Type 2 Diabetes Mellitus and Altered Immune System Leading to Susceptibility to Pathogens, Especially Mycobacterium tuberculosis



{kind=link}
Abstract
1. Introduction
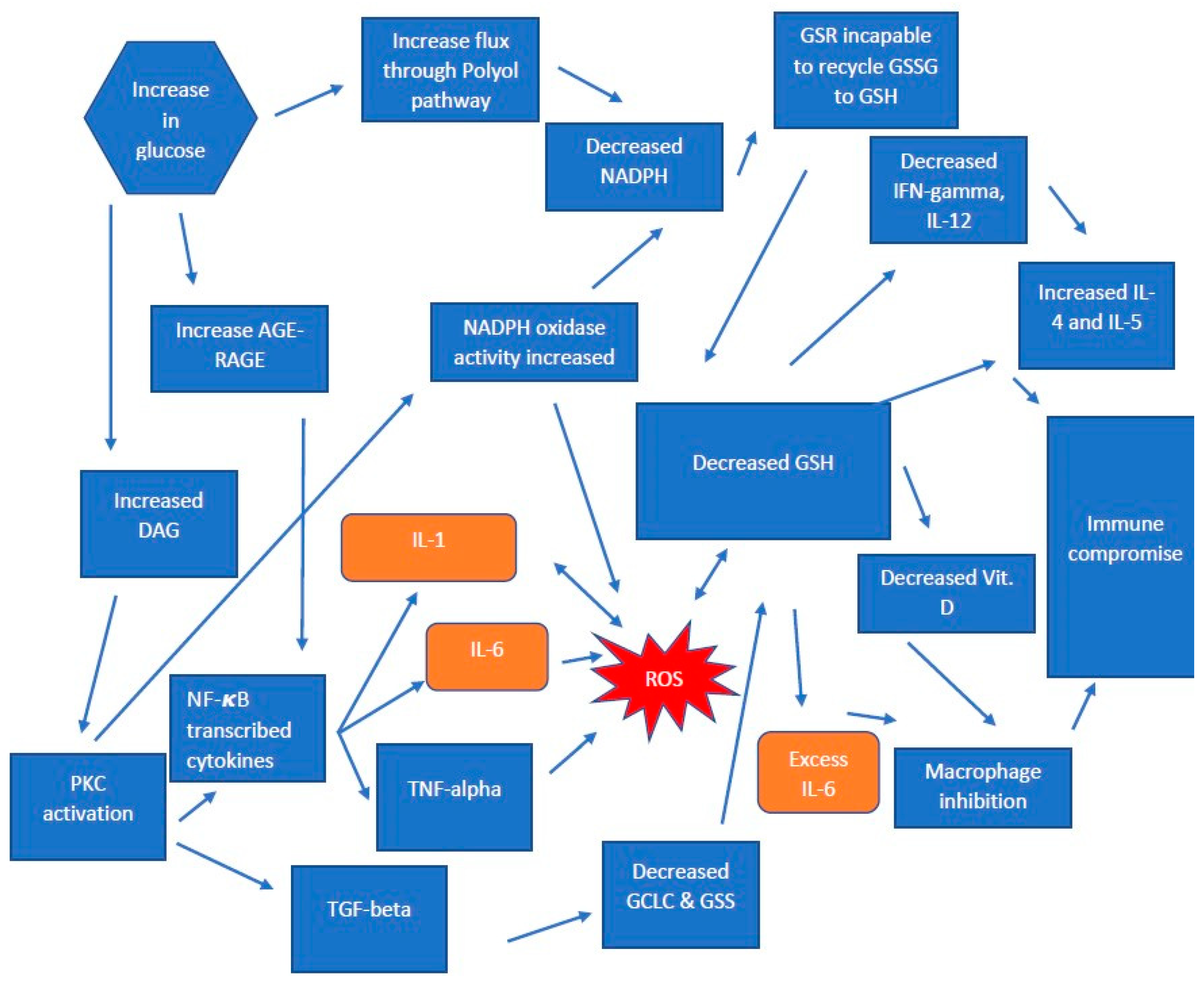
1.1. Type 2 Diabetes Mellitus
1.2. Pathogenesis in T2DM
1.3. The Production of Glutathione in T2DM
1.4. AGE-RAGE and GSH Deficiency in T2DM
1.5. T2DM and Tuberculosis
1.6. Cytokine Production and Immune Responses against M. tb Infection
1.7. Why GSH Is Important in a Functioning Immune System
1.8. Vitamin D and Macrophage Activation
2. Summary
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Diabetes Facts (Infographics); WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Centers for Disease Control and Prevention (CDC). National Diabetes Statistics Report, 2017 Estimates of Diabetes and Its Burden in the United States Background; CDC: Atlanta, GA, USA, 2018. [Google Scholar]
- World Health Organization. Tuberculosis. Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 13 November 2019).
