Y.C.; Lin, L.I.; Lin, Y.F.; Huang, H.Y.; Kuo, K.T. Ovarian and endometrial endometrioid adenocarcinomas have distinct profiles of microsatellite instability, PTEN expression, and ARID1A expression. Histopathology 2015, 66, 517–528. [Google Scholar] [CrossRef]
- Rambau, P.F.; McIntyre, J.B.; Taylor, J.; Lee, S.; Ogilvie, T.; Sienko, A.; Morris, D.; Duggan, M.A.; McCluggage, W.G.; Kobel, M. Morphologic Reproducibility, Genotyping, and Immunohistochemical Profiling Do Not Support a Category of Seromucinous Carcinoma of the Ovary. Am. J. Surg. Pathol. 2017, 41, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Stelloo, E.; Bosse, T.; Nout, R.A.; MacKay, H.J.; Church, D.N.; Nijman, H.W.; Leary, A.; Edmondson, R.J.; Powell, M.E.; Crosbie, E.J.; et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Mod. Pathol. 2015, 28, 836–844. [Google Scholar] [CrossRef]
- Teer, J.K.; Yoder, S.; Gjyshi, A.; Nicosia, S.V.; Zhang, C.; Monteiro, A.N.A. Mutational heterogeneity in non-serous ovarian cancers. Sci. Rep. 2017, 7, 9728. [Google Scholar] [CrossRef]
- McConechy, M.K.; Ding, J.; Cheang, M.C.; Wiegand, K.; Senz, J.; Tone, A.; Yang, W.; Prentice, L.; Tse, K.; Zeng, T.; et al. Use of mutation profiles to refine the classification of endometrial carcinomas. J. Pathol. 2012, 228, 20–30. [Google Scholar] [CrossRef]
- McConechy, M.K.; Ding, J.; Senz, J.; Yang, W.; Melnyk, N.; Tone, A.A.; Prentice, L.M.; Wiegand, K.C.; McAlpine, J.N.; Shah, S.P.; et al. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Mod. Pathol. 2014, 27, 128–134. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.J.; Bell, D.W. The genomics and genetics of endometrial cancer. Adv. Genom. Genet. 2012, 2012, 33–47. [Google Scholar]
- Harada, H.; Kizaka-Kondoh, S.; Li, G.; Itasaka, S.; Shibuya, K.; Inoue, M.; Hiraoka, M. Significance of HIF-1-active cells in angiogenesis and radioresistance. Oncogene 2007, 26, 7508–7516. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Berg, A.; Fasmer, K.E.; Mauland, K.K.; Ytre-Hauge, S.; Hoivik, E.A.; Husby, J.A.; Tangen, I.L.; Trovik, J.; Halle, M.K.; Woie, K.; et al. Tissue and imaging biomarkers for hypoxia predict poor outcome in endometrial cancer. Oncotarget 2016, 7, 69844–69856. [Google Scholar] [CrossRef]
- Kato, H.; Inoue, T.; Asanoma, K.; Nishimura, C.; Matsuda, T.; Wake, N. Induction of human endometrial cancer cell senescence through modulation of HIF-1alpha activity by EGLN1. Int. J. Cancer 2006, 118, 1144–1153. [Google Scholar] [CrossRef]
- Seeber, L.M.; Horree, N.; van der Groep, P.; van der Wall, E.; Verheijen, R.H.; van Diest, P.J. Necrosis related HIF-1alpha expression predicts prognosis in patients with endometrioid endometrial carcinoma. BMC Cancer 2010, 10, 307. [Google Scholar] [CrossRef]
- Koyasu, S.; Kobayashi, M.; Goto, Y.; Hiraoka, M.; Harada, H. Regulatory mechanisms of hypoxia-inducible factor 1 activity: Two decades of knowledge. Cancer Sci. 2018, 109, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Seeber, L.M.; Zweemer, R.P.; Verheijen, R.H.; van Diest, P.J. Hypoxia-inducible factor-1 as a therapeutic target in endometrial cancer management. Obstet. Gynecol. Int. 2010, 2010, 580971. [Google Scholar] [CrossRef]
- Sivridis, E. Angiogenesis and endometrial cancer. Anticancer Res. 2001, 21, 4383–4388. [Google Scholar]
