PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers


Abstract
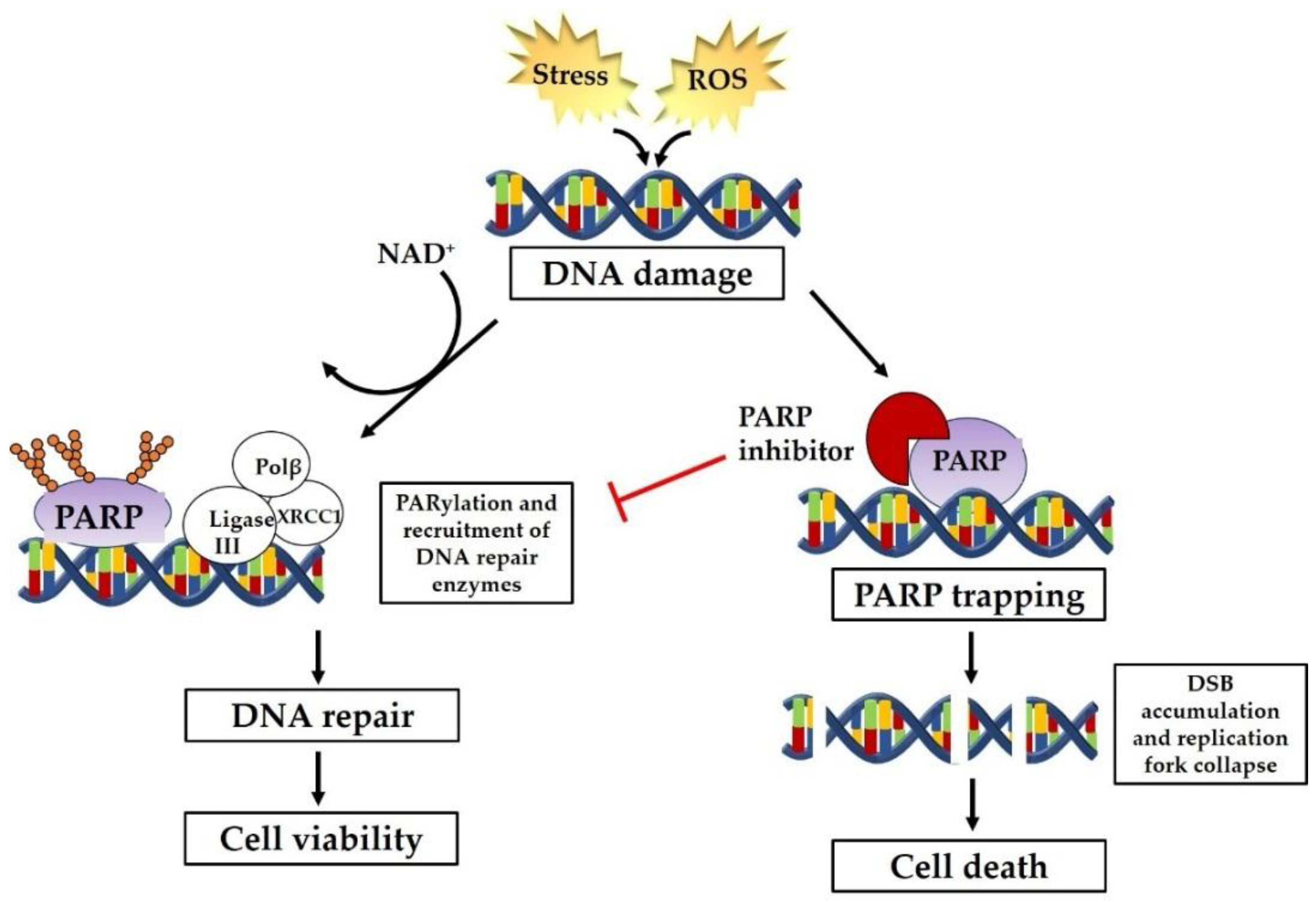
1. Introduction
2. PARP Inhibitors as Therapeutic Intervention
2.1. Selective Cytotoxicity of PARP Inhibitors
3. BRCAness and Homologous Recombination Deficiency (HRD)
4. Predictive Biomarkers in Breast Cancer
4.1. Sequencing-Based Methods
5. PARP Inhibitors as a Single Agent
6. Combination Strategies with PARP Inhibitors
7. Clinical Development of PARP Inhibitors in Breast Cancer
7.1. Veliparib (ABT-888)—AbbVie
7.1.1. Monotherapy
7.1.2. Combination Strategies
7.2. Niraparib (MK4827)—Tesaro
7.2.1. Monotherapy
7.2.2. Combination Strategies
7.3. Olaparib (AZD2281, Ku-0059436, Lynparza)—AstraZeneca
7.3.1. Monotherapy
7.3.2 Combination Strategies

{kind=link}
{kind=link}
| Therapeutic Strategy; Phase | Patient Population; Number of Breast Patients | RP2D | Results | Grade 3–5 Adverse Events | Identifier; References |
|---|---|---|---|---|---|
| Olaparib with carboplatin; I | BRCA1/2 mutation, sporadic TNBC; 8 | Olaparib 400 mg PO BID days 1–7, carboplatin AUC5 | CR, 23 months (2.4%, 1/8); PR, 10 months (75%, 6/8); SD, 14 months (12.5%, 1/8) | Neutropenia, thrombo-cytopenia, anemia | NCT01445418; [101] |
| Olaparib with carboplatin; I | TNBC; 10 (4 BRCA-mutant) | Olaparib 200 mg PO BID for 7 days; carboplatin AUC4 q21d | CR, 32 months (10%, 1/10); PR, ~9 months (30%, 3/10) | Neutropenia, anemia | NCT01237067; [102] |
| Olaparib with cisplatin; I | Metastatic, BRCA-mutant | Intermittent olaparib 50 mg PO BID days 1–5; cisplatin 60 mg/m2 | ORR (71%, 12/19) | Neutropenia, anemia, lipase elevation | NCT00782574; [96] |
| Olaparib with paclitaxel; I | Metastatic TNBC; 19 | Olaparib 200 mg PO BID; weekly paclitaxel, 3 weeks of 4-week cycle | PR (37%, 7/19); SD ≥ weeks (32%, 6/19) | Neutropenia | NCT00707707; [103] |
| Olaparib with cediranib; I | Recurrent TNBC; 8 | Olaparib 200 mg PO BID; Cediranib 30 mg PO QD | No CR or PR; SD > 24 weeks (25%, 2/8) | Hypertension, fatigue | NCT01116648; [100] |
7.4. Rucaparib (AG-014699, PF-01367338)—Clovis Oncology
7.4.1. Monotherapy
7.4.2. Combination Strategies
7.5. Talazoparib (BMN-673)—Pfizer, BioMarin, Medivation
Monotherapy
8. Acquired Resistance to PARP Inhibitors
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| aNHEJ | alternative non-homologous end-joining |
| BER | base excision repair |
| BID | bis in die, twice a day |
| BRCA1/2 | breast cancer type 1/type 2 susceptibility protein |
| CNA | copy number alterations |
| CAT | catalytic domain (of PARP-1) |
| CBR | clinical benefit rate |
| CGH | comparative genomic hybridization |
| CR | complete response |
| DLT | dose-limiting toxicities |
| DNMTi | DNA methyltransferase inhibitor |
| DSB | double-strand break |
| EMI1 | early mitotic inhibitor 1 |
| ER | estrogen receptor |
| EZH2 | enhancer of zeste homolog 2 |
| gBRCAm | germline BRCA mutation-associated |
| HD | helical domain |
| HER2 | human epidermal growth factor receptor 2 |
| HRD | homologous recombination deficiency/homologous recombination-deficient |
| HR | hormone receptor |
| HRR | homologous recombination repair |
| IRIF | ionizing radiation induced foci |
| IV | intravenous |
| LOH | loss of heterozygosity |
| LST | large-scale state transitions |
| miRNA | micro RNA |
| NAD+ | nicotinamide adenine dinucleotide |
| NHEJ | non-homologous end-joining |
| ORR | objective response rate |
