Progression-Related Loss of Stromal Caveolin 1 Levels Mediates Radiation Resistance in Prostate Carcinoma via the Apoptosis Inhibitor TRIAP1


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture Conditions
2.3. Irradiation of Cell Cultures
2.4. Colony Formation Assay
2.5. Flow Cytometry Analysis
2.6. Western Blotting
2.7. Real-Time Reverse Transcription PCR (qRT-PCR)
2.8. Conditioned Media
2.9. Mouse Tumour Model
2.10. Human Tumour Tissue
2.11. Immunohistochemistry and Immunofluorescence
2.12. Statistical Analysis
3. Results
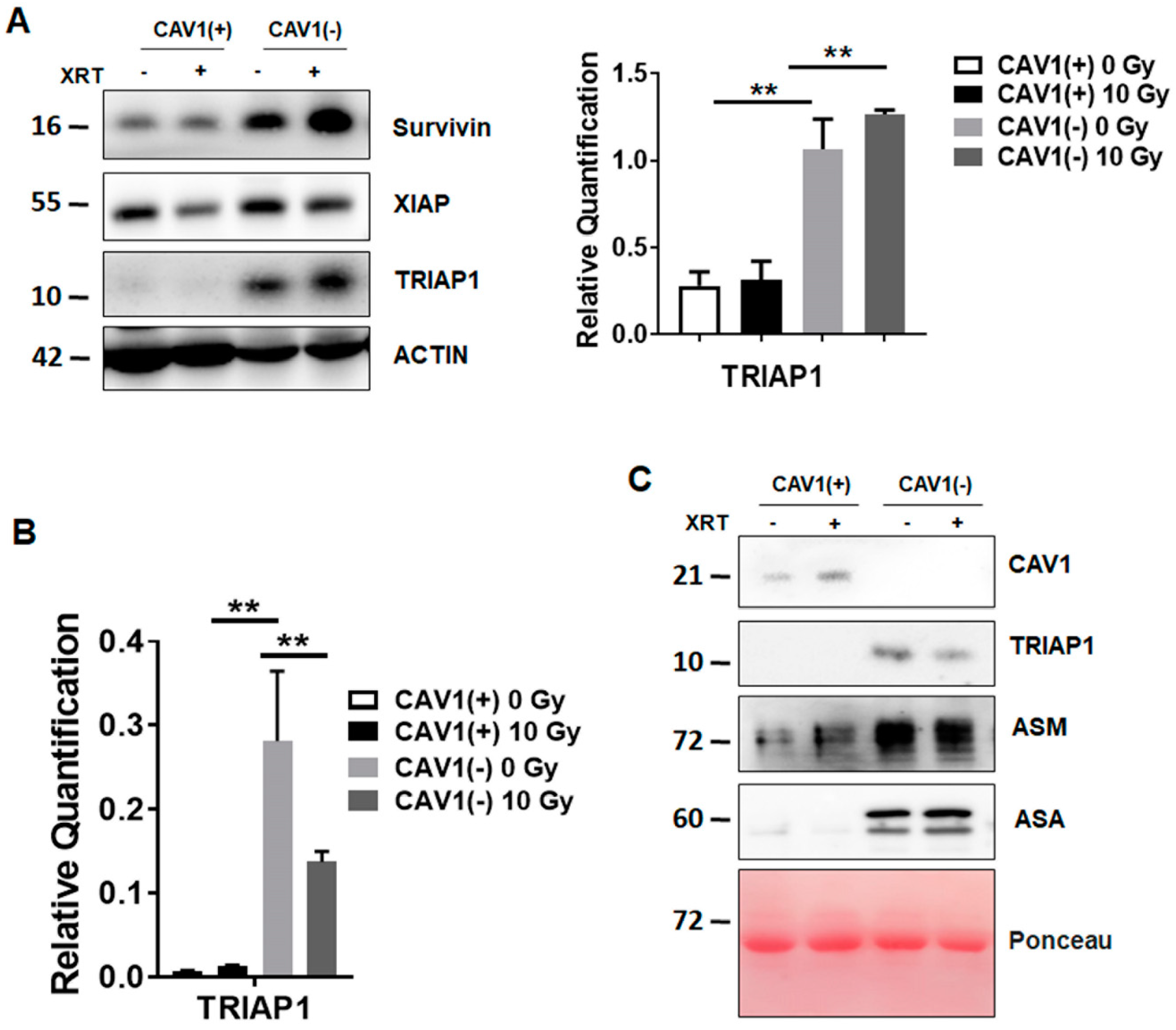
3.1. Radioresistant (CAV1-Silenced) Fibroblasts Express and Secrete Anti-Apoptotic TRIAP1
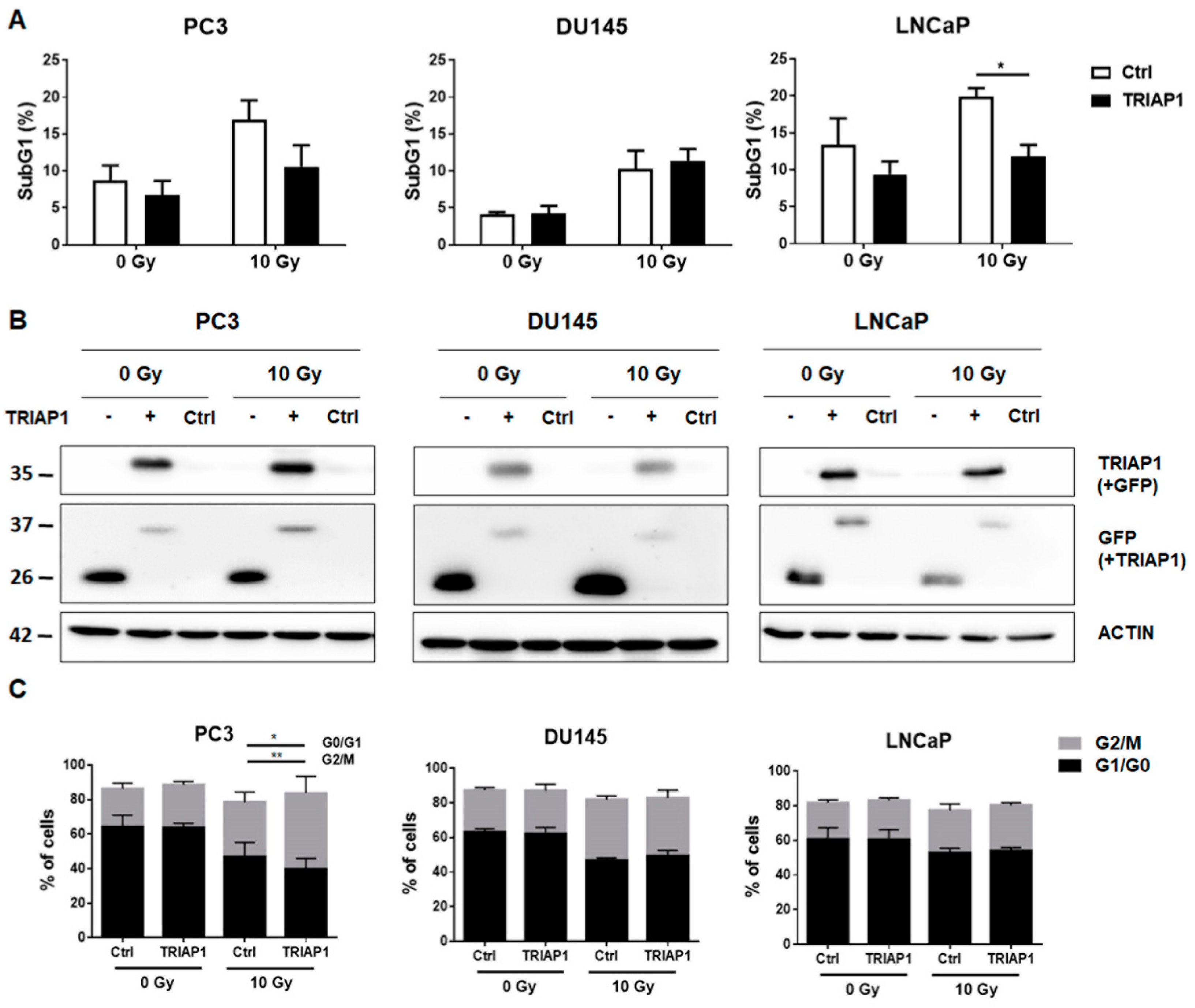
3.2. Ectopic TRIAP1 Expression in Prostate Carcinoma Cells Induces Radiation Resistance
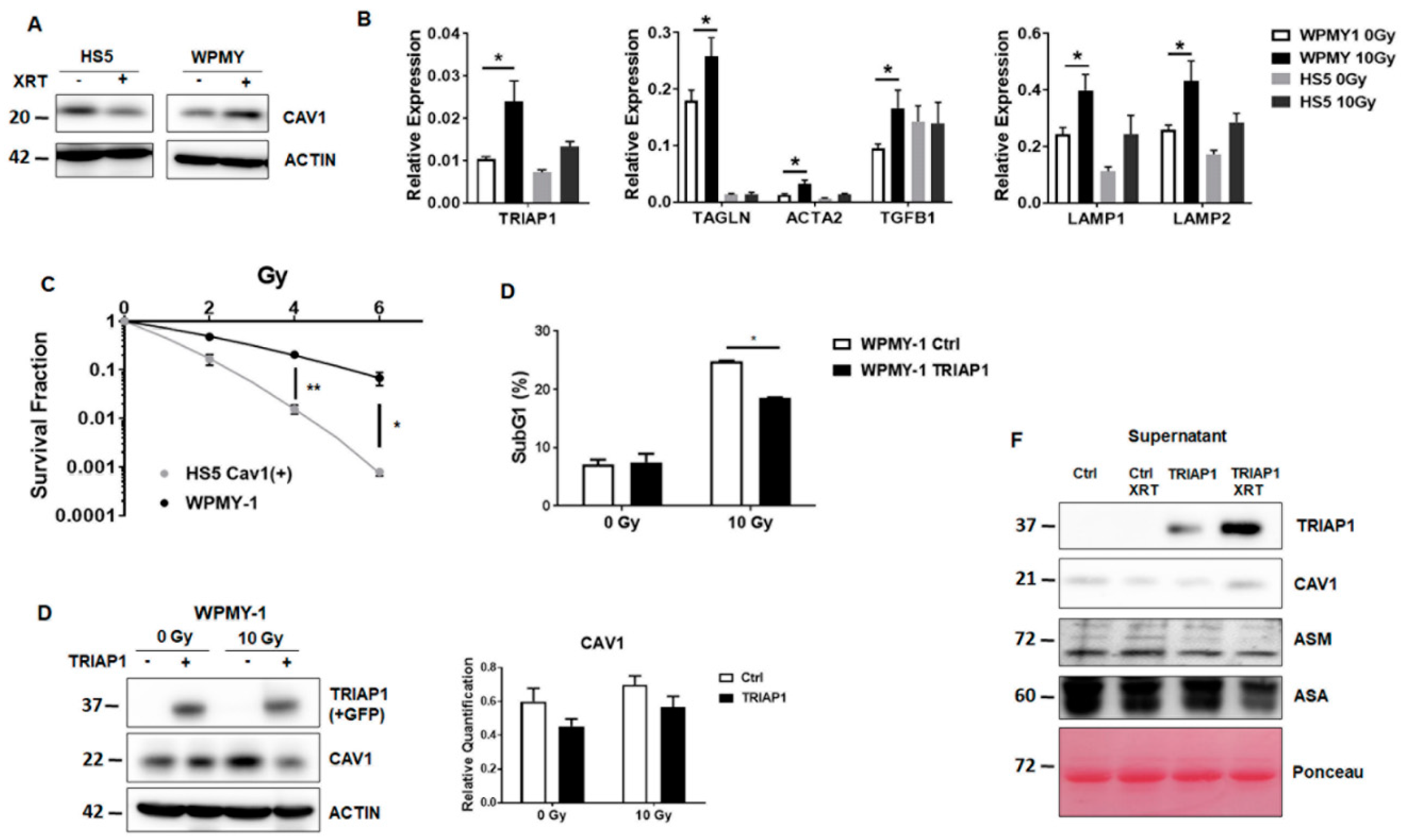
3.3. Generation of Stromal Prostate Fibroblasts with Stable TRIAP1 Expression
3.4. TRIAP1-Expressing Stromal Fibroblasts Mediate Radiation Resistance
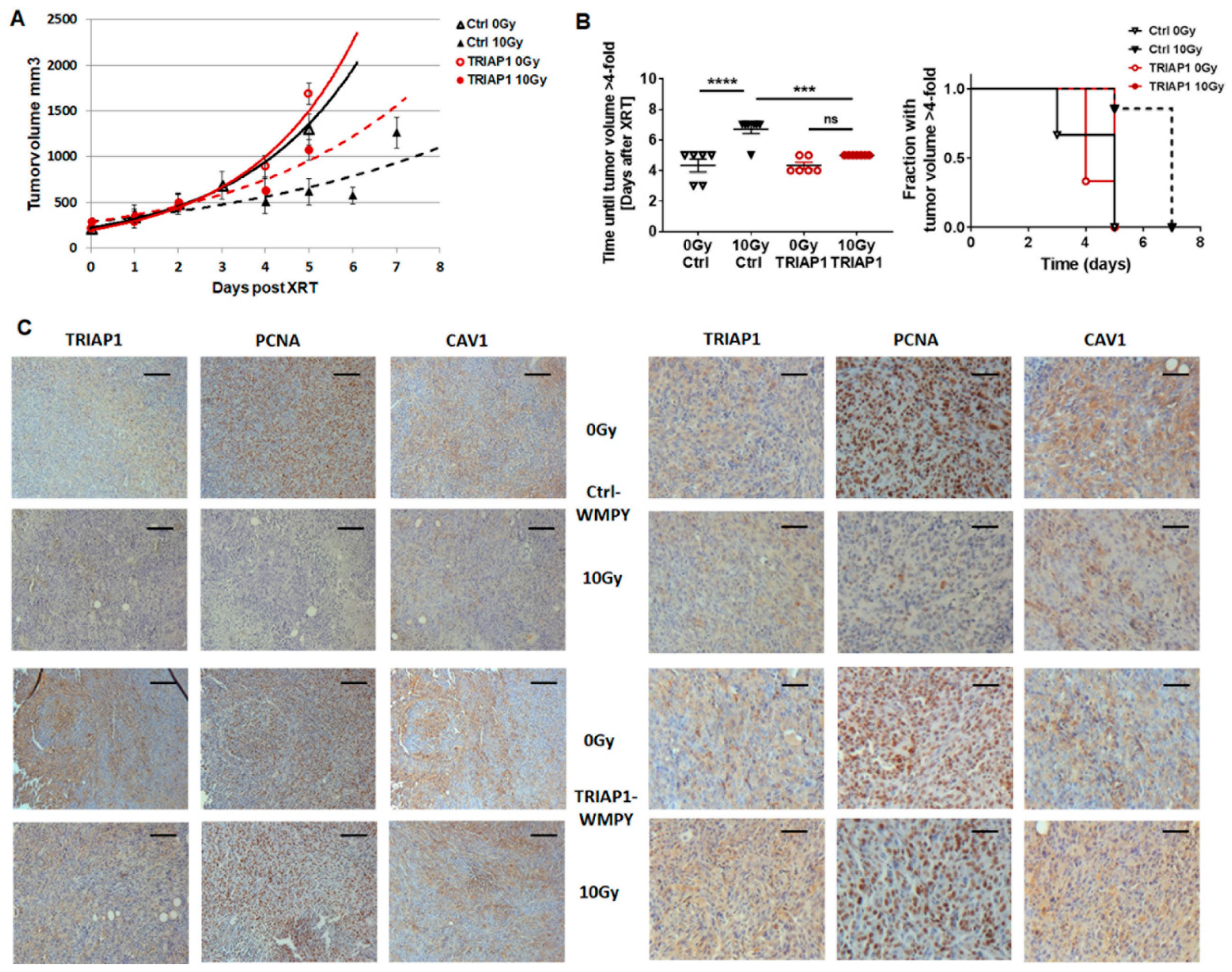
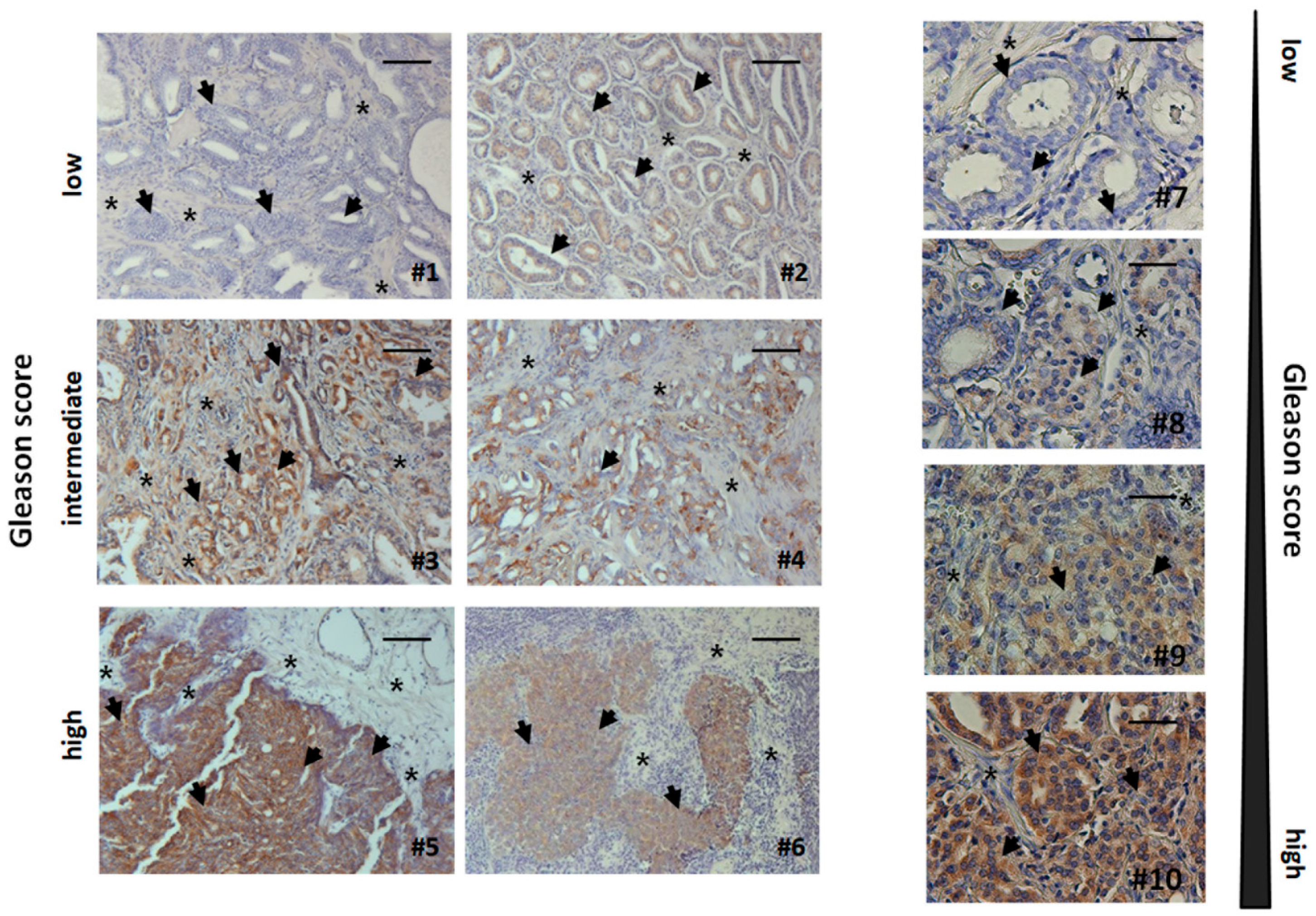
3.5. Human Advanced Prostate Cancer Specimens Were Characterized by an Increased TRIAP1-Immunoreactivity Indicating Radiation Resistance