- Liu, H.; Zhang, Z.; Xiong, W.; Zhang, L.; Xiong, Y.; Li, N.; He, H.; Du, Y.; Liu, Y. Hypoxia-inducible factor-1alpha promotes endometrial stromal cells migration and invasion by upregulating autophagy in endometriosis. Reproduction 2017, 153, 809–820. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef]
- Gordon, L.K.; Kiyohara, M.; Fu, M.; Braun, J.; Dhawan, P.; Chan, A.; Goodglick, L.; Wadehra, M. EMP2 regulates angiogenesis in endometrial cancer cells through induction of VEGF. Oncogene 2013, 32, 5369–5376. [Google Scholar] [CrossRef]
- Ge, Q.L.; Liu, S.H.; Ai, Z.H.; Tao, M.F.; Ma, L.; Wen, S.Y.; Dai, M.; Liu, F.; Liu, H.S.; Jiang, R.Z.; et al. RelB/NF-kappaB links cell cycle transition and apoptosis to endometrioid adenocarcinoma tumorigenesis. Cell Death Dis. 2016, 7, e2402. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, A.; Oda, K.; Ikeda, Y.; Sone, K.; Fukuda, T.; Inaba, K.; Makii, C.; Enomoto, A.; Hosoya, N.; Tanikawa, M.; et al. PI3K/mTOR pathway inhibition overcomes radioresistance via suppression of the HIF1-alpha/VEGF pathway in endometrial cancer. Gynecol. Oncol. 2015, 138, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Kotowicz, B.; Fuksiewicz, M.; Jonska-Gmyrek, J.; Berezowska, A.; Radziszewski, J.; Bidzinski, M.; Kowalska, M. Clinical significance of pretreatment serum levels of VEGF and its receptors, IL- 8, and their prognostic value in type I and II endometrial cancer patients. PLoS ONE 2017, 12, e0184576. [Google Scholar] [CrossRef]
- Mahecha, A.M.; Wang, H. The influence of vascular endothelial growth factor-A and matrix metalloproteinase-2 and -9 in angiogenesis, metastasis, and prognosis of endometrial cancer. Oncotargets Ther. 2017, 10, 4617–4624. [Google Scholar] [CrossRef]
- Horree, N.; van Diest, P.J.; van der Groep, P.; Sie-Go, D.M.; Heintz, A.P. Hypoxia and angiogenesis in endometrioid endometrial carcinogenesis. Cell. Oncol. 2007, 29, 219–227. [Google Scholar]
- Sahoo, S.S.; Tanwar, P.S. VEGF-mTOR signaling links obesity and endometrial cancer. Oncoscience 2018, 5, 150–151. [Google Scholar]
- Zhang, J.; Song, H.; Lu, Y.; Chen, H.; Jiang, S.; Li, L. Effects of estradiol on VEGF and bFGF by Akt in endometrial cancer cells are mediated through the NF-kappaB pathway. Oncol. Rep. 2016, 36, 705–714. [Google Scholar] [CrossRef]
- Donoghue, J.F.; Lederman, F.L.; Susil, B.J.; Rogers, P.A. Lymphangiogenesis of normal endometrium and endometrial adenocarcinoma. Hum. Reprod. 2007, 22, 1705–1713. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Zaccarelli, E.; Caruso, D.; Vici, P.; Benedetti Panici, P.; Tomao, F. Targeting angiogenesis in endometrial cancer—New agents for tailored treatments. Exp. Opin. Investig. Drugs 2016, 25, 31–49. [Google Scholar] [CrossRef]
- Sahoo, S.S.; Lombard, J.M.; Ius, Y.; O’Sullivan, R.; Wood, L.G.; Nahar, P.; Jaaback, K.; Tanwar, P.S. Adipose-Derived VEGF-mTOR Signaling Promotes Endometrial Hyperplasia and Cancer: Implications for Obese Women. Mol. Cancer Res. 2018, 16, 309–321. [Google Scholar] [CrossRef]
- Bian, X.; Gao, J.; Luo, F.; Rui, C.; Zheng, T.; Wang, D.; Wang, Y.; Roberts, T.M.; Liu, P.; Zhao, J.J.; et al. PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP-PI3K inhibition but not PARP inhibition as monotherapy. Oncogene 2018, 37, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.; Pawałowska, M.; Lubin, J.; Markowska, J. Signalling pathways in endometrial cancer. Contemp. Oncol. 2014, 18, 143–148. [Google Scholar]