| PARP | poly (ADP-ribose) polymerase |
| PARPi | poly (ADP-ribose) polymerase inhibitor |
| pCR | pathological complete response |
| PCT | physician’s choice of chemotherapy |
| PFS | progression-free survival |
| PO | per os, by mouth |
| PR | progesterone receptor |
| PRC2 | polycomb repressive complex 2 |
| PRR | partial response rate |
| QD | quaque die, once a day |
| RER | ribonucleotide excision repair |
| ROS | reactive oxygen species |
| RP2D | recommended phase 2 dose |
| SD | stable disease |
| SNP | single-nucleotide polymorphism |
| SSA | single-strand annealing |
| TAI | telomeric allelic imbalance |
| TNBC | triple negative breast cancer |
| TOP1 | topoisomerase 1 |
References
- Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; MacIntyre, M.F.; Allen, C.; Hansen, G.; Woodbrook, R.; Wolfe, C.; et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar] [CrossRef]
- Weigelt, B.; Geyer, F.C.; Reis-Filho, J.S. Histological Types of Breast Cancer: How Special Are They? Mol. Oncol. 2010, 4, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Arnedos, M.; Bihan, C.; Delaloge, S.; Andre, F. Triple-Negative Breast Cancer: Are We Making Headway at Least? Ther. Adv. Med. Oncol. 2012, 4, 195–210. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative Group. Effects of Chemotherapy and Hormonal Therapy for Early Breast Cancer on Recurrence and 15-Year Survival: An Overview of the Randomised Trials. Lancet 2005, 365, 1687–1717. [Google Scholar] [CrossRef]
- Cui, X.; Schiff, R.; Arpino, G.; Osborne, C.K.; Lee, A.V. Biology of Progesterone Receptor Loss in Breast Cancer and Its Implications for Endocrine Therapy. J. Clin. Oncol. 2005, 23, 7721–7735. [Google Scholar] [CrossRef] [PubMed]
- Kuukasjarvi, T.; Kononen, J.; Helin, H.; Holli, K.; Isola, J. Loss of Estrogen Receptor in Recurrent Breast Cancer Is Associated with Poor Response to Endocrine Therapy. J. Clin. Oncol. 1996, 14, 2584–2589. [Google Scholar] [CrossRef]
- Sharma, P.; Klemp, J.R.; Kimler, B.F.; Mahnken, J.D.; Geier, L.J.; Khan, Q.J.; Elia, M.; Connor, C.S.; McGinness, M.K.; Mammen, J.M.; et al. Germline BRCA Mutation Evaluation in a Prospective Triple-Negative Breast Cancer Registry: Implications for Hereditary Breast And/or Ovarian Cancer Syndrome Testing. Breast Cancer Res. Treat. 2014, 145, 707–714. [Google Scholar] [CrossRef]
- Lips, E.; Mulder, L.; Oonk, A.; Van Der Kolk, L.; Hogervorst, F.; Imholz, A.; Wesseling, J.; Rodenhuis, S.; Nederlof, P. Triple-Negative Breast Cancer: BRCAness and Concordance of Clinical Features with BRCA1-Mutation Carriers. Br. J. Cancer 2013, 108, 2172. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917. [Google Scholar] [CrossRef]
- Matulonis, U.A. PARP Inhibitors in BRCA-Related Ovarian Cancer—And Beyond! Available online: http://www.ascopost.com/issues/november-25-2017/parp-inhibitors-in-brca-related-ovarian-cancer-and-beyond/ (accessed on 8 June 2018).
- The U.S. Food and Drug Administration. FDA News Release: FDA Approves First Treatment for Breast Cancer with a Certain Inherited Genetic Mutation. Available online: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm592347.htm (accessed on 12 April 2018).
- The U.S. Food and Drug Administration. FDA News Release: FDA Approves Talazoparib for gBRCAm HER2-Negative Locally Advanced or Metastatic Breast Cancer. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm623540.htm (accessed on 11 January 2019).
