The Reeler Mouse: A Translational Model of Human Neurological Conditions, or Simply a Good Tool for Better Understanding Neurodevelopment?


Abstract
1. Introduction
2. The Reelin Gene and Protein
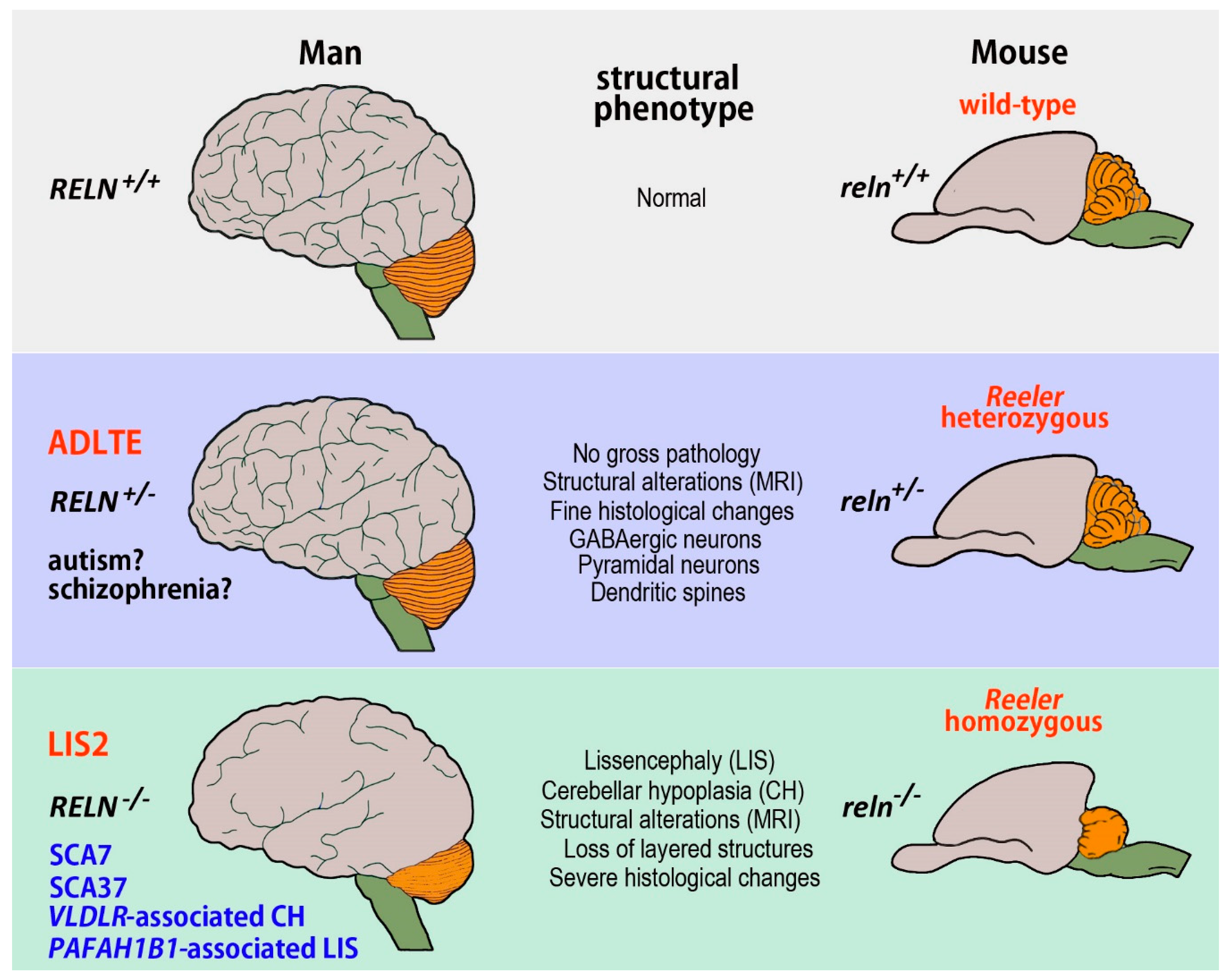
3. RELN-Related Human Neurological Conditions and Their Mouse Counterparts
3.1. Neurological Conditions Caused by RELN Mutations
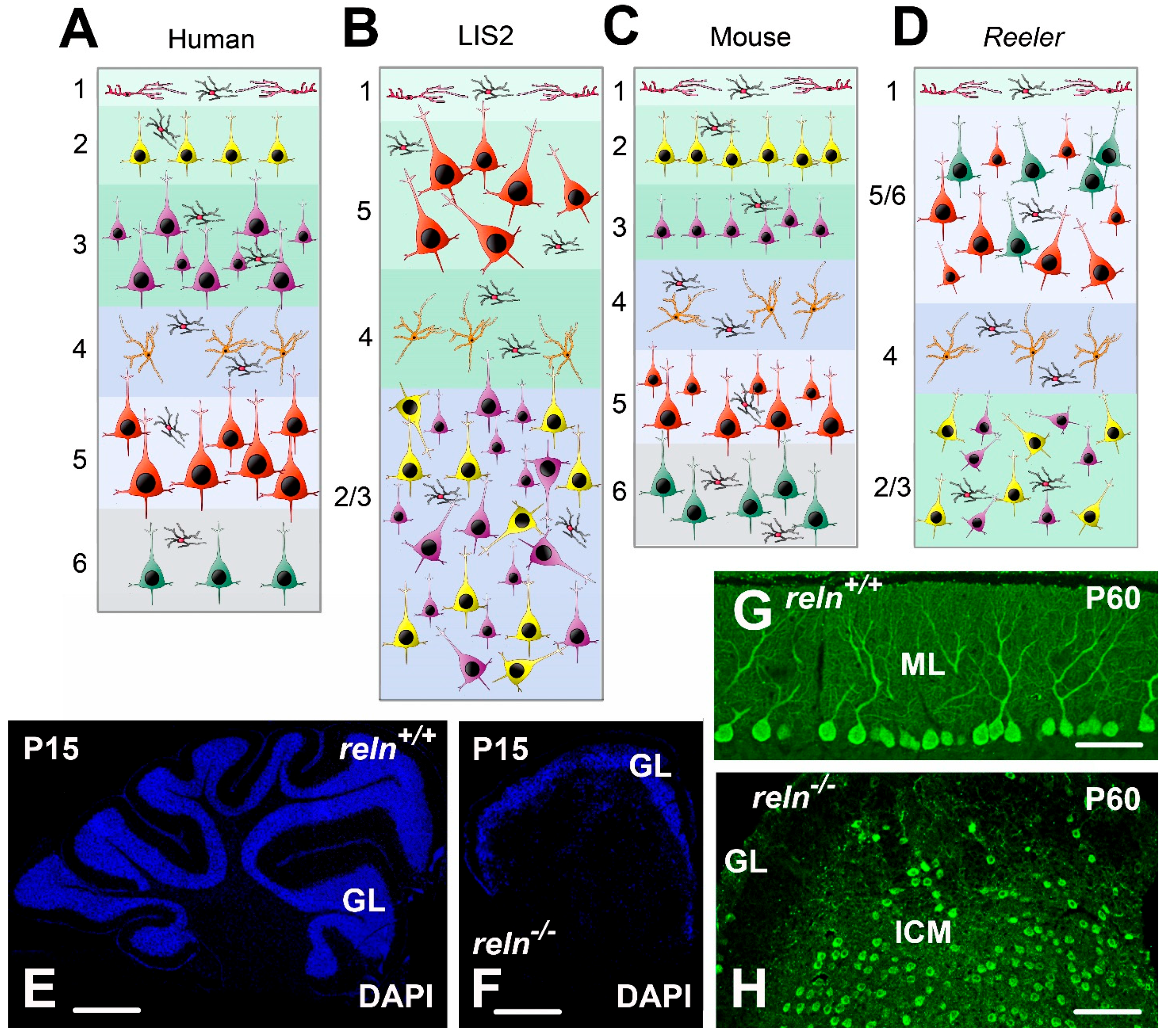
3.1.1. Human Lissencephalies and the Homozygous Reeler Mouse
Lissencephaly 2
Lissencephaly 3
3.1.2. Autosomal-Dominant Lateral Temporal Epilepsy and the Heterozygous Reeler Mouse
3.2. Human Conditions Caused by Mutations of Genes of the Reln Intracellular Pathway and Their Mouse Correlates
3.2.1. VLDLR-Associated Cerebellar Hypoplasia
3.2.2. Spinocerebellar Ataxia Type 37
3.2.3. PAFAH1B1-Associated Lissencephaly/Subcortical Band Heterotopia
3.3. Human Conditions Possibly Related to RELN Mutations and Their Mouse Correlates
3.3.1. Spinocerebellar Ataxia Type 7
3.3.2. Autism and the Heterozygous Reeler Mouse
Genetics
Imaging
Histopathology
3.3.3. Schizophrenia and the Heterozygous Reeler Mouse
Imaging
Histopathology
4. Does the Behavior of Heterozygous Reeler Mice Recall the Human Conditions Related to RELN?
5. Usefulness of the Reeler Mouse in Translational Studies: Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACC | anterior cingulate cortex |
| ADLTE | autosomal-dominant lateral temporal epilepsy |
| ApoER2 | apolipoprotein E2 |
| ARX | aristaless-related homeobox gene |
| ASD | autism spectrum disorders |
| ATXN7 | ataxin 7 |
| CCK | cholecystokinin |
| CNS | central nervous system |
| CNTN6 | contactin 6 |
| DCX | doublecortin |
| DTI | diffusion tractography imaging |
| ETL7 | temporal lobe epilepsy-7 |
| GFP | green fluorescent protein |
| HCN | hyperpolarization-activated cyclic nucleotide–gated |
| HdEEG | high-density electroencephalography |
| ILS | isolated lissencephaly sequence |
| IQ | intelligence quotient |
| LCH | lissencephaly with cerebellar hypoplasia |
| LGI1 | leucine-rich, glioma inactivated 1 gene |
| LIS1 | lissencephaly 1 |
| LIS2 | lissencephaly 2 |
| LIS3 | lissencephaly 3 |
| LTD | long-term depression |
| LTE | lateral temporal epilepsy |