- Philip, C.A.; Laskov, I.; Beauchamp, M.C.; Marques, M.; Amin, O.; Bitharas, J.; Kessous, R.; Kogan, L.; Baloch, T.; Gotlieb, W.H.; et al. Inhibition of PI3K-AKT-mTOR pathway sensitizes endometrial cancer cell lines to PARP inhibitors. BMC Cancer 2017, 17, 638. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, A.; Cornu, M.; Cybulski, N.; Polak, P.; Betz, C.; Trapani, F.; Terracciano, L.; Heim, M.H.; Ruegg, M.A.; Hall, M.N. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012, 15, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, K.N.; Li, R.; Shao, R.; Chen, C. Activation of PI3K/Akt/mTOR pathway and dual inhibitors of PI3K and mTOR in endometrial cancer. Curr. Med. Chem. 2014, 21, 3070–3080. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Konno, Y.; Watari, H.; Hosaka, M.; Noguchi, M.; Sakuragi, N. The impact of microRNA-mediated PI3K/AKT signaling on epithelial-mesenchymal transition and cancer stemness in endometrial cancer. J. Transl. Med. 2014, 12, 231. [Google Scholar] [CrossRef] [PubMed]
- Oda, K. Targeting Ras-PI3K/mTOR pathway and the predictive biomarkers in endometrial cancer. Gan Kagaku Ryoho. Cancer Chemother. 2011, 38, 1084–1087. [Google Scholar]
- Salvesen, H.B.; Carter, S.L.; Mannelqvist, M.; Dutt, A.; Getz, G.; Stefansson, I.M.; Raeder, M.B.; Sos, M.L.; Engelsen, I.B.; Trovik, J.; et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc. Natl. Acad. Sci. USA 2009, 106, 4834–4839. [Google Scholar] [CrossRef]
- Dedes, K.J.; Wetterskog, D.; Ashworth, A.; Kaye, S.B.; Reis-Filho, J.S. Emerging therapeutic targets in endometrial cancer. Nat. Rev. Clin. Oncol. 2011, 8, 261–271. [Google Scholar] [CrossRef]
- Risinger, J.I.; Hayes, K.; Maxwell, G.L.; Carney, M.E.; Dodge, R.K.; Barrett, J.C.; Berchuck, A. PTEN mutation in endometrial cancers is associated with favorable clinical and pathologic characteristics. Clin. Cancer Res. 1998, 4, 3005–3010. [Google Scholar]
- Levine, D.A.; Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67. [Google Scholar]
- Eritja, N.; Yeramian, A.; Chen, B.J.; Llobet-Navas, D.; Ortega, E.; Colas, E.; Abal, M.; Dolcet, X.; Reventos, J.; Matias-Guiu, X. Endometrial Carcinoma: Specific Targeted Pathways. Adv. Exp. Med. Biol. 2017, 943, 149–207. [Google Scholar]
- Wang, L.E.; Ma, H.; Hale, K.S.; Yin, M.; Meyer, L.A.; Liu, H.; Li, J.; Lu, K.H.; Hennessy, B.T.; Li, X.; et al. Roles of genetic variants in the PI3K and RAS/RAF pathways in susceptibility to endometrial cancer and clinical outcomes. J. Cancer Res. Clin. Oncol. 2012, 138, 377–385. [Google Scholar] [CrossRef]
- Wu, Y.L.; Maachani, U.B.; Schweitzer, M.; Singh, R.; Wang, M.; Chang, R.; Souweidane, M.M. Dual Inhibition of PI3K/AKT and MEK/ERK Pathways Induces Synergistic Antitumor Effects in Diffuse Intrinsic Pontine Glioma Cells. Transl. Oncol. 2017, 10, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, R.; Chatani, Y.; Yamori, T.; Tsuruo, T.; Oka, H.; Yoshida, O.; Shimada, Y.; Ari-i, S.; Wada, H.; Fujimoto, J.; et al. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene 1999, 18, 813. [Google Scholar] [CrossRef] [PubMed]
- Alexander-Sefre, F.; Salvesen, H.B.; Ryan, A.; Singh, N.; Akslen, L.A.; MacDonald, N.; Wilbanks, G.; Jacobs, I.J. Molecular assessment of depth of myometrial invasion in stage I endometrial cancer: A model based on K-ras mutation analysis. Gynecol. Oncol. 2003, 91, 218–225. [Google Scholar] [CrossRef]
