Synaptic Pruning by Microglia in Epilepsy

{kind=link}
Abstract
1. Introduction
2. Changes in Microglial Properties in the Epileptic Brain
2.1. Morphological Changes
2.2. Molecular Expression
2.3. Neurodegeneration
2.4. Neurogenesis
3. Synaptic Pruning by Microglia
4. The Complement System in the Epileptic Brain
5. Key Molecules that Support Complement-Dependent Inhibitory Synapse Elimination by Microglia
5.1. ATP
5.1.1. ATP-Mediated Microglia–Neuron Interaction
5.1.2. Engulfment of Inhibitory Synapses
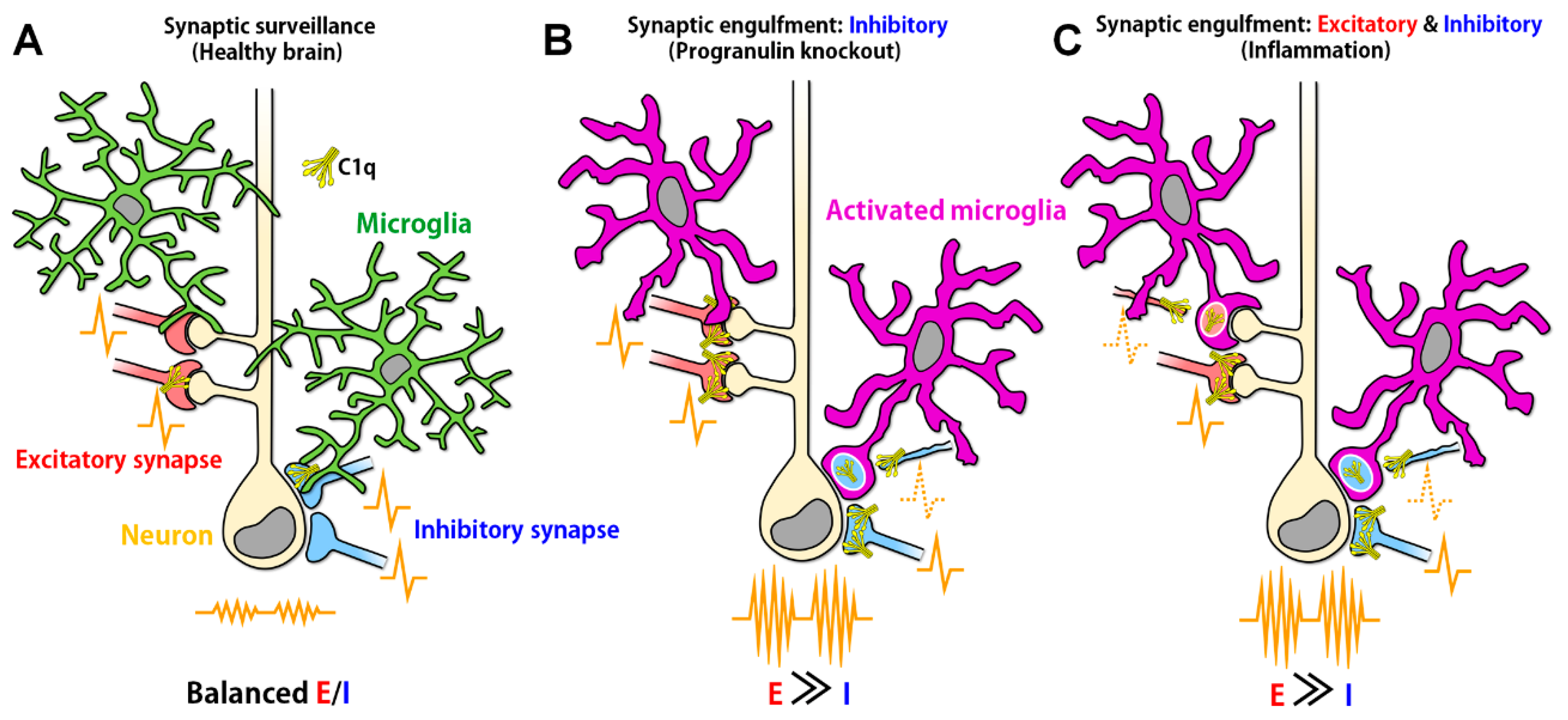
5.2. Progranulin
5.2.1. Synaptic E/I Imbalance Induced by Progranulin Mutation
5.2.2. Progranulin in the Epileptic Brain
5.3. SV2A
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Sillanpää, M.; Schmidt, D. Natural history of treated childhood-onset epilepsy: prospective, long-term population-based study. Brain 2006, 129, 617–624. [Google Scholar] [CrossRef]
- Brodie, M.J.; Barry, S.J.; Bamagous, G.A.; Norrie, J.D.; Kwan, P. Patterns of treatment response in newly diagnosed epilepsy. Neurology 2012, 78, 1548–1554. [Google Scholar] [CrossRef]
- Cope, D.W.; Di Giovanni, G.; Fyson, S.J.; Orbán, G.; Errington, A.C.; Lorincz, M.L.; Gould, T.M.; Carter, D.A.; Crunelli, V. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat. Med. 2009, 15, 1392–1398. [Google Scholar] [CrossRef]
- Shao, L.R.; Habela, C.W.; Stafstrom, C.E. Pediatric Epilepsy Mechanisms: Expanding the Paradigm of Excitation/Inhibition Imbalance. Children (Basel) 2019, 6. [Google Scholar] [CrossRef]
- Ferecskó, A.S.; Jiruska, P.; Foss, L.; Powell, A.D.; Chang, W.C.; Sik, A.; Jefferys, J.G. Structural and functional substrates of tetanus toxin in an animal model of temporal lobe epilepsy. Brain Struct. Funct. 2015, 220, 1013–1029. [Google Scholar] [CrossRef]
- Vossel, K.A.; Tartaglia, M.C.; Nygaard, H.B.; Zeman, A.Z.; Miller, B.L. Epileptic activity in Alzheimer’s disease: causes and clinical relevance. Lancet Neurol. 2017, 16, 311–322. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.Q.; Kreitzer, A.; et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef]
- Centonze, D.; Muzio, L.; Rossi, S.; Cavasinni, F.; De Chiara, V.; Bergami, A.; Musella, A.; D’Amelio, M.; Cavallucci, V.; Martorana, A.; et al. Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J. Neurosci. 2009, 29, 3442–3452. [Google Scholar] [CrossRef]
- Jáidar, O.; Carrillo-Reid, L.; Nakano, Y.; Lopez-Huerta, V.G.; Hernandez-Cruz, A.; Bargas, J.; Garcia-Munoz, M.; Arbuthnott, G.W. Synchronized activation of striatal direct and indirect pathways underlies the behavior in unilateral dopamine-depleted mice. Eur. J. Neurosci. 2019, 49, 1512–1528. [Google Scholar] [CrossRef]
- Andoh, M.; Shibata, K.; Okamoto, K.; Onodera, J.; Morishita, K.; Miura, Y.; Ikegaya, Y.; Koyama, R. Exercise Reverses Behavioral and Synaptic Abnormalities after Maternal Inflammation. Cell Rep. 2019, 27, 2817–2825.e5. [Google Scholar] [CrossRef]
- Testa, G.; Olimpico, F.; Pancrazi, L.; Borello, U.; Cattaneo, A.; Caleo, M.; Costa, M.; Mainardi, M. Cortical Seizures in. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Testa, G.; Mainardi, M.; Olimpico, F.; Pancrazi, L.; Cattaneo, A.; Caleo, M.; Costa, M. A triheptanoin-supplemented diet rescues hippocampal hyperexcitability and seizure susceptibility in FoxG1. Neuropharmacology 2019, 148, 305–310. [Google Scholar] [CrossRef]