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bissell, M.J. Thinking in three dimensions: Discovering reciprocal signaling between the extracellular matrix and nucleus and the wisdom of microenvironment and tissue architecture. Mol. Biol. Cell 2016, 27, 3205–3209. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Che, G. Value of caveolin-1 in cancer progression and prognosis: Emphasis on cancer-associated fibroblasts, human cancer cells and mechanism of caveolin-1 expression (Review). Oncol. Lett. 2014, 8, 1409–1421. [Google Scholar] [CrossRef]
- Ketteler, J.; Klein, D. Caveolin-1, cancer and therapy resistance. Int. J. Cancer 2018. [Google Scholar] [CrossRef]
- Di Vizio, D.; Morello, M.; Sotgia, F.; Pestell, R.G.; Freeman, M.R.; Lisanti, M.P. An absence of stromal caveolin-1 is associated with advanced prostate cancer, metastatic disease and epithelial Akt activation. Cell Cycle 2009, 8, 2420–2424. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Dasgupta, A.; Sotgia, F.; Mercier, I.; Pestell, R.G.; Sabel, M.; Kleer, C.G.; Brody, J.R.; Lisanti, M.P. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am. J. Pathol. 2009, 174, 2023–2034. [Google Scholar] [CrossRef]
- Ayala, G.; Morello, M.; Frolov, A.; You, S.; Li, R.; Rosati, F.; Bartolucci, G.; Danza, G.; Adam, R.M.; Thompson, T.C.; et al. Loss of caveolin-1 in prostate cancer stroma correlates with reduced relapse-free survival and is functionally relevant to tumour progression. J. Pathol. 2013, 231, 77–87. [Google Scholar] [CrossRef]