- Dobrzycka, B.; Terlikowski, S.J.; Mazurek, A.; Kowalczuk, O.; Niklińska, W.; Chyczewski, L.; Kulikowski, M. Mutations of the KRAS Oncogene in Endometrial Hyperplasia and Carcinoma. Folia Histochem Cytobiol. 2009, 47, 65–68. [Google Scholar] [CrossRef]
- Yang, Y. Wnt signaling in development and disease. Cell Biosci. 2012, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Kestler Hans, A.; Kühl, M. From individual Wnt pathways towards a Wnt signalling network. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 1333–1347. [Google Scholar] [CrossRef] [PubMed]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, T.H.; Planutis, K.; Tewari, K.S.; Holcombe, R.F. Role of canonical Wnt signaling in endometrial carcinogenesis. Expert Rev. Anticancer Ther. 2012, 12, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Coopes, A.; Henry, C.E.; Llamosas, E.; Ford, C.E. An update of Wnt signalling in endometrial cancer and its potential as a therapeutic target. Endocr. Relat. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, T.; Sakamoto, M.; Tsuda, H.; Maruyama, K.; Nozawa, S.; Hirohashi, S. Beta-catenin mutation in carcinoma of the uterine endometrium. Cancer Res. 1998, 58, 3526–3528. [Google Scholar] [PubMed]
- Saegusa, M.; Hashimura, M.; Yoshida, T.; Okayasu, I. Beta- Catenin mutations and aberrant nuclear expression during endometrial tumorigenesis. Br. J. Cancer 2001, 84, 209–217. [Google Scholar] [CrossRef]
- Liu, Y.; Meng, F.; Xu, Y.; Yang, S.; Xiao, M.; Chen, X.; Lou, G. Overexpression of Wnt7a is associated with tumor progression and unfavorable prognosis in endometrial cancer. Int. J. Gynecol. Cancer 2013, 23, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Gu, B.; Liu, N.; Nie, X.; Zhang, B.; Zhou, X.; Deng, M. Wnt/β-Catenin Pathway Regulates Cementogenic Differentiation of Adipose Tissue-Deprived Stem Cells in Dental Follicle Cell-Conditioned Medium. PLoS ONE 2014, 9, e93364. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef]
- Wang, Y.; Hanifi-Moghaddam, P.; Hanekamp, E.E.; Kloosterboer, H.J.; Franken, P.; Veldscholte, J.; van Doorn, H.C.; Ewing, P.C.; Kim, J.J.; Grootegoed, J.A.; et al. Progesterone Inhibition of Wnt/β-Catenin Signaling in Normal Endometrium and Endometrial Cancer. Clin. Cancer Res. 2009, 15, 5784–5793. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; van der Zee, M.; Fodde, R.; Blok, L.J. Wnt/Β-catenin and sex hormone signaling in endometrial homeostasis and cancer. Oncotarget 2010, 1, 674–684. [Google Scholar]
- Zhao, E.; Mu, Q. Phytoestrogen biological actions on Mammalian reproductive system and cancer growth. Sci. Pharm. 2011, 79, 1–20. [Google Scholar] [CrossRef]
- Hong, K.; Choi, Y. Role of estrogen and RAS signaling in repeated implantation failure. BMB Rep. 2018, 51, 225–229. [Google Scholar] [CrossRef]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef]
- Kim, J.J.; Kurita, T.; Bulun, S.E. Progesterone action in endometrial cancer, endometriosis, uterine fibroids, and breast cancer. Endocr. Rev. 2013, 34, 130–162. [Google Scholar] [CrossRef] [PubMed]
- De Marco, P.; Cirillo, F.; Vivacqua, A.; Malaguarnera, R.; Belfiore, A.; Maggiolini, M. Novel Aspects Concerning the Functional Cross-Talk between the Insulin/IGF-I System and Estrogen Signaling in Cancer Cells. Front. Endocrinol. 2015, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Celestino, J.; Schmandt, R.; McCampbell, A.S.; Urbauer, D.L.; Meyer, L.A.; Burzawa, J.K.; Huang, M.; Yates, M.S.; Iglesias, D.; et al. Chemopreventive effects of metformin on obesity-associated endometrial proliferation. Am. J. Obs. Gynecol. 2013, 209, 24e1–24e12. [Google Scholar] [CrossRef]