- Lee, E.; Lee, J.; Kim, E. Excitation/Inhibition Imbalance in Animal Models of Autism Spectrum Disorders. Biol. Psychiatry 2017, 81, 838–847. [Google Scholar] [CrossRef]
- Wu, Y.; Dissing-Olesen, L.; MacVicar, B.A.; Stevens, B. Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol. 2015, 36, 605–613. [Google Scholar] [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Stogsdill, J.A.; Eroglu, C. The interplay between neurons and glia in synapse development and plasticity. Curr. Opin. Neurobiol. 2017, 42, 1–8. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Miyamoto, A.; Wake, H.; Ishikawa, A.W.; Eto, K.; Shibata, K.; Murakoshi, H.; Koizumi, S.; Moorhouse, A.J.; Yoshimura, Y.; Nabekura, J. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 2016, 7, 12540. [Google Scholar] [CrossRef]
- Coulter, D.A.; Steinhäuser, C. Role of astrocytes in epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022434. [Google Scholar] [CrossRef]
- Morin-Brureau, M.; Milior, G.; Royer, J.; Chali, F.; LeDuigou, C.; Savary, E.; Blugeon, C.; Jourdren, L.; Akbar, D.; Dupont, S.; et al. Microglial phenotypes in the human epileptic temporal lobe. Brain 2018, 141, 3343–3360. [Google Scholar] [CrossRef]
- Avignone, E.; Ulmann, L.; Levavasseur, F.; Rassendren, F.; Audinat, E. Status epilepticus induces a particular microglial activation state characterized by enhanced purinergic signaling. J. Neurosci. 2008, 28, 9133–9144. [Google Scholar] [CrossRef]
- Rappold, P.M.; Lynd-Balta, E.; Joseph, S.A. P2X7 receptor immunoreactive profile confined to resting and activated microglia in the epileptic brain. Brain Res. 2006, 1089, 171–178. [Google Scholar] [CrossRef]
- Mainardi, M.; Pietrasanta, M.; Vannini, E.; Rossetto, O.; Caleo, M. Tetanus neurotoxin-induced epilepsy in mouse visual cortex. Epilepsia 2012, 53, e132–e136. [Google Scholar] [CrossRef]
- Vannini, E.; Restani, L.; Pietrasanta, M.; Panarese, A.; Mazzoni, A.; Rossetto, O.; Middei, S.; Micera, S.; Caleo, M. Altered sensory processing and dendritic remodeling in hyperexcitable visual cortical networks. Brain Struct. Funct. 2016, 221, 2919–2936. [Google Scholar] [CrossRef]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef]
- Janz, R.; Goda, Y.; Geppert, M.; Missler, M.; Südhof, T.C. SV2A and SV2B function as redundant Ca2+ regulators in neurotransmitter release. Neuron 1999, 24, 1003–1016. [Google Scholar] [CrossRef]
- Venkatesan, K.; Alix, P.; Marquet, A.; Doupagne, M.; Niespodziany, I.; Rogister, B.; Seutin, V. Altered balance between excitatory and inhibitory inputs onto CA1 pyramidal neurons from SV2A-deficient but not SV2B-deficient mice. J. Neurosci. Res. 2012, 90, 2317–2327. [Google Scholar] [CrossRef]
- Bialas, A.R.; Presumey, J.; Das, A.; van der Poel, C.E.; Lapchak, P.H.; Mesin, L.; Victora, G.; Tsokos, G.C.; Mawrin, C.; Herbst, R.; et al. Microglia-dependent synapse loss in type I interferon-mediated lupus. Nature 2017, 546, 539–543. [Google Scholar] [CrossRef]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef]