| LTP | long-term potentiation |
| MAP2 | microtubule-associated protein 2 |
| MDS | Miller-Dieker syndrome |
| MEMRI | manganese-enhanced MRI |
| NCAM2 | neural cell adhesion molecule 2 |
| PAFAH1B1 | platelet-activating factor acetylhydrolase IB subunit α |
| PDD-NOS | pervasive developmental disorder-not otherwise specified |
| PFC | prefrontal cortex |
| PPI | pre-pulse inhibition |
| PTP | post tetanic potentiation |
| PV | parvalbumin |
| RELN | Reelin gene (human) |
| Reln | Reelin gene (mouse) |
| RELN | Reelin glycoprotein (human) |
| Reln | Reelin glycoprotein (mouse) |
| ROI | region-of-interest |
| SBH | subcortical band heterotopia |
| SCA37 | spinocerebellar ataxia type 37 |
| SCA7 | spinocerebellar ataxia type 7 |
| SNP | single nucleotide polymorphism |
| SYP1 | synaptophysin 1 |
| TUBA1A | α tubulin 1A |
| VENs | von Economo neurons |
| VLDLR | very low-density lipoprotein receptor |
References
- Andersen, T.E.; Finsen, B.; Goffinet, A.M.; Issinger, O.G.; Boldyreff, B. A reeler mutant mouse with a new, spontaneous mutation in the reelin gene. Brain Res. Mol. Brain Res. 2002, 105, 153–156. [Google Scholar] [CrossRef]
- Tissir, F.; Goffinet, A.M. Reelin and brain development. Nat. Rev. Neurosci. 2003, 4, 496–505. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, G.; Miao, G.G.; Chen, S.C.; Soares, H.D.; Morgan, J.I.; Curran, T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 1995, 374, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Falconer, D.S. Two new mutants, ‘trembler’ and ‘reeler’, with neurological actions in the house mouse (Mus musculus L.). J. Genet. 1951, 50, 192–201. [Google Scholar] [CrossRef]
- Caviness, V.S.; Rakic, P. Mechanisms of Cortical Development: A View from Mutations in Mice. Annu. Rev. Neurosci. 1978, 1, 297–326. [Google Scholar] [CrossRef]
- Goffinet, A.M. Events governing organization of postmigratory neurons: Studies on brain development in normal and reeler mice. Brain Res. Rev. 1984, 7, 261–296. [Google Scholar] [CrossRef]
- Folsom, T.D.; Fatemi, S.H. The involvement of Reelin in neurodevelopmental disorders. Neuropharmacology 2013, 68, 122–135. [Google Scholar] [CrossRef]