- Eliyatkin, N.; Aktas, S.; Diniz, G.; Ozgur, H.H.; Ekin, Z.Y.; Kupelioglu, A. Expression of Stromal Caveolin- 1 May Be a Predictor for Aggressive Behaviour of Breast Cancer. Pathol. Oncol. Res. 2018, 24, 59–65. [Google Scholar] [CrossRef]
- Shan-Wei, W.; Kan-Lun, X.; Shu-Qin, R.; Li-Li, Z.; Li-Rong, C. Overexpression of caveolin-1 in cancer-associated fibroblasts predicts good outcome in breast cancer. Breast Care 2012, 7, 477–483. [Google Scholar] [CrossRef]
- Panic, A.; Ketteler, J.; Reis, H.; Sak, A.; Herskind, C.; Maier, P.; Rubben, H.; Jendrossek, V.; Klein, D. Progression-related loss of stromal Caveolin 1 levels fosters the growth of human PC3 xenografts and mediates radiation resistance. Sci. Rep. 2017, 7, 41138. [Google Scholar] [CrossRef] [PubMed]
- Hammarsten, P.; Dahl Scherdin, T.; Hagglof, C.; Andersson, P.; Wikstrom, P.; Stattin, P.; Egevad, L.; Granfors, T.; Bergh, A. High Caveolin-1 Expression in Tumor Stroma Is Associated with a Favourable Outcome in Prostate Cancer Patients Managed by Watchful Waiting. PLoS ONE 2016, 11, e0164016. [Google Scholar] [CrossRef]
- Antognelli, C.; Ferri, I.; Bellezza, G.; Siccu, P.; Love, H.D.; Talesa, V.N.; Sidoni, A. Glyoxalase 2 drives tumorigenesis in human prostate cells in a mechanism involving androgen receptor and p53-p21 axis. Mol. Carcinog. 2017, 56, 2112–2126. [Google Scholar] [CrossRef]