- Frieden, T.R.; Sterling, T.R.; Munsiff, S.S.; Watt, C.J.; Dye, C. Tuberculosis. Lancet 2003, 362, 887–899. [Google Scholar] [CrossRef]
- Ai, J.-W.; Ruan, Q.-L.; Liu, Q.-H.; Zhang, W.-H. Updates on the risk factors for latent tuberculosis reactivation and their managements. Emerg. Microbes Infect. 2016, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar] [PubMed]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef]
- Wu, Y.; Ding, Y.; Tanaka, Y.; Zhang, W. Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int. J. Med. Sci. 2014, 11, 1185–1200. [Google Scholar] [CrossRef] [PubMed]
- Schemmel, K.E.; Padiyara, R.S.; D’Souza, J.J. Aldose reductase inhibitors in the treatment of diabetic peripheral neuropathy: A review. J. Diabetes Complicat. 2010, 24, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Kato, N. Aldose Reductase Inhibitors. J. Enzyme Inhib. 2001, 16, 465–473. [Google Scholar] [CrossRef]
- Oates, P.J. Aldose reductase, still a compelling target for diabetic neuropathy. Curr. Drug Targets 2008, 9, 14–36. [Google Scholar] [CrossRef] [PubMed]
- Morré, D.M.; Lenaz, G.; Morré, D.J. Surface oxidase and oxidative stress propagation in aging. J. Exp. Biol. 2000, 203 Pt 10, 1513–1521. [Google Scholar]
- Pollreisz, A.; Schmidt-Erfurth, U. Diabetic Cataract—Pathogenesis, Epidemiology and Treatment. J. Ophthalmol. 2010, 2010, 1–8. [Google Scholar] [CrossRef]
- Cheng, H.-M.; González, R.G. The effect of high glucose and oxidative stress on lens metabolism, aldose reductase, and senile cataractogenesis. Metabolism 1986, 35, 10–14. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D.; Stasinopoulos, M.; Zelenko, Z.; Shiloach, J. Polyol accumulation in muscle and liver in a mouse model of type 2 diabetes. J. Diabetes Complicat. 2016, 30, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef]
- Lagman, M.; Ly, J.; Saing, T.; Singh, M.K.; Tudela, E.V.; Morris, D.; Chi, P.-T.; Ochoa, C.; Sathananthan, A.; Venketaraman, V. Investigating the causes for decreased levels of glutathione in individuals with type II diabetes. PLoS ONE 2015, 10, e0118436. [Google Scholar] [CrossRef]
- Bakin, A.V.; Stourman, N.V.; Sekhar, K.R.; Rinehart, C.; Yan, X.; Meredith, M.J.; Arteaga, C.L.; Freeman, M.L. Smad3-ATF3 signaling mediates TGF-β suppression of genes encoding Phase II detoxifying proteins. Free Radic. Biol. Med. 2005, 38, 375–387. [Google Scholar] [CrossRef]
- Franklin, C.C.; Rosenfeld-Franklin, M.E.; White, C.; Kavanagh, T.J.; Fausto, N. TGFbeta1-induced suppression of glutathione antioxidant defenses in hepatocytes: Caspase-dependent post-translational and caspase-independent transcriptional regulatory mechanisms. FASEB J. 2003, 17, 1535–1537. [Google Scholar] [CrossRef]
- Yan, Z.; Garg, S.K.; Kipnis, J.; Banerjee, R. Extracellular redox modulation by regulatory T cells. Nat. Chem. Biol. 2009, 5, 721–723. [Google Scholar] [CrossRef]
- World Health Organization. ‘Tuberculosis’ Fact Sheet; WHO: Geneva, Switzerland, 2010; Volume 104. [Google Scholar]