- DeSilva, U.; D’Arcangelo, G.; Braden, V.V.; Chen, J.; Miao, G.G.; Curran, T.; Green, E.D. The human reelin gene: Isolation, sequencing, and mapping on chromosome 7. Genome Res. 1997, 7, 157–164. [Google Scholar] [CrossRef]
- Hong, S.E.; Shugart, Y.Y.; Huang, D.T.; Shahwan, S.A.; Grant, P.E.; Hourihane, J.O.; Martin, N.D.; Walsh, C.A. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 2000, 26, 93–96. [Google Scholar] [CrossRef]
- Fatemi, S.H. Reelin glycoprotein in autism and schizophrenia. Int. Rev. Neurobiol. 2005, 71, 179–187. [Google Scholar]
- Fatemi, S.H.; Stary, J.M.; Halt, A.R.; Realmuto, G.R. Dysregulation of Reelin and Bcl-2 proteins in autistic cerebellum. J. Autism Dev. Disord. 2001, 31, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.R.; Feng, G. Annual Research Review: Transgenic mouse models of childhood-onset psychiatric disorders. J. Child Psychol. Psychiatry 2011, 52, 442–475. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.H. Autism spectrum disorders—A genetics review. Genet. Med. 2011, 13, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Vorstman, J.A.S.; Parr, J.R.; Moreno-De-Luca, D.; Anney, R.J.L.; Nurnberger, J.I., Jr.; Hallmayer, J.F. Autism genetics: Opportunities and challenges for clinical translation. Nat. Rev. Genet. 2017, 18, 362–376. [Google Scholar] [CrossRef]
- Geyer, M.A. Developing translational animal models for symptoms of schizophrenia or bipolar mania. Neurotox. Res. 2008, 14, 71–78. [Google Scholar] [CrossRef]
- Chadman, K.K. Animal models for autism in 2017 and the consequential implications to drug discovery. Exp. Opin. Drug Discov. 2017, 12, 1187–1194. [Google Scholar] [CrossRef]
- Zaki, M.; Shehab, M.; El-Aleem, A.A.; Abdel-Salam, G.; Koeller, H.B.; Ilkin, Y.; Ross, M.E.; Dobyns, W.B.; Gleeson, J.G. Identification of a novel recessive RELN mutation using a homozygous balanced reciprocal translocation. Am. J. Med. Genet. A 2007, 143, 939–944. [Google Scholar] [CrossRef]
- Hirotsune, S.; Takahara, T.; Sasaki, N.; Hirose, K.; Yoshiki, A.; Ohashi, T.; Kusakabe, M.; Murakami, Y.; Muramatsu, M.; Watanabe, S. The reeler gene encodes a protein with an EGF-like motif expressed by pioneer neurons. Nat. Genet. 1995, 10, 77–83. [Google Scholar] [CrossRef]