- De Marco, P.; Bartella, V.; Vivacqua, A.; Lappano, R.; Santolla, M.F.; Morcavallo, A.; Pezzi, V.; Belfiore, A.; Maggiolini, M. Insulin-like growth factor-I regulates GPER expression and function in cancer cells. Oncogene 2013, 32, 678–688. [Google Scholar] [CrossRef] [PubMed]
- McCampbell, A.S.; Broaddus, R.R.; Loose, D.S.; Davies, P.J. Overexpression of the insulin-like growth factor I receptor and activation of the AKT pathway in hyperplastic endometrium. Clin. Cancer Res. 2006, 12, 6373–6378. [Google Scholar] [CrossRef]
- Laskov, I.; Abou-Nader, P.; Amin, O.; Philip, C.A.; Beauchamp, M.C.; Yasmeen, A.; Gotlieb, W.H. Metformin Increases E-cadherin in Tumors of Diabetic Patients With Endometrial Cancer and Suppresses Epithelial-Mesenchymal Transition in Endometrial Cancer Cell Lines. Int. J. Gynecol. Cancer 2016, 26, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Kent, C.N.; Guttilla Reed, I.K. Regulation of epithelial-mesenchymal transition in endometrial cancer: Connecting PI3K, estrogen signaling, and microRNAs. Clin. Transl. Oncol. 2016, 18, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Makker, A.; Goel, M.M. Tumor progression, metastasis, and modulators of epithelial-mesenchymal transition in endometrioid endometrial carcinoma: An update. Endocr. Relat. Cancer 2016, 23, R85–R111. [Google Scholar] [CrossRef]
- Colas, E.; Pedrola, N.; Devis, L.; Ertekin, T.; Campoy, I.; Martinez, E.; Llaurado, M.; Rigau, M.; Olivan, M.; Garcia, M.; et al. The EMT signaling pathways in endometrial carcinoma. Clin. Transl. Oncol. 2012, 14, 715–720. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef]
- Chiu, H.-C.; Wu, M.-Y.; Li, C.-H.; Huang, S.-C.; Yiang, G.-T.; Yen, H.-S.; Liu, W.-L.; Li, C.-J.; Kao, W.-Y. Epithelial-Mesenchymal Transition with Malignant Transformation Leading Multiple Metastasis from Disseminated Peritoneal Leiomyomatosis. J. Clin. Med. 2018, 7, 207. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Tian, M.; Schiemann, W.P. Deconstructing the mechanisms and consequences of TGF-beta-induced EMT during cancer progression. Cell Tissue Res. 2012, 347, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, K.S.; Omar, I.S.; Kwong, S.C.; Mohamed, Z.; Woo, Y.L.; Mat Adenan, N.A.; Chung, I. Cancer-associated fibroblasts promote endometrial cancer growth via activation of interleukin-6/STAT-3/c-Myc pathway. Am. J. Cancer Res. 2016, 6, 200–213. [Google Scholar]
- Sahoo, S.S.; Zhang, X.D.; Hondermarck, H.; Tanwar, P.S. The Emerging Role of the Microenvironment in Endometrial Cancer. Cancers 2018, 10, 408. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, K.S.; Tham, S.T.; Mohamed, Z.; Woo, Y.L.; Mat Adenan, N.A.; Chung, I. Cancer-associated fibroblasts promote proliferation of endometrial cancer cells. PLoS ONE 2013, 8, e68923. [Google Scholar] [CrossRef]
- Teng, F.; Tian, W.Y.; Wang, Y.M.; Zhang, Y.F.; Guo, F.; Zhao, J.; Gao, C.; Xue, F.X. Cancer-associated fibroblasts promote the progression of endometrial cancer via the SDF-1/CXCR4 axis. J. Hematol. Oncol. 2016, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Franco, O.E.; Shaw, A.K.; Strand, D.W.; Hayward, S.W. Cancer associated fibroblasts in cancer pathogenesis. Semin. Cell Dev. Biol. 2010, 21, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, W.; Sun, X.; Lin, Y.; Chen, W. Cancer-associated fibroblasts induce epithelial-mesenchymal transition through secreted cytokines in endometrial cancer cells. Oncol. Lett. 2018, 15, 5694–5702. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef]