- Cotter, T.G. Apoptosis and cancer: The genesis of a research field. Nat. Rev. Cancer 2009, 9, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.; Cazzanelli, G.; Rasul, S.; Hitchinson, B.; Hu, Y.; Coombes, R.C.; Raguz, S.; Yague, E. Apoptosis inhibitor TRIAP1 is a novel effector of drug resistance. Oncol. Rep. 2015, 34, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Park, W.R.; Nakamura, Y. p53CSV, a novel p53-inducible gene involved in the p53-dependent cell-survival pathway. Cancer Res. 2005, 65, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Andrysik, Z.; Kim, J.; Tan, A.C.; Espinosa, J.M. A genetic screen identifies TCF3/E2A and TRIAP1 as pathway-specific regulators of the cellular response to p53 activation. Cell Rep. 2013, 3, 1346–1354. [Google Scholar] [CrossRef]
- Klein, D.; Schmandt, T.; Muth-Kohne, E.; Perez-Bouza, A.; Segschneider, M.; Gieselmann, V.; Brustle, O. Embryonic stem cell-based reduction of central nervous system sulfatide storage in an animal model of metachromatic leukodystrophy. Gene Ther. 2006, 13, 1686–1695. [Google Scholar] [CrossRef]
- Lansmann, S.; Ferlinz, K.; Hurwitz, R.; Bartelsen, O.; Glombitza, G.; Sandhoff, K. Purification of acid sphingomyelinase from human placenta: Characterization and N-terminal sequence. FEBS Lett. 1996, 399, 227–231. [Google Scholar] [CrossRef]
- Barzan, D.; Maier, P.; Zeller, W.J.; Wenz, F.; Herskind, C. Overexpression of caveolin-1 in lymphoblastoid TK6 cells enhances proliferation after irradiation with clinically relevant doses. Strahlenther. Onkol. 2010, 186, 99–106. [Google Scholar] [CrossRef]