- Ogawa, M.; Miyata, T.; Nakajima, K.; Yagyu, K.; Seike, M.; Ikenaka, K.; Yamamoto, H.; Mikoshiba, K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron 1995, 14, 899–912. [Google Scholar] [CrossRef]
- D’Arcangelo, G. Reelin in the years: Controlling neuronal migration and maturation in the mammalian brain. Adv. Neurosci. 2014, 2014, 597395. [Google Scholar] [CrossRef]
- Hiesberger, T.; Trommsdorff, M.; Howell, B.W.; Goffinet, A.; Mumby, M.C.; Cooper, J.A.; Herz, J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 1999, 24, 481–489. [Google Scholar] [CrossRef]
- Assadi, A.H.; Zhang, G.; Beffert, U.; McNeil, R.S.; Renfro, A.L.; Niu, S.; Quattrocchi, C.C.; Antalffy, B.A.; Sheldon, M.; Armstrong, D.D.; et al. Interaction of reelin signaling and Lis1 in brain development. Nat. Genet. 2003, 35, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Bahi-Buisson, N.; Cavallin, M. Tubulinopathies overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2016. [Google Scholar]
- Lo, N.C.; Chong, C.S.; Smith, A.C.; Dobyns, W.B.; Carrozzo, R.; Ledbetter, D.H. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum. Mol. Genet. 1997, 6, 157–164. [Google Scholar]
- Kato, M.; Dobyns, W.B. Lissencephaly and the molecular basis of neuronal migration. Hum. Mol. Genet. 2003, 12, R89–R96. [Google Scholar] [CrossRef]
- Lecourtois, M.; Poirier, K.; Friocourt, G.; Jaglin, X.; Goldenberg, A.; Saugier-Veber, P.; Chelly, J.; Laquerriere, A. Human lissencephaly with cerebellar hypoplasia due to mutations in TUBA1A: Expansion of the foetal neuropathological phenotype. Acta Neuropathol. 2010, 119, 779–789. [Google Scholar] [CrossRef]
- Ross, M.E.; Swanson, K.; Dobyns, W.B. Lissencephaly with cerebellar hypoplasia (LCH): A heterogeneous group of cortical malformations. Neuropediatrics 2001, 32, 256–263. [Google Scholar] [CrossRef]
- Sergi, C.; Zoubaa, S.; Schiesser, M. Norman-Roberts syndrome: Prenatal diagnosis and autopsy findings. Prenat. Diagn. 2000, 20, 505–509. [Google Scholar] [CrossRef]
- Badea, A.; Nicholls, P.J.; Johnson, G.A.; Wetsel, W.C. Neuroanatomical phenotypes in the reeler mouse. Neuroimage 2007, 34, 1363–1374. [Google Scholar] [CrossRef][Green Version]