- Papageorgis, P.; Stylianopoulos, T. Role of TGFβ in regulation of the tumor microenvironment and drug delivery (review). Int. J. Oncol. 2015, 46, 933–943. [Google Scholar] [CrossRef]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Muinelo-Romay, L.; Colas, E.; Barbazan, J.; Alonso-Alconada, L.; Alonso-Nocelo, M.; Bouso, M.; Curiel, T.; Cueva, J.; Anido, U.; Forteza, J.; et al. High-risk endometrial carcinoma profiling identifies TGF-beta1 as a key factor in the initiation of tumor invasion. Mol. Cancer Ther. 2011, 10, 1357–1366. [Google Scholar] [CrossRef]
- Parekh, T.V.; Gama, P.; Wen, X.; Demopoulos, R.; Munger, J.S.; Carcangiu, M.L.; Reiss, M.; Gold, L.I. Transforming growth factor beta signaling is disabled early in human endometrial carcinogenesis concomitant with loss of growth inhibition. Cancer Res. 2002, 62, 2778–2790. [Google Scholar]
- Lei, X.; Wang, L.; Yang, J.; Sun, L.Z. TGFbeta signaling supports survival and metastasis of endometrial cancer cells. Cancer Manag. Res. 2009, 2009, 15–24. [Google Scholar]
- Yu, L.; Hu, R.; Sullivan, C.; Swanson, R.J.; Oehninger, S.; Sun, Y.P.; Bocca, S. MFGE8 regulates TGF-beta-induced epithelial mesenchymal transition in endometrial epithelial cells in vitro. Reproduction 2016, 152, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Bokhari, A.A.; Lee, L.R.; Raboteau, D.; Hamilton, C.A.; Maxwell, G.L.; Rodriguez, G.C.; Syed, V. Progesterone inhibits endometrial cancer invasiveness by inhibiting the TGFbeta pathway. Cancer Prev. Res. 2014, 7, 1045–1055. [Google Scholar] [CrossRef]
- Tanaka, Y.; Terai, Y.; Kawaguchi, H.; Fujiwara, S.; Yoo, S.; Tsunetoh, S.; Takai, M.; Kanemura, M.; Tanabe, A.; Ohmichi, M. Prognostic impact of EMT (epithelial-mesenchymal-transition)-related protein expression in endometrial cancer. Cancer Biol. Ther. 2013, 14, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen signaling multiple pathways to impact gene transcription. Curr. Genom. 2006, 7, 497–508. [Google Scholar] [CrossRef]
- Pietras, R.J.; Márquez-Garbán, D.C. Membrane-Associated Estrogen Receptor Signaling Pathways in Human Cancers. Clin. Cancer Res. 2007, 13, 4672–4676. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.E.; Tarulli, G.A.; Serandour, A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone receptor modulates ERα action in breast cancer. Nature 2015, 523, 313. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Thiel, K.W.; Leslie, K.K. Progesterone: The ultimate endometrial tumor suppressor. Trends Endocrinol. Metab. 2011, 22, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.X.; Wei, L.H.; Tu, Z.; Sun, P.M.; Wang, J.L.; Zhao, D.; Li, X.P.; Tang, J.M. 17 beta-estradiol activates PI3K/Akt signaling pathway by estrogen receptor (ER)-dependent and ER-independent mechanisms in endometrial cancer cells. J. Steroid Biochem. Mol. Biol. 2006, 99, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zhao, M.; Wang, T.; Zhang, G. Upregulation of estrogen receptor mediates migration, invasion and proliferation of endometrial carcinoma cells by regulating the PI3K/AKT/mTOR pathway. Oncol. Rep. 2014, 31, 1175–1182. [Google Scholar] [CrossRef]