- Amé, J.-C.; Spenlehauer, C.; de Murcia, G. The PARP Superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef]
- Wei, H.; Yu, X. Functions of PARylation in DNA Damage Repair Pathways. Genom. Proteom. Bioinform. 2016, 14, 131–139. [Google Scholar] [CrossRef]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-Ribosyl)ation Reactions in the Regulation of Nuclear Functions. Biochem. J. 1999, 342 Pt 2, 249–268. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Kraus, W.L. The PARP Side of the Nucleus: Molecular Actions, Physiological Outcomes, and Clinical Targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef]
- Michels, J.; Vitale, I.; Saparbaev, M.; Castedo, M.; Kroemer, G. Predictive Biomarkers for Cancer Therapy with PARP Inhibitors. Oncogene 2014, 33, 3894. [Google Scholar] [CrossRef] [PubMed]
- Shall, S.; de Murcia, G. Poly(ADP-Ribose) Polymerase-1: What Have We Learned from the Deficient Mouse Model? Mutat. Res. 2000, 460, 1–15. [Google Scholar] [CrossRef]
- Langelier, M.-F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural Basis for DNA Damage-Dependent poly(ADP-Ribosyl)ation by Human PARP-1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Gibson, B.A.; Kraus, W.L. New Insights into the Molecular and Cellular Functions of Poly (ADP-Ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411. [Google Scholar] [CrossRef]
- Kraus, W.L. Transcriptional Control by PARP-1: Chromatin Modulation, Enhancer-Binding, Coregulation, and Insulation. Curr. Opin. Cell Biol. 2008, 20, 294–302. [Google Scholar] [CrossRef]
- Beck, C.; Robert, I.; Reina-San-Martin, B.; Schreiber, V.; Dantzer, F. Poly (ADP-Ribose) Polymerases in Double-Strand Break Repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014, 329, 18–25. [Google Scholar] [CrossRef]
- Langelier, M.-F.; Riccio, A.A.; Pascal, J.M. PARP-2 and PARP-3 Are Selectively Activated by 5′ Phosphorylated DNA Breaks through an Allosteric Regulatory Mechanism Shared with PARP-1. Nucleic Acids Res. 2014, 42, 7762–7775. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, D.J. DNA Double Strand Break Repair via Non-Homologous End-Joining. Transl. Cancer Res. 2013, 2, 130. [Google Scholar]
- Bétermier, M.; Bertrand, P.; Lopez, B.S. Is Non-Homologous End-Joining Really an Inherently Error-Prone Process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef] [PubMed]
- Simsek, D.; Jasin, M. Alternative End-Joining Is Suppressed by the Canonical NHEJ Component Xrcc4-Ligase IV during Chromosomal Translocation Formation. Nat. Struct. Mol. Biol. 2010, 17, 410. [Google Scholar] [CrossRef]
- Chen, J.-M.; Cooper, D.N.; Férec, C.; Kehrer-Sawatzki, H.; Patrinos, G.P. Genomic Rearrangements in Inherited Disease and Cancer. Semin. Cancer Biol. 2010, 20, 222–233. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a Trap to Kill Cancer Cells: PARP Inhibitors and Their Mechanisms of Action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef] [PubMed]
- Dawicki-McKenna, J.M.; Langelier, M.-F.; DeNizio, J.E.; Riccio, A.A.; Cao, C.D.; Karch, K.R.; McCauley, M.; Steffen, J.D.; Black, B.E.; Pascal, J.M. PARP-1 Activation Requires Local Unfolding of an Autoinhibitory Domain. Mol. Cell 2015, 60, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Langelier, M.-F.; Zandarashvili, L.; Aguiar, P.M.; Black, B.E.; Pascal, J.M. NAD+ Analog Reveals PARP-1 Substrate-Blocking Mechanism and Allosteric Communication from Catalytic Center to DNA-Binding Domains. Nat. Commun. 2018, 9, 844. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic Dissection of PARP1 Trapping and the Impact on in Vivo Tolerability and Efficacy of PARP Inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477. [Google Scholar] [CrossRef]
- Hopkins, T.A.; Ainsworth, W.B.; Ellis, P.A.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Abraham, V.C.; Algire, M.A.; Shi, Y.; Olson, A.M.; et al. PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity Both in Cancer Cells and Healthy Bone Marrow. Mol. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Das, B.B.; Huang, S.N.; Murai, J.; Rehman, I.; Ame, J.-C.; Sengupta, S.; Das, S.K.; Majumdar, P.; Zhang, H.; Biard, D.; et al. PARP1-TDP1 Coupling for the Repair of Topoisomerase I-Induced DNA Damage. Nucleic Acids Res. 2014, 42, 4435–4449. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Zhang, Y.; Morris, J.; Ji, J.; Takeda, S.; Doroshow, J.H.; Pommier, Y. Rationale for Poly (ADP-Ribose) Polymerase (PARP) Inhibitors in Combination Therapy with Camptothecins or Temozolomide Based on PARP Trapping versus Catalytic Inhibition. J. Pharmacol. Exp. Ther. 2014, 349, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Murina, O.; Reijns, M.A.; Agathanggelou, A.; Challis, R.; Tarnauskaite, Z.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR Screens Identify Genomic Ribonucleotides as a Source of PARP-Trapping Lesions. Nature 2018, 559, 285. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.J.; Kruse, T.A.; Tan, Q.; Lænkholm, A.-V.; Bak, M.; Lykkesfeldt, A.E.; Sørensen, K.P.; Hansen, T.V.O.; Ejlertsen, B.; Gerdes, A.