- Klein, D.; Schmitz, T.; Verhelst, V.; Panic, A.; Schenck, M.; Reis, H.; Drab, M.; Sak, A.; Herskind, C.; Maier, P.; et al. Endothelial Caveolin-1 regulates the radiation response of epithelial prostate tumors. Oncogenesis 2015, 4, e148. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.; Steens, J.; Wiesemann, A.; Schulz, F.C.; Kaschani, F.; Roeck, K.; Yamaguchi, M.; Wirsdorfer, F.; Kaiser, M.; Fischer, J.; et al. Mesenchymal stem cell therapy protects lungs from radiation-induced endothelial cell loss by restoring superoxide dismutase 1 expression. Antioxid. Redox Signal. 2016. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef]
- Singh, S.R.; Rameshwar, P.; Siegel, P. Targeting tumor microenvironment in cancer therapy. Cancer Lett. 2016. [Google Scholar] [CrossRef]
- Sun, Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2015. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.J.; Bahal, R.; Babar, I.A.; Pincus, Z.; Barrera, F.; Liu, C.; Svoronos, A.; Braddock, D.T.; Glazer, P.M.; Engelman, D.M.; et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 2015, 518, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Izpisua Belmonte, J.C. The microenvironment and resistance to personalized cancer therapy. Nat. Rev. Clin. Oncol. 2013, 10. [Google Scholar] [CrossRef]