- Tarozzo, G.; Bortolazzi, S.; Crochemore, C.; Chen, S.C.; Lira, A.S.; Abrams, J.S.; Beltramo, M. Fractalkine protein localization and gene expression in mouse brain. J. Neurosci. Res. 2003, 73, 81–88. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bisht, K.; Tremblay, M. Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front. Cell Neurosci. 2014, 8, 129. [Google Scholar] [CrossRef]
- Xu, Y.; Zeng, K.; Han, Y.; Wang, L.; Chen, D.; Xi, Z.; Wang, H.; Wang, X.; Chen, G. Altered expression of CX3CL1 in patients with epilepsy and in a rat model. Am. J. Pathol. 2012, 180, 1950–1962. [Google Scholar] [CrossRef]
- Yeo, S.I.; Kim, J.E.; Ryu, H.J.; Seo, C.H.; Lee, B.C.; Choi, I.G.; Kim, D.S.; Kang, T.C. The roles of fractalkine/CX3CR1 system in neuronal death following pilocarpine-induced status epilepticus. J. Neuroimmunol. 2011, 234, 93–102. [Google Scholar] [CrossRef]
- Del Puerto, A.; Wandosell, F.; Garrido, J.J. Neuronal and glial purinergic receptors functions in neuron development and brain disease. Front. Cell Neurosci. 2013, 7, 197. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Pankratov, Y.; Lalo, U.; Nedergaard, M. P2X receptors in neuroglia. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1. [Google Scholar] [CrossRef]
- Jimenez-Pacheco, A.; Diaz-Hernandez, M.; Arribas-Blázquez, M.; Sanz-Rodriguez, A.; Olivos-Oré, L.A.; Artalejo, A.R.; Alves, M.; Letavic, M.; Miras-Portugal, M.T.; Conroy, R.M.; et al. Transient P2X7 Receptor Antagonism Produces Lasting Reductions in Spontaneous Seizures and Gliosis in Experimental Temporal Lobe Epilepsy. J. Neurosci. 2016, 36, 5920–5932. [Google Scholar] [CrossRef]
- Kim, J.E.; Kang, T.C. The P2X7 receptor-pannexin-1 complex decreases muscarinic acetylcholine receptor-mediated seizure susceptibility in mice. J. Clin. Invest. 2011, 121, 2037–2047. [Google Scholar] [CrossRef]
- Beggs, S.; Trang, T.; Salter, M.W. P2X4R+ microglia drive neuropathic pain. Nat. Neurosci. 2012, 15, 1068–1073. [Google Scholar] [CrossRef]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef]
- Ulmann, L.; Levavasseur, F.; Avignone, E.; Peyroutou, R.; Hirbec, H.; Audinat, E.; Rassendren, F. Involvement of P2X4 receptors in hippocampal microglial activation after status epilepticus. Glia 2013, 61, 1306–1319. [Google Scholar] [CrossRef] [PubMed]
- Akahoshi, N.; Murashima, Y.L.; Himi, T.; Ishizaki, Y.; Ishii, I. Increased expression of the lysosomal protease cathepsin S in hippocampal microglia following kainate-induced seizures. Neurosci. Lett. 2007, 429, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Sasse, V.A.; Wang, Y.; Maulik, M.; Kar, S. Increased levels and activity of cathepsins B and D in kainate-induced toxicity. Neuroscience 2015, 284, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.E.; Nichols, N.R.; Pasinetti, G.M.; Finch, C.E. TGF-beta 1 mRNA increases in macrophage/microglial cells of the hippocampus in response to deafferentation and kainic acid-induced neurodegeneration. Exp. Neurol. 1993, 120, 291–301. [Google Scholar] [CrossRef]
- Makwana, M.; Jones, L.L.; Cuthill, D.; Heuer, H.; Bohatschek, M.; Hristova, M.; Friedrichsen, S.; Ormsby, I.; Bueringer, D.; Koppius, A.; et al. Endogenous transforming growth factor beta 1 suppresses inflammation and promotes survival in adult CNS. J. Neurosci. 2007, 27, 11201–11213. [Google Scholar] [CrossRef]