-M.; et al. Classifications within Molecular Subtypes Enables Identification of BRCA1/BRCA2 Mutation Carriers by RNA Tumor Profiling. PLoS ONE 2013, 8, e64268. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of Breast and Ovarian Cancers among BRCA1 and BRCA2 Mutation Carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Prev. Biomark. 2012, 21, 134–147. [Google Scholar] [CrossRef]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of’BRCAness’ in Sporadic Cancers. Nat. Rev. Cancer 2004, 4, 814. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering Signatures of Mutational Processes Operative in Human Cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of Somatic Mutations in 560 Breast Cancer Whole-Genome Sequences. Nature 2016, 534, 47. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A Mutational Signature Reveals Alterations Underlying Deficient Homologous Recombination Repair in Breast Cancer. Nat. Genet. 2017, 49, 1476. [Google Scholar] [CrossRef]
- Couch, F.J.; Hu, C.; Lilyquist, J.; Shimelis, H.; Akinhanmi, M.; Na, J.; Polley, E.C.; Hart, S.N.; McFarland, R.; LaDuca, H.; et al. Abstract S2-01: Breast Cancer Risks Associated with Mutations in Cancer Predisposition Genes Identified by Clinical Genetic Testing of 60,000 Breast Cancer Patients. In San Antonio Breast Cancer Symposium; Springer: San Antonio, TX, USA, 2016; Abstract S2–01. [Google Scholar]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 Defects with Genomic Scores Predictive of DNA Damage Repair Deficiency among Breast Cancer Subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef]
- Bergamaschi, A.; Kim, Y.H.; Wang, P.; Sørlie, T.; Hernandez-Boussard, T.; Lonning, P.E.; Tibshirani, R.; Børresen-Dale, A.-L.; Pollack, J.R. Distinct Patterns of DNA Copy Number Alteration Are Associated with Different Clinicopathological Features and Gene-Expression Subtypes of Breast Cancer. Genes Chromosomes Cancer 2006, 45, 1033–1040. [Google Scholar] [CrossRef]
- Carter, N.P. Methods and Strategies for Analyzing Copy Number Variation Using DNA Microarrays. Nat. Genet. 2007, 39 (Suppl. 7), S16–S21. [Google Scholar] [CrossRef]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N. Genomic Scars as Biomarkers of Homologous Recombination Deficiency and Drug Response in Breast and Ovarian Cancers. Breast Cancer Res. 2014, 16, 211. [Google Scholar] [CrossRef]
- Moskwa, P.; Buffa, F.M.; Pan, Y.; Panchakshari, R.; Gottipati, P.; Muschel, R.J.; Beech, J.; Kulshrestha, R.; Abdelmohsen, K.; Weinstock, D.M.; et al. miR-182-Mediated Downregulation of BRCA1 Impacts DNA Repair and Sensitivity to PARP Inhibitors. Mol. Cell 2011, 41, 210–220. [Google Scholar] [CrossRef]
- Tommasi, S.; Pinto, R.; Danza, K.; Pilato, B.; Palumbo, O.; Micale, L.; De Summa, S. miR-151-5p, Targeting Chromatin Remodeler SMARCA5, as a Marker for the BRCAness Phenotype. Oncotarget 2016, 7, 80363. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhou, W.; Cheng, C.-T.; Ren, X.; Somlo, G.; Fong, M.Y.; Chin, A.R.; Li, H.; Yu, Y.; Xu, Y.; et al. TGFbeta Induces’ BRCAness’ and Sensitivity to PARP Inhibition in Breast Cancer by Regulating DNA Repair Genes. Mol. Cancer Res. 2014, 12, 1597–1609. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.; Fiskus, W.; Choi, D.S.; Bhaskara, S.; Cerchietti, L.; Devaraj, S.G.; Shah, B.; Sharma, S.; Chang, J.C.; Melnick, A.M.; et al. Histone Deacetylase Inhibitor Treatment Induces “BRCAness” and Synergistic Lethality with PARP Inhibitor and Cisplatin against Human Triple Negative Breast Cancer Cells. Oncotarget 2014, 5, 5637. [Google Scholar] [CrossRef] [PubMed]
- Wiegmans, A.P.; Yap, P.-Y.; Ward, A.; Lim, Y.C.; Khanna, K.K. Differences in Expression of Key DNA Damage Repair Genes after Epigenetic-Induced BRCAness Dictate Synthetic Lethality with PARP1 Inhibition. Mol. Cancer Ther. 2015, 14, 2321–2331. [Google Scholar] [CrossRef] [PubMed]
- Oplustilova, L.; Wolanin, K.; Mistrik, M.; Korinkova, G.; Simkova, D.; Bouchal, J.; Lenobel, R.; Bartkova, J.; Lau, A.; O’Connor, M.J.; et al. Evaluation of Candidate Biomarkers to Predict Cancer Cell Sensitivity or Resistance to PARP-1 Inhibitor Treatment. Cell Cycle 2012, 11, 3837–3850. [Google Scholar] [CrossRef] [PubMed]
- Naipal, K.A.; Verkaik, N.S.; Ameziane, N.; van Deurzen, C.H.; Ter Brugge, P.; Meijers, M.; Sieuwerts, A.M.; Martens, J.; O’Connor, M.J.; Vrieling, H.; et al. Functional Ex Vivo Assay to Select Homologous Recombination Deficient Breast Tumors for PARP Inhibitor Treatment. Clin. Cancer Res. 2014, 20, 4816–4826. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enriquez, S.; Llop-Guevara, A.; Ibrahim, Y.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.; et al. RAD51 Foci as a Functional Biomarker of Homologous Recombination Repair and PARP Inhibitor Resistance in Germline BRCA-Mutated Breast Cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Graeser, M.K.; McCarthy, A.; Lord, C.J.; Savage, K.; Hills, M.; Salter, J.; Orr, N.; Parton, M.; Smith, I.E.; Reis-Filho, J.; et al. A Marker of Homologous Recombination Predicts Pathological Complete Response to Neoadjuvant Chemotherapy in Primary Breast Cancer. Clin. Cancer Res. 2010, 16, 6159–6168. [Google Scholar] [CrossRef]