- Swartz, M.A.; Iida, N.; Roberts, E.W.; Sangaletti, S.; Wong, M.H.; Yull, F.E.; Coussens, L.M.; DeClerck, Y.A. Tumor microenvironment complexity: Emerging roles in cancer therapy. Cancer Res. 2012, 72, 2473–2480. [Google Scholar] [CrossRef]
- Wang, S.; Wang, N.; Zheng, Y.; Zhang, J.; Zhang, F.; Wang, Z. Caveolin-1: An Oxidative Stress-Related Target for Cancer Prevention. Oxidative Med. Cell. Longev. 2017, 2017, 7454031. [Google Scholar] [CrossRef]
- Kamposioras, K.; Tsimplouli, C.; Verbeke, C.; Anthoney, A.; Daoukopoulou, A.; Papandreou, C.N.; Sakellaridis, N.; Vassilopoulos, G.; Potamianos, S.P.; Liakouli, V.; et al. Silencing of caveolin-1 in fibroblasts as opposed to epithelial tumor cells results in increased tumor growth rate and chemoresistance in a human pancreatic cancer model. Int. J. Oncol. 2018. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010, 9, 2423–2433. [Google Scholar] [CrossRef]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Kallunki, T.; Olsen, O.D.; Jaattela, M. Cancer-associated lysosomal changes: Friends or foes? Oncogene 2013, 32, 1995–2004. [Google Scholar] [CrossRef]
- Gocheva, V.; Zeng, W.; Ke, D.; Klimstra, D.; Reinheckel, T.; Peters, C.; Hanahan, D.; Joyce, J.A. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006, 20, 543–556. [Google Scholar] [CrossRef]