- Revankar, C.M.; Mitchell, H.D.; Field, A.S.; Burai, R.; Corona, C.; Ramesh, C.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. Synthetic Estrogen Derivatives Demonstrate the Functionality of Intracellular GPR30. ACS Chem. Biol. 2007, 2, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Petrie, W.K.; Dennis, M.K.; Hu, C.; Dai, D.; Arterburn, J.B.; Smith, H.O.; Hathaway, H.J.; Prossnitz, E.R. G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth. Obstet. Gynecol. Int. 2013, 2013, 472720. [Google Scholar] [CrossRef]
- Ehrlich, C.E.; Young, P.C.; Stehman, F.B.; Sutton, G.P.; Alford, W.M. Steroid receptors and clinical outcome in patients with adenocarcinoma of the endometrium. Am. J. Obstet. Gynecol. 1988, 158, 796–807. [Google Scholar] [CrossRef]
- Jeon, Y.T.; Park, I.A.; Kim, Y.B.; Kim, J.W.; Park, N.H.; Kang, S.B.; Lee, H.P.; Song, Y.S. Steroid receptor expressions in endometrial cancer: Clinical significance and epidemiological implication. Cancer Lett. 2006, 239, 198–204. [Google Scholar] [CrossRef]
- Thigpen, J.T.; Brady, M.F.; Alvarez, R.D.; Adelson, M.D.; Homesley, H.D.; Manetta, A.; Soper, J.T.; Given, F.T. Oral medroxyprogesterone acetate in the treatment of advanced or recurrent endometrial carcinoma: A dose-response study by the Gynecologic Oncology Group. J. Clin. Oncol. 1999, 17, 1736–1744. [Google Scholar] [CrossRef]
- Hanekamp, E.E.; Kuhne, E.C.; Smid-Koopman, E.; de Ruiter, P.E.; Chadha-Ajwani, S.; Brinkmann, A.O.; Burger, C.W.; Grootegoed, J.A.; Huikeshoven, F.J.; Blok, L.J. Loss of progesterone receptor may lead to an invasive phenotype in human endometrial cancer. Eur. J. Cancer 2002, 38, S71–S72. [Google Scholar] [CrossRef]
- Van der Horst, P.H.; Wang, Y.; Vandenput, I.; Kuhne, L.C.; Ewing, P.C.; van Ijcken, W.F.; van der Zee, M.; Amant, F.; Burger, C.W.; Blok, L.J. Progesterone inhibits epithelial-to-mesenchymal transition in endometrial cancer. PLoS ONE 2012, 7, e30840. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| When T | When N | When M | FIGO Stage |
|---|---|---|---|
| T1 | N0 | M0 | I |
| T1a | N0 | M0 | IA |
| T1b | N0 | M0 | IB |
| T2 | N0 | M0 | II |
| T3 | N0 | M0 | III |
| T3a | N0 | M0 | IIIA |
| T3b | N0 | M0 | IIIB |
| T1-3 | N1/N1mi/N1a | M0 | IIIC1 |
| T1-3 | N2/N2mi/N2a | M0 | IIIC2 |
| T4 | Any N | M0 | IVA |
| Any T | Any N | M1 | IVB |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, H.-C.; Li, C.-J.; Yiang, G.-T.; Tsai, A.P.-Y.; Wu, M.-Y. Epithelial to Mesenchymal Transition and Cell Biology of Molecular Regulation in Endometrial Carcinogenesis. J. Clin. Med. 2019, 8, 439. https://doi.org/10.3390/jcm8040439
Chiu H-C, Li C-J, Yiang G-T, Tsai AP-Y, Wu M-Y. Epithelial to Mesenchymal Transition and Cell Biology of Molecular Regulation in Endometrial Carcinogenesis. Journal of Clinical Medicine. 2019; 8(4):439. https://doi.org/10.3390/jcm8040439
Chicago/Turabian StyleChiu, Hsiao-Chen, Chia-Jung Li, Giou-Teng Yiang, Andy Po-Yi Tsai, and Meng-Yu Wu. 2019. "Epithelial to Mesenchymal Transition and Cell Biology of Molecular Regulation in Endometrial Carcinogenesis" Journal of Clinical Medicine 8, no. 4: 439. https://doi.org/10.3390/jcm8040439
APA StyleChiu, H.-C., Li, C.-J., Yiang, G.-T., Tsai, A. P.-Y., & Wu, M.-Y. (2019). Epithelial to Mesenchymal Transition and Cell Biology of Molecular Regulation in Endometrial Carcinogenesis. Journal of Clinical Medicine, 8(4), 439. https://doi.org/10.3390/jcm8040439