- Roig, B.; Rodriguez-Balada, M.; Samino, S.; Lam, E.W.-F.; Guaita-Esteruelas, S.; Gomes, A.R.; Correig, X.; Borràs, J.; Yanes, O.; Gumà, J. Metabolomics Reveals Novel Blood Plasma Biomarkers Associated to the BRCA1-Mutated Phenotype of Human Breast Cancer. Sci. Rep. 2017, 7, 17831. [Google Scholar] [CrossRef] [PubMed]
- Abkevich, V.; Timms, K.; Hennessy, B.; Potter, J.; Carey, M.; Meyer, L.; Smith-McCune, K.; Broaddus, R.; Lu, K.; Chen, J.; et al. Patterns of Genomic Loss of Heterozygosity Predict Homologous Recombination Repair Defects in Epithelial Ovarian Cancer. Br. J. Cancer 2012, 107, 1776. [Google Scholar] [CrossRef] [PubMed]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef]
- Telli, M.L.; Hellyer, J.; Audeh, W.; Jensen, K.C.; Bose, S.; Timms, K.M.; Gutin, A.; Abkevich, V.; Peterson, R.N.; Neff, C.; et al. Homologous Recombination Deficiency (HRD) Status Predicts Response to Standard Neoadjuvant Chemotherapy in Patients with Triple-Negative or BRCA1/2 Mutation-Associated Breast Cancer. Breast Cancer Res. Treat. 2018, 168, 625–630. [Google Scholar] [CrossRef]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.-Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric Allelic Imbalance Indicates Defective DNA Repair and Sensitivity to DNA-Damaging Agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.A.; Longy, M.; et al. Ploidy and Large-Scale Genomic Instability Consistently Identify Basal-like Breast Carcinomas with BRCA1/2 Inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect Is a Predictor of BRCA1 and BRCA2 Deficiency Based on Mutational Signatures. Nat. Med. 2017, 23, 517. [Google Scholar] [CrossRef]
- Gross, E.; Tinteren, H.; Li, Z.; Raab, S.; Meul, C.; Avril, S.; Laddach, N.; Aubele, M.; Propping, C.; Gkazepis, A.; et al. Identification of BRCA1-like Triple-Negative Breast Cancers by Quantitative Multiplex-Ligation-Dependent Probe Amplification (MLPA) Analysis of BRCA1-Associated Chromosomal Regions: A Validation Study. BMC Cancer 2016, 16, 811. [Google Scholar] [CrossRef]
- Peng, G.; Lin, C.C.-J.; Mo, W.; Dai, H.; Park, Y.-Y.; Kim, S.M.; Peng, Y.; Mo, Q.; Siwko, S.; Hu, R.; et al. Genome-Wide Transcriptome Profiling of Homologous Recombination DNA Repair. Nat. Commun. 2014, 5, 3361. [Google Scholar] [CrossRef]
- Buchtel, K.M.; Postula, K.J.V.; Weiss, S.; Williams, C.; Pineda, M.; Weissman, S.M. FDA Approval of PARP Inhibitors and the Impact on Genetic Counseling and Genetic Testing Practices. J. Genet. Couns. 2018, 27, 131–139. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib Maintenance Treatment for Recurrent Ovarian Carcinoma after Response to Platinum Therapy (ARIEL3): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Nagahashi, M.; Shimada, Y.; Ichikawa, H.; Kameyama, H.; Takabe, K.; Okuda, S.; Wakai, T. Next Generation Sequencing-Based Gene Panel Tests for the Management of Solid Tumors. Cancer Sci. 2019, 110, 6. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational Landscape of Metastatic Cancer Revealed from Prospective Clinical Sequencing of 10,000 Patients. Nat. Med. 2017, 23, 703. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific Killing of BRCA2-Deficient Tumours with Inhibitors of Poly (ADP-Ribose) Polymerase. Nature 2005, 434, 913. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly (ADP)-Ribose Polymerase Inhibition: Frequent Durable Responses in BRCA Carrier Ovarian Cancer Correlating with Platinum-Free Interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’connor, M.J.; et al. Inhibition of Poly (ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents-a Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Fernandez-Capetillo, O.; Carrera, A.C. Nuclear Phosphoinositide 3-Kinase β Controls Double-Strand Break DNA Repair. Proc. Natl. Acad. Sci. USA 2010, 107, 7491–7496. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in Relapsed, Platinum-Sensitive High-Grade Ovarian Carcinoma (ARIEL2 Part 1): An International, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Pahuja, S.; Beumer, J.H.; Appleman, L.J.; Tawbi, H.A.; Stoller, R.G.; Lee, J.J.; Lin, Y.; Kiesel, B.; Yu, J.; Tan, A.R.; et al. Outcome of BRCA 1/2- Mutated (BRCA+) and Triple-Negative, BRCA Wild Type (BRCA-Wt) Breast Cancer Patients in a Phase I Study of Single-Agent Veliparib (V). J. Clin. Oncol. 2014, 32 (Suppl. 26), 135. [Google Scholar] [CrossRef]
- Appleman, L.J.; Beumer, J.H.; Jiang, Y.; Puhalla, S.; Lin, Y.; Owonikoko, T.K.; Harvey, R.D.; Stoller, R.; Pietro, D.P.; Tawbi, H.A.; et al. A Phase I Study of Veliparib (ABT-888) in Combination with Carboplatin and Paclitaxel in Advanced Solid Malignancies. J. Clin. Oncol. 2012, 30 (Suppl. 15), 3049. [Google Scholar]
- Tan, A.R.; Toppmeyer, D.; Stein, M.N.; Moss, R.A.; Gounder, M.; Lindquist, D.C.; Ji, J.J.; Chen, A.P.; Egorin, M.J.; Kiesel, B.; et al. Phase I Trial of Veliparib, (ABT-888), a poly(ADP-Ribose) Polymerase (PARP) Inhibitor, in Combination with Doxorubicin and Cyclophosphamide in Breast Cancer and Other Solid Tumors. J. Clin. Oncol. 2011, 29 (Suppl. 15), 3041. [Google Scholar] [CrossRef]