- Fehrenbacher, N.; Bastholm, L.; Kirkegaard-Sorensen, T.; Rafn, B.; Bottzauw, T.; Nielsen, C.; Weber, E.; Shirasawa, S.; Kallunki, T.; Jaattela, M. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res. 2008, 68, 6623–6633. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2016, 24, 23–33. [Google Scholar] [CrossRef]
- Siu, M.K.; Abou-Kheir, W.; Yin, J.J.; Chang, Y.S.; Barrett, B.; Suau, F.; Casey, O.; Chen, W.Y.; Fang, L.; Hynes, P.; et al. Correction: Loss of EGFR signaling-regulated miR-203 promotes prostate cancer bone metastasis and tyrosine kinase inhibitors resistance. Oncotarget 2018, 9, 32403. [Google Scholar] [CrossRef]
- Liu, P.; Qi, X.; Bian, C.; Yang, F.; Lin, X.; Zhou, S.; Xie, C.; Zhao, X.; Yi, T. MicroRNA-18a inhibits ovarian cancer growth via directly targeting TRIAP1 and IPMK. Oncol. Lett. 2017, 13, 4039–4046. [Google Scholar] [CrossRef] [PubMed]
- Fook-Alves, V.L.; de Oliveira, M.B.; Zanatta, D.B.; Strauss, B.E.; Colleoni, G.W. TP53 Regulated Inhibitor of Apoptosis 1 (TRIAP1) stable silencing increases late apoptosis by upregulation of caspase 9 and APAF1 in RPMI8226 multiple myeloma cell line. Biochim. Biophys. Acta 2016, 1862, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tang, X.; He, Q.; Yang, X.; Ren, X.; Wen, X.; Zhang, J.; Wang, Y.; Liu, N.; Ma, J. Overexpression of Mitochondria Mediator Gene TRIAP1 by miR-320b Loss Is Associated with Progression in Nasopharyngeal Carcinoma. PLoS Genet. 2016, 12, e1006183. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ketteler, J.; Panic, A.; Reis, H.; Wittka, A.; Maier, P.; Herskind, C.; Yagüe, E.; Jendrossek, V.; Klein, D. Progression-Related Loss of Stromal Caveolin 1 Levels Mediates Radiation Resistance in Prostate Carcinoma via the Apoptosis Inhibitor TRIAP1. J. Clin. Med. 2019, 8, 348. https://doi.org/10.3390/jcm8030348
Ketteler J, Panic A, Reis H, Wittka A, Maier P, Herskind C, Yagüe E, Jendrossek V, Klein D. Progression-Related Loss of Stromal Caveolin 1 Levels Mediates Radiation Resistance in Prostate Carcinoma via the Apoptosis Inhibitor TRIAP1. Journal of Clinical Medicine. 2019; 8(3):348. https://doi.org/10.3390/jcm8030348
Chicago/Turabian StyleKetteler, Julia, Andrej Panic, Henning Reis, Alina Wittka, Patrick Maier, Carsten Herskind, Ernesto Yagüe, Verena Jendrossek, and Diana Klein. 2019. "Progression-Related Loss of Stromal Caveolin 1 Levels Mediates Radiation Resistance in Prostate Carcinoma via the Apoptosis Inhibitor TRIAP1" Journal of Clinical Medicine 8, no. 3: 348. https://doi.org/10.3390/jcm8030348
APA StyleKetteler, J., Panic, A., Reis, H., Wittka, A., Maier, P., Herskind, C., Yagüe, E., Jendrossek, V., & Klein, D. (2019). Progression-Related Loss of Stromal Caveolin 1 Levels Mediates Radiation Resistance in Prostate Carcinoma via the Apoptosis Inhibitor TRIAP1. Journal of Clinical Medicine, 8(3), 348. https://doi.org/10.3390/jcm8030348