- Silva, A.C.; Lee, J.H.; Wu, C.W.H.; Tucciarone, J.; Pelled, G.; Aoki, I.; Koretsky, A.P. Detection of cortical laminar architecture using manganese-enhanced MRI. J. Neurosci. Meth. 2008, 167, 246–257. [Google Scholar] [CrossRef]
- Harsan, L.A.; David, C.; Reisert, M.; Schnell, S.; Hennig, J.; von Elverfeld, D.; Staiger, J.F. Mapping remodeling of thalamocortical projections in the living reeler mouse brain by diffusion tractography. Proc. Natl. Acad. Sci. USA 2013, 110, E1797–E1806. [Google Scholar] [CrossRef]
- Pappas, G.D.; Kriho, V.; Liu, W.S.; Tremolizzo, L.; Lugli, G.; Larson, J. Immunocytochemical localization of reelin in the olfactory bulb of the heterozygous reeler mouse: an animal model for schizophrenia. Neurol. Res. 2003, 25, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, Y.; Terashima, T. Developmental anatomy of reeler mutant mouse. Dev. Growth Differ. 2009, 51, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Wyss, J.M.; Stanfield, B.B.; Cowan, W.M. Structural abnormalities in the olfactory bulb of the Reeler mouse. Brain Res. 1980, 188, 566–571. [Google Scholar] [CrossRef]
- Marrone, M.C.; Marinelli, S.; Biamonte, F.; Keller, F.; Sgobio, C.A.; Ammassari-Teule, M.; Bernardi, G.; Mercuri, N.B. Altered cortico-striatal synaptic plasticity and related behavioural impairments in reeler mice. Eur. J. Neurosci. 2006, 24, 2061–2070. [Google Scholar] [CrossRef]
- Cariboni, A.; Rakic, S.; Liapi, A.; Maggi, R.; Goffinet, A.; Parnavelas, J.G. Reelin provides an inhibitory signal in the migration of gonadotropin-releasing hormone neurons. Development 2005, 132, 4709–4718. [Google Scholar] [CrossRef]
- Stanfield, B.B.; Wyss, J.M.; Cowan, W.M. The projection of the supramammillary region upon the dentate gyrus in normal and reeler mice. Brain Res. 1980, 198, 196–203. [Google Scholar] [CrossRef]
- Baba, K.; Sakakibara, S.; Setsu, T.; Terashima, T. The superficial layers of the superior colliculus are cytoarchitectually and myeloarchitectually disorganized in the reelin-deficient mouse, reeler. Brain Res. 2007, 1140, 205–215. [Google Scholar] [CrossRef]
- Terashima, T. Distribution of mesencephalic trigeminal nucleus neurons in the reeler mutant mouse. Anat. Rec. 1996, 244, 563–571. [Google Scholar] [CrossRef]