- Rodler, E.T.; Gralow, J.; Kurland, B.F.; Griffin, M.; Yeh, R.; Thompson, J.A.; Porter, P.; Swisher, E.M.; Gadi, V.K.; Korde, L.A.; et al. Phase I: Veliparib with Cisplatin (CP) and Vinorelbine (VNR) in Advanced Triple-Negative Breast Cancer (TNBC) And/or BRCA Mutation-Associated Breast Cancer. J. Clin. Oncol. 2014, 32 (Suppl. 15), 2569. [Google Scholar] [CrossRef]
- Wesolowski, R.; Zhao, M.; Geyer, S.M.; Lustberg, M.B.; Mrozek, E.; Layman, R.M.; Macrae, E.M.; Zhang, J.; Hall, N.; Schregel, K.; et al. Phase I Trial of the PARP Inhibitor Veliparib (V) in Combination with Carboplatin (C) in Metastatic Breast Cancer (MBC). J. Clin. Oncol. 2014, 32 (Suppl. 15), 1074. [Google Scholar] [CrossRef]
- Isakoff, S.J.; Overmoyer, B.; Tung, N.M.; Gelman, R.S.; Giranda, V.L.; Bernhard, K.M.; Habin, K.R.; Ellisen, L.W.; Winer, E.P.; Goss, P.E. A Phase II Trial of the PARP Inhibitor Veliparib (ABT888) and Temozolomide for Metastatic Breast Cancer. J. Clin. Oncol. 2010, 28 (Suppl. 15), 1019. [Google Scholar] [CrossRef]
- Loibl, S.; O’Shaughnessy, J.; Untch, M.; Sikov, W.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; von Minckwitz, G.; Maag, D.; et al. Addition of the PARP Inhibitor Veliparib plus Carboplatin or Carboplatin Alone to Standard Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer (BrighTNess): A Randomised, Phase 3 Trial. Lancet Oncol. 2018, 19, 497–509. [Google Scholar] [CrossRef]
- Jones, P.; Wilcoxen, K.; Rowley, M.; Toniatti, C. Niraparib: A Poly (ADP-Ribose) Polymerase (PARP) Inhibitor for the Treatment of Tumors with Defective Homologous Recombination. J. Med. Chem. 2015, 58, 3302–3314. [Google Scholar] [CrossRef]
- Jones, P.; Altamura, S.; Boueres, J.; Ferrigno, F.; Fonsi, M.; Giomini, C.; Lamartina, S.; Monteagudo, E.; Ontoria, J.M.; Orsale, M.V.; et al. Discovery of 2-4-[(3S)-Piperidin-3-Yl]phenyl-2H-Indazole-7-Carboxamide (MK-4827): A Novel Oral Poly (ADP-Ribose) Polymerase (PARP) Inhibitor Efficacious in BRCA-1 and-2 Mutant Tumors. J. Med. Chem. 2009, 52, 7170–7185. [Google Scholar] [CrossRef] [PubMed]
- The U.S. Food and Drug Administration. FDA Approves Maintenance Treatment for Recurrent Epithelial Ovarian, Fallopian Tube or Primary Peritoneal Cancers. Available online: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm548948.htm (accessed on 15 May 2018).
- Sandhu, S.K.; Schelman, W.R.; Wilding, G.; Moreno, V.; Baird, R.D.; Miranda, S.; Hylands, L.; Riisnaes, R.; Forster, M.; Omlin, A.; et al. The Poly (ADP-Ribose) Polymerase Inhibitor Niraparib (MK4827) in BRCA Mutation Carriers and Patients with Sporadic Cancer: A Phase 1 Dose-Escalation Trial. Lancet Oncol. 2013, 14, 882–892. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral Poly (ADP-Ribose) Polymerase Inhibitor Olaparib in Patients with BRCA1 or BRCA2 Mutations and Advanced Breast Cancer: A Proof-of-Concept Trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in Patients with Recurrent High-Grade Serous or Poorly Differentiated Ovarian Carcinoma or Triple-Negative Breast Cancer: A Phase 2, Multicentre, Open-Label, Non-Randomised Study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib Monotherapy in Patients with Advanced Cancer and a Germline BRCA1/2 Mutation. J. Clin. Oncol. 2014, 33, 244–250. [Google Scholar] [CrossRef]
- Balmana, J.; Tung, N.; Isakoff, S.; Grana, B.; Ryan, P.; Saura, C.; Lowe, E.; Frewer, P.; Winer, E.; Baselga, J.; et al. Phase I Trial of Olaparib in Combination with Cisplatin for the Treatment of Patients with Advanced Breast, Ovarian and Other Solid Tumors. Ann. Oncol. 2014, 25, 1656–1663. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Djamgoz, M.B. Triple Negative Breast Cancer: Emerging Therapeutic Modalities and Novel Combination Therapies. Cancer Treat. Rev. 2018, 62, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Osoegawa, A.; Gills, J.J.; Kawabata, S.; Dennis, P.A. Rapamycin Sensitizes Cancer Cells to Growth Inhibition by the PARP Inhibitor Olaparib. Oncotarget 2017, 8, 87044. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.; Wulf, G.M.; Birrer, M.J.; Westin, S.N.; Quy, P.; Whalen, C.; Aghajanian, C.; Solit, D.B.; Mills, G.B.; Cantley, L.; et al. Phase I Study of Oral BKM120 and Oral Olaparib for High-Grade Serous Ovarian Cancer (HGSC) or Triple-Negative Breast Cancer (TNBC). J. Clin. Oncol. 2014, 32 (Suppl. 15), 2510. [Google Scholar] [CrossRef]
- Liu, J.F.; Tolaney, S.M.; Birrer, M.; Fleming, G.F.; Buss, M.K.; Dahlberg, S.E.; Lee, H.; Whalen, C.; Tyburski, K.; Winer, E.; et al. A Phase 1 Trial of the PARP Inhibitor Olaparib (AZD2281) in Combination with the Anti-Angiogenic Cediranib (AZD2171) in Recurrent Epithelial Ovarian or Triple-Negative Breast Cancer. Eur. J. Cancer 2013, 49, 2972. [Google Scholar] [CrossRef]