- Brionne, T.C.; Tesseur, I.; Masliah, E.; Wyss-Coray, T. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron 2003, 40, 1133–1145. [Google Scholar] [CrossRef]
- Zöller, T.; Schneider, A.; Kleimeyer, C.; Masuda, T.; Potru, P.S.; Pfeifer, D.; Blank, T.; Prinz, M.; Spittau, B. Silencing of TGFβ signalling in microglia results in impaired homeostasis. Nat. Commun. 2018, 9, 4011. [Google Scholar] [CrossRef]
- Eriksson, C.; Zou, L.P.; Ahlenius, S.; Winblad, B.; Schultzberg, M. Inhibition of kainic acid induced expression of interleukin-1 beta and interleukin-1 receptor antagonist mRNA in the rat brain by NMDA receptor antagonists. Brain Res. Mol. Brain Res. 2000, 85, 103–113. [Google Scholar] [CrossRef]
- Turrin, N.P.; Rivest, S. Innate immune reaction in response to seizures: implications for the neuropathology associated with epilepsy. Neurobiol. Dis. 2004, 16, 321–334. [Google Scholar] [CrossRef]
- Benson, M.J.; Manzanero, S.; Borges, K. Complex alterations in microglial M1/M2 markers during the development of epilepsy in two mouse models. Epilepsia 2015, 56, 895–905. [Google Scholar] [CrossRef]
- Wang, N.; Mi, X.; Gao, B.; Gu, J.; Wang, W.; Zhang, Y.; Wang, X. Minocycline inhibits brain inflammation and attenuates spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neuroscience 2015, 287, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.N.; Jeon, G.S.; Kim, D.W.; Cho, I.H.; Cho, S.S. Expression of adenomatous polyposis coli protein in reactive astrocytes in hippocampus of kainic acid-induced rat. Neurochem. Res. 2010, 35, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Heo, K.; Cho, Y.J.; Cho, K.J.; Kim, H.W.; Kim, H.J.; Shin, H.Y.; Lee, B.I.; Kim, G.W. Minocycline inhibits caspase-dependent and -independent cell death pathways and is neuroprotective against hippocampal damage after treatment with kainic acid in mice. Neurosci. Lett. 2006, 398, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Parent, J.M.; Yu, T.W.; Leibowitz, R.T.; Geschwind, D.H.; Sloviter, R.S.; Lowenstein, D.H. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J. Neurosci. 1997, 17, 3727–3738. [Google Scholar] [CrossRef] [PubMed]
- Jessberger, S.; Römer, B.; Babu, H.; Kempermann, G. Seizures induce proliferation and dispersion of doublecortin-positive hippocampal progenitor cells. Exp. Neurol. 2005, 196, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Pun, R.Y.; Rolle, I.J.; Lasarge, C.L.; Hosford, B.E.; Rosen, J.M.; Uhl, J.D.; Schmeltzer, S.N.; Faulkner, C.; Bronson, S.L.; Murphy, B.L.; et al. Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron 2012, 75, 1022–1034. [Google Scholar] [CrossRef]
- Parent, J.M.; Elliott, R.C.; Pleasure, S.J.; Barbaro, N.M.; Lowenstein, D.H. Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Ann. Neurol. 2006, 59, 81–91. [Google Scholar] [CrossRef]
- Luo, C.; Ikegaya, Y.; Koyama, R. Microglia and neurogenesis in the epileptic dentate gyrus. Neurogenesis (Austin) 2016, 3, e1235525. [Google Scholar] [CrossRef]
- Yang, F.; Liu, Z.R.; Chen, J.; Zhang, S.J.; Quan, Q.Y.; Huang, Y.G.; Jiang, W. Roles of astrocytes and microglia in seizure-induced aberrant neurogenesis in the hippocampus of adult rats. J. Neurosci. Res. 2010, 88, 519–529. [Google Scholar] [CrossRef]