- Park, S.W.; Shin, J.W.; Kim, J.Y.; Park, I.W.; Choi, B.W.; Choi, J.C.; Kim, Y.S. The effect of diabetic control status on the clinical features of pulmonary tuberculosis. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 1305–1310. [Google Scholar] [CrossRef]
- Baker, M.A.; Harries, A.D.; Jeon, C.Y.; Hart, J.E.; Kapur, A.; Lönnroth, K.; Ottmani, S.-E.; Goonesekera, S.D.; Murray, M.B. The impact of diabetes on tuberculosis treatment outcomes: A systematic review. BMC Med. 2011, 9, 81. [Google Scholar] [CrossRef]
- Dinarello, C.A. Historical insights into cytokines. Eur. J. Immunol. 2007, 37 (Suppl. 1), 34–45. [Google Scholar] [CrossRef]
- Domingo-Gonzalez, R.; Prince, O.; Cooper, A. Cytokines and Chemokines in Mycobacterium tuberculosis Infection. In Tuberculosis and the Tubercle Bacillus, 2nd ed.; American Society of Microbiology: Washington, DC, USA, 2016; pp. 33–72. [Google Scholar]
- Kaufmann, S.H.E. Protection against tuberculosis: Cytokines, T cells, and macrophages. Ann. Rheum. Dis. 2002, 61 (Suppl. 2), ii54–ii58. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Plessner, H.L.; Voitenok, N.N.; Flynn, J.L. Tumor Necrosis Factor and Tuberculosis. J. Investig. Dermatol. Symp. Proc. 2007, 12, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.M.; Mayer-Barber, K.D.; Sher, A. Role of innate cytokines in mycobacterial infection. Mucosal Immunol. 2011, 4, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Philips, J.A.; Ernst, J.D. Tuberculosis Pathogenesis and Immunity. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 353–384. [Google Scholar] [CrossRef] [PubMed]
- Wickremasinghe, M.I.; Thomas, L.H.; Friedland, J.S. Pulmonary epithelial cells are a source of IL-8 in the response to Mycobacterium tuberculosis: Essential role of IL-1 from infected monocytes in a NF-κB-dependent network. J. Immunol. 1999, 163, 3936–3947. [Google Scholar] [PubMed]
- Alexander, Y.; Persson, Z.; Blomgran-Julinder, R.; Rahman, S.; Zheng, L.; Stendahl, O. Mycobacterium tuberculosis-induced apoptotic neutrophils trigger a pro-inflammatory response in macrophages through release of heat shock protein 72, acting in synergy with the bacteria. Microbes Infect. 2008, 10, 233–240. [Google Scholar]
- Flynn, J.L.; Chan, J. I Mmunology of T Uberculosis. Annu. Rev. Immunol. 2001, 19, 93–129. [Google Scholar] [CrossRef]
- Liew, F.Y.; Li, Y.; Millott, S. Tumor necrosis factor-alpha synergizes with IFN-gamma in mediating killing of Leishmania major through the induction of nitric oxide. J. Immunol. 1990, 145, 4306–4310. [Google Scholar]
- Beamer, G.L.; Flaherty, D.K.; Assogba, B.D.; Stromberg, P.; Gonzalez-Juarrero, M.; de Waal Malefyt, R.; Vesosky, B.; Turner, J. Interleukin-10 promotes Mycobacterium tuberculosis disease progression in CBA/J mice. J. Immunol. 2008, 181, 5545–5550. [Google Scholar] [CrossRef]
- O’Leary, S.; O’Sullivan, M.P.; Keane, J. IL-10 Blocks Phagosome Maturation in Mycobacterium tuberculosis– Infected Human Macrophages. Am. J. Respir. Cell Mol. Biol. 2011, 45, 172–180. [Google Scholar] [CrossRef]
- Gong, J.H.; Zhang, M.; Modlin, R.L.; Linsley, P.S.; Iyer, D.; Lin, Y.; Barnes, P.F. Interleukin-10 downregulates Mycobacterium tuberculosis-induced Th1 responses and CTLA-4 expression. Infect. Immun. 1996, 64, 913–918. [Google Scholar] [PubMed]