- Lee, J.-M.; Hays, J.L.; Annunziata, C.M.; Noonan, A.M.; Minasian, L.; Zujewski, J.A.; Yu, M.; Gordon, N.; Ji, J.; Sissung, T.M.; et al. Phase I/Ib Study of Olaparib and Carboplatin in BRCA1 or BRCA2 Mutation-Associated Breast or Ovarian Cancer with Biomarker Analyses. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Chiou, V.L.; Annunziata, C.; Lipkowitz, S.; Minasian, L.; Gordon, N.; Yu, M.; Steinberg, S.; Houston, N.; Kohn, E.; Lee, J. Abstract CT326: Pharmacokinetic/pharmacodynamic Study of Sequence Specificity of the PARP Inhibitor, Olaparib and Carboplatin in Recurrent Women’s Cancers. In Proceedings of the AACR 106th Annual Meeting 2015, Philadelphia, PA, USA, 18–22 April 2015. AM2015–CT326. [Google Scholar]
- Dent, R.A.; Lindeman, G.J.; Clemons, M.; Wildiers, H.; Chan, A.; McCarthy, N.J.; Singer, C.F.; Lowe, E.S.; Watkins, C.L.; Carmichael, J. Phase I Trial of the Oral PARP Inhibitor Olaparib in Combination with Paclitaxel for First-or Second-Line Treatment of Patients with Metastatic Triple-Negative Breast Cancer. Breast Cancer Res. 2013, 15, R88. [Google Scholar] [CrossRef]
- Patsouris, A.; Vicier, C.; Campion, L.; Gouraud, W.; Jimenez, M.; Pezzella, V.; Loehr, A.; Raponi, M.; Bieche, I.; Callens, C.; et al. An Open-Label, Phase II Study of Rucaparib, a PARP Inhibitor, in HER2- Metastatic Breast Cancer Patients with High Genomic Loss of Heterozygosity: RUBY. J. Clin. Oncol. 2017, 35 (Suppl. 15). [Google Scholar] [CrossRef]
- Drew, Y.; Ledermann, J.; Hall, G.; Rea, D.; Glasspool, R.; Highley, M.; Jayson, G.; Sludden, J.; Murray, J.; Jamieson, D.; et al. Phase 2 Multicentre Trial Investigating Intermittent and Continuous Dosing Schedules of the Poly (ADP-Ribose) Polymerase Inhibitor Rucaparib in Germline BRCA Mutation Carriers with Advanced Ovarian and Breast Cancer. Br. J. Cancer 2016, 114, 723. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.; Thomas, H.; Berry, P.; Kyle, S.; Patterson, M.; Jones, C.; Los, G.; Hostomsky, Z.; Plummer, E.; Boddy, A.; et al. Tumour Cell Retention of Rucaparib, Sustained PARP Inhibition and Efficacy of Weekly as Well as Daily Schedules. Br. J. Cancer 2014, 110, 1977. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.H.; Evans, T.J.; Middleton, M.R.; Molife, L.R.; Spicer, J.; Dieras, V.; Roxburgh, P.; Giordano, H.; Jaw-Tsai, S.; Goble, S.; et al. A Phase I Study of Intravenous and Oral Rucaparib in Combination with Chemotherapy in Patients with Advanced Solid Tumours. Br. J. Cancer 2017, 116, 884. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Rehman, F.L.; Feng, Y.; Boshuizen, J.; Bajrami, I.; Elliott, R.; Wang, B.; Lord, C.J.; Post, L.E.; Ashworth, A. BMN 673, a Novel and Highly Potent PARP1/2 Inhibitor for the Treatment of Human Cancers with DNA Repair Deficiency. Clin. Cancer Res. 2013, 19, 5003–5015. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Rafii, S.; Ramanathan, R.K.; Mina, L.A.; Byers, L.A.; Chugh, R.; Goldman, J.W.; Sachdev, J.C.; Matei, D.E.; Wheler, J.J.; et al. Safety and Antitumor Activity of the PARP Inhibitor BMN673 in a Phase 1 Trial Recruiting Metastatic Small-Cell Lung Cancer (SCLC) and Germline BRCA-Mutation Carrier Cancer Patients. J. Clin. Oncol. 2014. [Google Scholar] [CrossRef]
- De Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017, 7, 620–629. [Google Scholar] [CrossRef]
- Turner, N.C.; Telli, M.L.; Rugo, H.S.; Mailliez, A.; Ettl, J.; Grischke, E.-M.; Mina, L.A.; Balmana Gelpi, J.; Fasching, P.A.; Hurvitz, S.A.; et al. Final Results of a Phase 2 Study of Talazoparib (TALA) Following Platinum or Multiple Cytotoxic Regimens in Advanced Breast Cancer Patients (pts) with Germline BRCA1/2 Mutations (ABRAZO). J. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Litton, J.; Scoggins, M.; Ramirez, D.; Murthy, R.; Whitman, G.; Hess, K.; Adrada, B.; Moulder, S.; Barcenas, C.; Valero, V.; et al. A Feasibility Study of Neoadjuvant Talazoparib for Operable Breast Cancer Patients with a Germline BRCA Mutation Demonstrates Marked Activity. NPJ Breast Cancer 2017, 3, 49. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Litton, J.; Rugo, H.S.; Ettl, J.; Hurvitz, S.; Gonçalves, A.; Lee, K.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. A Phase 3 Trial Comparing Talazoparib, an Oral PARP Inhibitor, to Physician’s Choice of Therapy in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. In Proceedings of the San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 5–9 December 2017; pp. 5–9. [Google Scholar]
- Ettl, J.; Quek, R.; Lee, K.-H.; Rugo, H.; Hurvitz, S.; Gonçalves, A.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.; Martin, M.; et al. Quality of Life with Talazoparib versus Physician’s Choice of Chemotherapy in Patients with Advanced Breast Cancer and Germline BRCA1/2 Mutation: Patient-Reported Outcomes from the EMBRACA Phase III Trial. Ann. Oncol. 2018, 29, 1939–1947. [Google Scholar] [CrossRef]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 Causes PARP Inhibitor Resistance in Brca1-Mutated Mouse Mammary Tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, L.O.; Rulten, S.L.; Cranston, A.N.; Odedra, R.; Brown, H.; Jaspers, J.E.; Jones, L.; Knights, C.; Evers, B.; Ting, A.; et al. The PARP Inhibitor AZD2461 Provides Insights into the Role of PARP3 Inhibition for Both Synthetic Lethality and Tolerability with Chemotherapy in Preclinical Models. Cancer Res. 2016, 76, 6084–6094. [Google Scholar]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 Loss Rescues BRCA1 Deficiency and Is Associated with Triple-Negative and BRCA-Mutated Breast Cancers. Nat. Struct. Mol. Biol. 2010, 17, 688. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.-T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 Regulates DSB Repair Using Rif1 to Control 5′ End Resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef]