- Ali, I.; Chugh, D.; Ekdahl, C.T. Role of fractalkine-CX3CR1 pathway in seizure-induced microglial activation, neurodegeneration, and neuroblast production in the adult rat brain. Neurobiol. Dis. 2015, 74, 194–203. [Google Scholar] [CrossRef]
- Luo, C.; Koyama, R.; Ikegaya, Y. Microglia engulf viable newborn cells in the epileptic dentate gyrus. Glia 2016, 64, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Murao, N.; Katano, Y.; Juliandi, B.; Kohyama, J.; Akira, S.; Kawai, T.; Nakashima, K. TLR9 signalling in microglia attenuates seizure-induced aberrant neurogenesis in the adult hippocampus. Nat. Commun. 2015, 6, 6514. [Google Scholar] [CrossRef] [PubMed]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; de Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Huttenlocher, P.R. Morphometric study of human cerebral cortex development. Neuropsychologia 1990, 28, 517–527. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The complement system: An unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Aono, H.; Choudhury, M.E.; Higaki, H.; Miyanishi, K.; Kigami, Y.; Fujita, K.; Akiyama, J.I.; Takahashi, H.; Yano, H.; Kubo, M.; et al. Microglia may compensate for dopaminergic neuron loss in experimental Parkinsonism through selective elimination of glutamatergic synapses from the subthalamic nucleus. Glia 2017, 65, 1833–1847. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, I.; Willems, J.G.; Kooi, E.J.; van Eden, C.; Gold, S.M.; Geurts, J.J.; Baas, F.; Huitinga, I.; Ramaglia, V. Complement C1q-C3-associated synaptic changes in multiple sclerosis hippocampus. Ann. Neurol. 2015, 77, 1007–1026. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, S.K.; Witt, T.; Barbaro, N.M.; Cohen-Gadol, A.A.; Brewster, A.L. Enhanced classical complement pathway activation and altered phagocytosis signaling molecules in human epilepsy. Exp. Neurol. 2017, 295, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Boer, K.; van Vliet, E.A.; Redeker, S.; Baayen, J.C.; Spliet, W.G.; van Rijen, P.C.; Troost, D.; da Silva, F.H.; Wadman, W.J.; et al. Complement activation in experimental and human temporal lobe epilepsy. Neurobiol. Dis. 2007, 26, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Schartz, N.D.; Wyatt-Johnson, S.K.; Price, L.R.; Colin, S.A.; Brewster, A.L. Status epilepticus triggers long-lasting activation of complement C1q-C3 signaling in the hippocampus that correlates with seizure frequency in experimental epilepsy. Neurobiol. Dis. 2018, 109, 163–173. [Google Scholar] [CrossRef]
- Drexel, M.; Preidt, A.P.; Sperk, G. Sequel of spontaneous seizures after kainic acid-induced status epilepticus and associated neuropathological changes in the subiculum and entorhinal cortex. Neuropharmacology 2012, 63, 806–817. [Google Scholar] [CrossRef]
- Abiega, O.; Beccari, S.; Diaz-Aparicio, I.; Nadjar, A.; Layé, S.; Leyrolle, Q.; Gómez-Nicola, D.; Domercq, M.; Pérez-Samartín, A.; Sánchez-Zafra, V.; et al. Neuronal Hyperactivity Disturbs ATP Microgradients, Impairs Microglial Motility, and Reduces Phagocytic Receptor Expression Triggering Apoptosis/Microglial Phagocytosis Uncoupling. PLoS Biol. 2016, 14, e1002466. [Google Scholar] [CrossRef]