- Lowe, D.M.; Redford, P.S.; Wilkinson, R.J.; O’Garra, A.; Martineau, A.R. Neutrophils in tuberculosis: Friend or foe? Trends Immunol. 2012, 33, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, E.; Iona, E.; Ferroni, L.; Miettinen, M.; Fattorini, L.; Orefici, G.; Julkunen, I.; Coccia, E.M. Infection of Human Macrophages and Dendritic Cells with Mycobacterium tuberculosis Induces a Differential Cytokine Gene Expression That Modulates T Cell Response. J. Immunol. 2001, 166, 7033–7041. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Haneda, K.; Tamura, G.; Shirato, K. Ovalbumin (OVA) and Mycobacterium tuberculosis Bacilli Cooperatively Polarize Anti-OVA T-helper (Th) Cells toward a Th1-Dominant Phenotype and Ameliorate Murine Tracheal Eosinophilia. Am. J. Respir. Cell Mol. Biol. 1999, 20, 1260–1267. [Google Scholar] [CrossRef]
- Boom, W.H.; Canaday, D.H.; Fulton, S.A.; Gehring, A.J.; Rojas, R.E.; Torres, M. Human immunity to M. tuberculosis: T cell subsets and antigen processing. Tuberculosis (Edinb) 2003, 83, 98–106. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Coffman, R.L. TH1 and TH2 Cells: Different Patterns of Lymphokine Secretion Lead to Different Functional Properties. Annu. Rev. Immunol. 1989, 7, 145–173. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.M. T cells in mycobacterial infection and disease. Curr. Opin. Immunol. 2009, 21, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Ottenhoff, T.H.M.; Kaufmann, S.H.E. Vaccines against Tuberculosis: Where Are We and Where Do We Need to Go? PLoS Pathog. 2012, 8, e1002607. [Google Scholar] [CrossRef]
- VanHeyningen, T.K.; Collins, H.L.; Russell, D.G. IL-6 produced by macrophages infected with Mycobacterium species suppresses T cell responses. J. Immunol. 1997, 158, 330–337. [Google Scholar]
- Venketaraman, V.; Millman, A.; Salman, M.; Swaminathan, S.; Goetz, M.; Lardizabal, A.; Hom, D.; Connell, N.D. Glutathione levels and immune responses in tuberculosis patients. Microb. Pathog. 2008, 44, 255–261. [Google Scholar] [CrossRef]
- Venketaraman, V.; Dayaram, Y.K.; Talaue, M.T.; Connell, N.D. Glutathione and Nitrosoglutathione in Macrophage Defense against Mycobacterium tuberculosis. Infect. Immun. 2005, 73, 1886–1889. [Google Scholar] [CrossRef] [PubMed]
- Afzali, B.; Mitchell, P.; Lechler, R.I.; John, S.; Lombardi, G. Translational Mini-Review Series on Th17 Cells: Induction of interleukin-17 production by regulatory T cells. Clin. Exp. Immunol. 2010, 159, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Millman, A.C.; Salman, M.; Dayaram, Y.K.; Connell, N.D.; Venketaraman, V. Natural Killer Cells, Glutathione, Cytokines, and Innate Immunity Against Mycobacterium tuberculosis. J. Interf. Cytokine Res. 2008, 28, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Kasai, M.; Yoneda, T.; Habu, S.; Maruyama, Y.; Okumura, K.; Tokunaga, T. In vivo effect of anti-asialo GM1 antibody on natural killer activity. Nature 1981, 291, 334–335. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Travers, P.; Walport, M. Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Garg, S.K.; Yan, Z.; Vitvitsky, V.; Banerjee, R. Differential dependence on cysteine from transsulfuration versus transport during T cell activation. Antioxid. Redox Signal. 2011, 15, 39–47. [Google Scholar] [CrossRef]