- Xie, A.; Hartlerode, A.; Stucki, M.; Odate, S.; Puget, N.; Kwok, A.; Nagaraju, G.; Yan, C.; Alt, F.W.; Chen, J.; et al. Distinct Roles of Chromatin-Associated Proteins MDC1 and 53BP1 in Mammalian Double-Strand Break Repair. Mol. Cell 2007, 28, 1045–1057. [Google Scholar] [CrossRef]
- Li, M.; Cole, F.; Patel, D.S.; Misenko, S.M.; Her, J.; Malhowski, A.; Alhamza, A.; Zheng, H.; Baer, R.; Ludwig, T.; et al. 53BP1 Ablation Rescues Genomic Instability in Mice Expressing “RING-Less” BRCA1. EMBO Rep. 2016, 17, 1532–1541. [Google Scholar] [CrossRef]
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Marzio, A.; Puccini, J.; Kwon, Y.; Maverakis, N.K.; Arbini, A.; Sung, P.; Bar-Sagi, D.; Pagano, M. The F-Box Domain-Dependent Activity of EMI1 Regulates PARPi Sensitivity in Triple-Negative Breast Cancers. Mol. Cell 2019, 73, 224–237.e6. [Google Scholar] [CrossRef] [PubMed]
- Skaar, J.R.; Pagan, J.K.; Pagano, M. SCF Ubiquitin Ligase-Targeted Therapies. Nat. Rev. Drug Discov. 2014, 13, 889. [Google Scholar] [CrossRef]
- Skaar, J.R.; Pagan, J.K.; Pagano, M. Mechanisms and Function of Substrate Recruitment by F-Box Proteins. Nat. Rev. Mol. Cell Biol. 2013, 14, 369. [Google Scholar] [CrossRef] [PubMed]
- Barber, L.J.; Sandhu, S.; Chen, L.; Campbell, J.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Rodrigues, D.N.; Reis-Filho, J.S.; Moreno, V.; et al. Secondary Mutations in BRCA2 Associated with Clinical Resistance to a PARP Inhibitor. J. Pathol. 2013, 229, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Du, Y.; Nakai, K.; Ding, M.; Chang, S.; Hsu, J.; Yao, J.; Wei, Y.; Nie, L.; Jiao, S.; et al. EZH2 Contributes to the Response to PARP Inhibitors through Its PARP-Mediated Poly-ADP Ribosylation in Breast Cancer. Oncogene 2018, 37, 208. [Google Scholar] [CrossRef]
- Bae, W.K.; Yoo, K.H.; Lee, J.S.; Kim, Y.; Chung, I.-J.; Park, M.H.; Yoon, J.H.; Furth, P.A.; Hennighausen, L. The Methyltransferase EZH2 Is Not Required for Mammary Cancer Development, Although High EZH2 and Low H3K27me3 Correlate with Poor Prognosis of ER-Positive Breast Cancers. Mol. Carcinog. 2015, 54, 1172–1180. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Drean, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-Wide and High-Density CRISPR-Cas9 Screens Identify Point Mutations in PARP1 Causing PARP Inhibitor Resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Du, Y.; Yamaguchi, H.; Wei, Y.; Hsu, J.L.; Wang, H.-L.; Hsu, Y.-H.; Lin, W.-C.; Yu, W.-H.; Leonard, P.G.; Lee, G.R., IV; et al. Blocking c-Met-Mediated PARP1 Phosphorylation Enhances Anti-Tumor Effects of PARP Inhibitors. Nat. Med. 2016, 22, 194. [Google Scholar] [CrossRef]
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication Fork Stability Confers Chemoresistance in BRCA-Deficient Cells. Nature 2016, 535, 382. [Google Scholar] [CrossRef]
- Haynes, B.; Murai, J.; Lee, J.-M. Restored Replication Fork Stabilization, a Mechanism of PARP Inhibitor Resistance, Can Be Overcome by Cell Cycle Checkpoint Inhibition. Cancer Treat. Rev. 2018, 71, 1–7. [Google Scholar] [CrossRef]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 Promotes Degradation of Stalled Replication Forks by Recruiting MUS81 through Histone H3 Trimethylation. Nat. Cell Biol. 2017, 19, 1371. [Google Scholar] [CrossRef]
- Meghani, K.; Fuchs, W.; Detappe, A.; Drané, P.; Gogola, E.; Rottenberg, S.; Jonkers, J.; Matulonis, U.; Swisher, E.M.; Konstantinopoulos, P.A.; et al. Multifaceted Impact of MicroRNA 493-5p on Genome-Stabilizing Pathways Induces Platinum and PARP Inhibitor Resistance in BRCA2-Mutated Carcinomas. Cell Rep. 2018, 23, 100–111. [Google Scholar] [CrossRef]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Llobet, S.G.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e12. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keung, M.Y.T.; Wu, Y.; Vadgama, J.V. PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. J. Clin. Med. 2019, 8, 435. https://doi.org/10.3390/jcm8040435
Keung MYT, Wu Y, Vadgama JV. PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. Journal of Clinical Medicine. 2019; 8(4):435. https://doi.org/10.3390/jcm8040435
Chicago/Turabian StyleKeung, Man Yee T., Yanyuan Wu, and Jaydutt V. Vadgama. 2019. "PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers" Journal of Clinical Medicine 8, no. 4: 435. https://doi.org/10.3390/jcm8040435
APA StyleKeung, M. Y. T., Wu, Y., & Vadgama, J. V. (2019). PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. Journal of Clinical Medicine, 8(4), 435. https://doi.org/10.3390/jcm8040435