- Fonseca, M.I.; Chu, S.H.; Hernandez, M.X.; Fang, M.J.; Modarresi, L.; Selvan, P.; MacGregor, G.R.; Tenner, A.J. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J. Neuroinflamm. 2017, 14, 48. [Google Scholar] [CrossRef]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Li, Y.; Du, X.F.; Liu, C.S.; Wen, Z.L.; Du, J.L. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev. Cell 2012, 23, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Eyo, U.B.; Peng, J.; Swiatkowski, P.; Mukherjee, A.; Bispo, A.; Wu, L.J. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J. Neurosci. 2014, 34, 10528–10540. [Google Scholar] [CrossRef] [PubMed]
- Dissing-Olesen, L.; LeDue, J.M.; Rungta, R.L.; Hefendehl, J.K.; Choi, H.B.; MacVicar, B.A. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J. Neurosci. 2014, 34, 10511–10527. [Google Scholar] [CrossRef] [PubMed]
- Santiago, M.F.; Veliskova, J.; Patel, N.K.; Lutz, S.E.; Caille, D.; Charollais, A.; Meda, P.; Scemes, E. Targeting pannexin1 improves seizure outcome. PLoS ONE 2011, 6, e25178. [Google Scholar] [CrossRef]
- Wu, P.H.; Phillis, J.W. Distribution and release of adenosine triphosphate in rat brain. Neurochem. Res. 1978, 3, 563–571. [Google Scholar] [CrossRef]
- Wieraszko, A.; Goldsmith, G.; Seyfried, T.N. Stimulation-dependent release of adenosine triphosphate from hippocampal slices. Brain Res. 1989, 485, 244–250. [Google Scholar] [CrossRef]
- Madry, C.; Kyrargyri, V.; Arancibia-Cárcamo, I.L.; Jolivet, R.; Kohsaka, S.; Bryan, R.M.; Attwell, D. Microglial Ramification, Surveillance, and Interleukin-1β Release Are Regulated by the Two-Pore Domain K. Neuron 2018, 97, 299–312.e6. [Google Scholar] [CrossRef]
- Chekeni, F.B.; Elliott, M.R.; Sandilos, J.K.; Walk, S.F.; Kinchen, J.M.; Lazarowski, E.R.; Armstrong, A.J.; Penuela, S.; Laird, D.W.; Salvesen, G.S.; et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 2010, 467, 863–867. [Google Scholar] [CrossRef]
- Huang, Y.J.; Maruyama, Y.; Dvoryanchikov, G.; Pereira, E.; Chaudhari, N.; Roper, S.D. The role of pannexin 1 hemichannels in ATP release and cell-cell communication in mouse taste buds. Proc. Natl. Acad. Sci. USA 2007, 104, 6436–6441. [Google Scholar] [CrossRef]
- Thompson, R.J.; Jackson, M.F.; Olah, M.E.; Rungta, R.L.; Hines, D.J.; Beazely, M.A.; MacDonald, J.F.; MacVicar, B.A. Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science 2008, 322, 1555–1559. [Google Scholar] [CrossRef]
- Scemes, E.; Velíšek, L.; Velíšková, J. Astrocyte and Neuronal Pannexin1 Contribute Distinctly to Seizures. ASN Neuro 2019, 11, 1759091419833502. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, R.; Li Puma, D.D.; Mainardi, M.; Lazzarino, G.; Tavazzi, B.; Arancio, O.; Grassi, C. Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons. Glia 2017, 65, 1302–1316. [Google Scholar] [CrossRef] [PubMed]
- Anselmi, F.; Hernandez, V.H.; Crispino, G.; Seydel, A.; Ortolano, S.; Roper, S.D.; Kessaris, N.; Richardson, W.; Rickheit, G.; Filippov, M.A.; et al. ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proc. Natl. Acad. Sci. USA 2008, 105, 18770–18775. [Google Scholar] [CrossRef] [PubMed]
- Nualart-Marti, A.; del Molino, E.M.; Grandes, X.; Bahima, L.; Martin-Satué, M.; Puchal, R.; Fasciani, I.; González-Nieto, D.; Ziganshin, B.; Llobet, A.; et al. Role of connexin 32 hemichannels in the release of ATP from peripheral nerves. Glia 2013, 61, 1976–1989. [Google Scholar] [CrossRef] [PubMed]