- Verhasselt, V.; Berghe, W.V.; Vanderheyde, N.; Willems, F.; Haegeman, G.; Goldman, M. N-acetyl-L-cysteine inhibits primary human T cell responses at the dendritic cell level: Association with NF-kappaB inhibition. J. Immunol. 1999, 162, 2569–2574. [Google Scholar]
- Bernal-Fernandez, G.; Espinosa-Cueto, P.; Leyva-Meza, R.; Mancilla, N.; Mancilla, R. Decreased Expression of T-Cell Costimulatory Molecule CD28 on CD4 and CD8 T Cells of Mexican Patients with Pulmonary Tuberculosis. Tuberc. Res. Treat. 2010, 2010, 1–8. [Google Scholar] [CrossRef][Green Version]
- Haraguchi, S.; Day, N.K.; Nelson, R.P.; Emmanuel, P.; Duplantier, J.E.; Christodoulou, C.S.; Good, R.A. Interleukin 12 deficiency associated with recurrent infections. Proc. Natl. Acad. Sci. USA 1998, 95, 13125–13129. [Google Scholar] [CrossRef]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Tan, K.S.; Lee, K.O.; Low, K.C.; Gamage, A.M.; Liu, Y.; Tan, G.Y.G.; Koh, H.Q.V.; Alonso, S.; Gan, Y.H. Glutathione deficiency in type 2 diabetes impairs cytokine responses and control of intracellular bacteria. J. Clin. Investig. 2012, 122, 2289–2300. [Google Scholar] [CrossRef]
- Kumar, N.P.; Sridhar, R.; Banurekha, V.V.; Jawahar, M.S.; Fay, M.P.; Nutman, T.B.; Babu, S. Type 2 diabetes mellitus coincident with pulmonary tuberculosis is associated with heightened systemic type 1, type 17, and other proinflammatory cytokines. Ann. Am. Thorac. Soc. 2013, 10, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wen, F.; Zhang, X.; Su, S.B. Expression of T-helper-associated cytokines in patients with type 2 diabetes mellitus with retinopathy. Mol. Vis. 2012, 18, 219–226. [Google Scholar] [PubMed]
- Sutaria, N.; Liu, C.-T.; Chen, T.C. Vitamin D Status, Receptor Gene Polymorphisms, and Supplementation on Tuberculosis: A Systematic Review of Case-Control Studies and Randomized Controlled Trials. J. Clin. Transl. Endocrinol. 2014, 1, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yuan, Y.; Lin, Y.; Zhang, T.; Bai, Y.; Kang, D.; Li, X.; Kang, W.; Dlodlo, R.A.; Harries, A.D. Vitamin D status of tuberculosis patients with diabetes mellitus in different economic areas and associated factors in China. PLoS ONE 2018, 13, e0206372. [Google Scholar] [CrossRef]
- Jain, S.K.; Micinski, D. Vitamin D upregulates glutamate cysteine ligase and glutathione reductase, and GSH formation, and decreases ROS and MCP-1 and IL-8 secretion in high-glucose exposed U937 monocytes. Biochem. Biophys. Res. Commun. 2013, 437, 7–11. [Google Scholar] [CrossRef]
- Farrokhian, A.; Raygan, F.; Bahmani, F.; Talari, H.R.; Esfandiari, R.; Esmaillzadeh, A.; Asemi, Z. Long-Term Vitamin D Supplementation Affects Metabolic Status in Vitamin D–Deficient Type 2 Diabetic Patients with Coronary Artery Disease. J. Nutr. 2017, 147, 384–389. [Google Scholar] [CrossRef]