- Schock, S.C.; Leblanc, D.; Hakim, A.M.; Thompson, C.S. ATP release by way of connexin 36 hemichannels mediates ischemic tolerance in vitro. Biochem. Biophys. Res. Commun. 2008, 368, 138–144. [Google Scholar] [CrossRef]
- Zappalà, A.; Cicero, D.; Serapide, M.F.; Paz, C.; Catania, M.V.; Falchi, M.; Parenti, R.; Pantò, M.R.; La Delia, F.; Cicirata, F. Expression of pannexin1 in the CNS of adult mouse: cellular localization and effect of 4-aminopyridine-induced seizures. Neuroscience 2006, 141, 167–178. [Google Scholar] [CrossRef]
- Venance, L.; Rozov, A.; Blatow, M.; Burnashev, N.; Feldmeyer, D.; Monyer, H. Connexin expression in electrically coupled postnatal rat brain neurons. Proc. Natl. Acad. Sci. USA 2000, 97, 10260–10265. [Google Scholar] [CrossRef]
- Muldoon, S.F.; Villette, V.; Tressard, T.; Malvache, A.; Reichinnek, S.; Bartolomei, F.; Cossart, R. GABAergic inhibition shapes interictal dynamics in awake epileptic mice. Brain 2015, 138, 2875–2890. [Google Scholar] [CrossRef]
- Peng, Z.; Houser, C.R. Temporal patterns of fos expression in the dentate gyrus after spontaneous seizures in a mouse model of temporal lobe epilepsy. J. Neurosci. 2005, 25, 7210–7220. [Google Scholar] [CrossRef]
- Trapp, B.D.; Wujek, J.R.; Criste, G.A.; Jalabi, W.; Yin, X.; Kidd, G.J.; Stohlman, S.; Ransohoff, R. Evidence for synaptic stripping by cortical microglia. Glia 2007, 55, 360–368. [Google Scholar] [CrossRef]
- Chen, Z.; Jalabi, W.; Hu, W.; Park, H.J.; Gale, J.T.; Kidd, G.J.; Bernatowicz, R.; Gossman, Z.C.; Chen, J.T.; Dutta, R.; et al. Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat. Commun. 2014, 5, 4486. [Google Scholar] [CrossRef] [PubMed]
- Cerri, C.; Genovesi, S.; Allegra, M.; Pistillo, F.; Püntener, U.; Guglielmotti, A.; Perry, V.H.; Bozzi, Y.; Caleo, M. The Chemokine CCL2 Mediates the Seizure-enhancing Effects of Systemic Inflammation. J. Neurosci. 2016, 36, 3777–3788. [Google Scholar] [CrossRef] [PubMed]
- Di Liberto, G.; Pantelyushin, S.; Kreutzfeldt, M.; Page, N.; Musardo, S.; Coras, R.; Steinbach, K.; Vincenti, I.; Klimek, B.; Lingner, T.; et al. Neurons under T Cell Attack Coordinate Phagocyte-Mediated Synaptic Stripping. Cell 2018, 175, 458–471.e19. [Google Scholar] [CrossRef] [PubMed]
- Plata, A.; Lebedeva, A.; Denisov, P.; Nosova, O.; Postnikova, T.Y.; Pimashkin, A.; Brazhe, A.; Zaitsev, A.V.; Rusakov, D.A.; Semyanov, A. Astrocytic Atrophy Following. Front. Mol. Neurosci. 2018, 11, 215. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef]
- Cruts, M.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Wils, H.; Pirici, D.; Rademakers, R.; Vandenberghe, R.; Dermaut, B.; Martin, J.J.; et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006, 442, 920–924. [Google Scholar] [CrossRef]
- Martens, L.H.; Zhang, J.; Barmada, S.J.; Zhou, P.; Kamiya, S.; Sun, B.; Min, S.W.; Gan, L.; Finkbeiner, S.; Huang, E.J.; et al. Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J. Clin. Invest. 2012, 122, 3955–3959. [Google Scholar] [CrossRef]
- Stephan, A.H.; Madison, D.V.; Mateos, J.M.; Fraser, D.A.; Lovelett, E.A.; Coutellier, L.; Kim, L.; Tsai, H.H.; Huang, E.J.; Rowitch, D.H.; et al. A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 2013, 33, 13460–13474. [Google Scholar] [CrossRef]