- Parsanathan, R.; Jain, S.K. Glutathione deficiency alters the vitamin D-metabolizing enzymes CYP27B1 and CYP24A1 in human renal proximal tubule epithelial cells and kidney of HFD-fed mice. Free Radic. Biol. Med. 2019, 131, 376–381. [Google Scholar] [CrossRef]
- Jain, S.K.; Parsanathan, R.; Achari, A.E.; Kanikarla-Marie, P.; Bocchini, J.A. Glutathione Stimulates Vitamin D Regulatory and Glucose-Metabolism Genes, Lowers Oxidative Stress and Inflammation, and Increases 25-Hydroxy-Vitamin D Levels in Blood: A Novel Approach to Treat 25-Hydroxyvitamin D Deficiency. Antioxidants Redox Signal. 2018, 29, 1792–1807. [Google Scholar] [CrossRef]
- Jain, S.K.; Kahlon, G.; Bass, P.; Levine, S.N.; Warden, C. Can l-cysteine and Vitamin D rescue Vitamin D and Vitamin D binding protein levels in blood Plasma of African American type 2 diabetic patients? Antioxid. Redox Signal. 2015, 23, 688–693. [Google Scholar] [CrossRef]
- Chesdachai, S.; Zughaier, S.M.; Hao, L.; Kempker, R.R.; Blumberg, H.M.; Ziegler, T.R.; Tangpricha, V. The effects of first-line anti-tuberculosis drugs on the actions of vitamin D in human macrophages. J. Clin. Transl. Endocrinol. 2016, 6, 23–29. [Google Scholar] [CrossRef]
- Hewison, M. Vitamin D and the intracrinology of innate immunity. Mol. Cell. Endocrinol. 2010, 321, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Ashenafi, S.; Mazurek, J.; Rehn, A.; Lemma, B.; Aderaye, G.; Bekele, A.; Assefa, G.; Chanyalew, M.; Aseffa, A.; Andersson, J.; et al. Vitamin D3 status and the association with human cathelicidin expression in patients with different clinical forms of active tuberculosis. Nutrients 2018, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.T.; Gonzalez, Y.; Hernández-Sánchez, F.; Miguel, G.F.; Torres, M. Low serum vitamin D levels in type 2 diabetes patients are associated with decreased mycobacterial activity. BMC Infect. Dis. 2017, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferlita, S.; Yegiazaryan, A.; Noori, N.; Lal, G.; Nguyen, T.; To, K.; Venketaraman, V. Type 2 Diabetes Mellitus and Altered Immune System Leading to Susceptibility to Pathogens, Especially Mycobacterium tuberculosis. J. Clin. Med. 2019, 8, 2219. https://doi.org/10.3390/jcm8122219
Ferlita S, Yegiazaryan A, Noori N, Lal G, Nguyen T, To K, Venketaraman V. Type 2 Diabetes Mellitus and Altered Immune System Leading to Susceptibility to Pathogens, Especially Mycobacterium tuberculosis. Journal of Clinical Medicine. 2019; 8(12):2219. https://doi.org/10.3390/jcm8122219
Chicago/Turabian StyleFerlita, Steve, Aram Yegiazaryan, Navid Noori, Gagandeep Lal, Timothy Nguyen, Kimberly To, and Vishwanath Venketaraman. 2019. "Type 2 Diabetes Mellitus and Altered Immune System Leading to Susceptibility to Pathogens, Especially Mycobacterium tuberculosis" Journal of Clinical Medicine 8, no. 12: 2219. https://doi.org/10.3390/jcm8122219
APA StyleFerlita, S., Yegiazaryan, A., Noori, N., Lal, G., Nguyen, T., To, K., & Venketaraman, V. (2019). Type 2 Diabetes Mellitus and Altered Immune System Leading to Susceptibility to Pathogens, Especially Mycobacterium tuberculosis. Journal of Clinical Medicine, 8(12), 2219. https://doi.org/10.3390/jcm8122219