- Huchtemann, T.; Körtvélyessy, P.; Feistner, H.; Heinze, H.J.; Bittner, D. Progranulin levels in status epilepticus as a marker of neuronal recovery and neuroprotection. Epilepsy Behav. 2015, 49, 170–172. [Google Scholar] [CrossRef]
- Zhu, S.; Tai, C.; Petkau, T.L.; Zhang, S.; Liao, C.; Dong, Z.; Wen, W.; Chang, Q.; Tian Wang, Y.; MacVicar, B.A.; et al. Progranulin promotes activation of microglia/macrophage after pilocarpine-induced status epilepticus. Brain Res. 2013, 1530, 54–65. [Google Scholar] [CrossRef]
- Canafoglia, L.; Morbin, M.; Scaioli, V.; Pareyson, D.; D’Incerti, L.; Fugnanesi, V.; Tagliavini, F.; Berkovic, S.F.; Franceschetti, S. Recurrent generalized seizures, visual loss, and palinopsia as phenotypic features of neuronal ceroid lipofuscinosis due to progranulin gene mutation. Epilepsia 2014, 55, e56–e59. [Google Scholar] [CrossRef] [PubMed]
- Bartholome, O.; Van den Ackerveken, P.; Sánchez Gil, J.; de la Brassinne Bonardeaux, O.; Leprince, P.; Franzen, R.; Rogister, B. Puzzling Out Synaptic Vesicle 2 Family Members Functions. Front. Mol. Neurosci. 2017, 10, 148. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Xiao, F.; Lu, Y.; Huang, Z.; Yuan, J.; Xiao, Z.; Xi, Z.; Wang, X. Down-regulation synaptic vesicle protein 2A in the anterior temporal neocortex of patients with intractable epilepsy. J. Mol. Neurosci. 2009, 39, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, E.A.; Aronica, E.; Redeker, S.; Boer, K.; Gorter, J.A. Decreased expression of synaptic vesicle protein 2A, the binding site for levetiracetam, during epileptogenesis and chronic epilepsy. Epilepsia 2009, 50, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Cesca, F.; Baldelli, P.; Valtorta, F.; Benfenati, F. The synapsins: key actors of synapse function and plasticity. Prog. Neurobiol. 2010, 91, 313–348. [Google Scholar] [CrossRef]
- Rosahl, T.W.; Spillane, D.; Missler, M.; Herz, J.; Selig, D.K.; Wolff, J.R.; Hammer, R.E.; Malenka, R.C.; Südhof, T.C. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 1995, 375, 488–493. [Google Scholar] [CrossRef]
- Farisello, P.; Boido, D.; Nieus, T.; Medrihan, L.; Cesca, F.; Valtorta, F.; Baldelli, P.; Benfenati, F. Synaptic and extrasynaptic origin of the excitation/inhibition imbalance in the hippocampus of synapsin I/II/III knockout mice. Cereb Cortex 2013, 23, 581–593. [Google Scholar] [CrossRef]
- Fassio, A.; Patry, L.; Congia, S.; Onofri, F.; Piton, A.; Gauthier, J.; Pozzi, D.; Messa, M.; Defranchi, E.; Fadda, M.; et al. SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum. Mol. Genet. 2011, 20, 2297–2307. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andoh, M.; Ikegaya, Y.; Koyama, R. Synaptic Pruning by Microglia in Epilepsy. J. Clin. Med. 2019, 8, 2170. https://doi.org/10.3390/jcm8122170
Andoh M, Ikegaya Y, Koyama R. Synaptic Pruning by Microglia in Epilepsy. Journal of Clinical Medicine. 2019; 8(12):2170. https://doi.org/10.3390/jcm8122170
Chicago/Turabian StyleAndoh, Megumi, Yuji Ikegaya, and Ryuta Koyama. 2019. "Synaptic Pruning by Microglia in Epilepsy" Journal of Clinical Medicine 8, no. 12: 2170. https://doi.org/10.3390/jcm8122170
APA StyleAndoh, M., Ikegaya, Y., & Koyama, R. (2019). Synaptic Pruning by Microglia in Epilepsy. Journal of Clinical Medicine, 8(12), 2170. https://doi.org/10.3390/jcm8122170