- Nishikawa, S.; Goto, S.; Yamada, K.; Hamasaki, T.; Ushio, Y. Lack of Reelin causes malpositioning of nigral dopaminergic neurons: Evidence from comparison of normal and Reln (rl) mutant mice. J. Comp. Neurol. 2003, 461, 166–173. [Google Scholar] [CrossRef]
- Takaoka, Y.; Setsu, T.; Misaki, K.; Yamauchi, T.; Terashima, T. Expression of reelin in the dorsal cochlear nucleus of the mouse. Brain Res. Dev. Brain Res. 2005, 159, 127–134. [Google Scholar] [CrossRef]
- Goffinet, A.M. The embryonic development of the inferior olivary complex in normal and reeler (rlORL) mutant mice. J. Comp. Neurol. 1983, 219, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Terashima, T.; Inoue, K.; Inoue, Y.; Mikoshiba, K.; Tsukada, Y. Observations on the brainstem-spinal descending systems of normal and reeler mutant mice by the retrograde HRP method. J. Comp. Neurol. 1984, 225, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Okado, H.; Terashima, T. Retrograde infection of precerebellar nuclei neurons by injection of a recombinant adenovirus into the cerebellar cortex of normal and reeler mice. Arch. Histol. Cytol. 2007, 70, 51–62. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Villeda, S.A.; Akopians, A.L.; Babayan, A.H.; Basbaum, A.I.; Phelps, P.E. Absence of Reelin results in altered nociception and aberrant neuronal positioning in the dorsal spinal cord. Neuroscience 2006, 139, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Yip, J.W.; Yip, Y.P.; Nakajima, K.; Capriotti, C. Reelin controls position of autonomic neurons in the spinal cord. Proc. Natl. Acad. Sci. USA 2000, 97, 8612–8616. [Google Scholar] [CrossRef]
- Phelps, P.E.; Rich, R.; Dupuy-Davies, S.; Rios, Y.; Wong, T. Evidence for a cell-specific action of Reelin in the spinal cord. Dev. Biol. 2002, 244, 180–198. [Google Scholar] [CrossRef]
- Stanfield, B.B.; Cowan, W.M. The morphology of the hippocampus and dentate gyrus in normal and reeler mice. J. Comp. Neurol. 1979, 185, 393–422. [Google Scholar] [CrossRef]
- Pinto Lord, M.C.; Caviness, V.S., Jr. Determinants of cell shape and orientation: A comparative Golgi analysis of cell-axon interrelationships in the developing neocortex of normal and reeler mice. J. Comp. Neurol. 1979, 187, 49–69. [Google Scholar] [CrossRef]
- Niu, S.; Yabut, O.; D’Arcangelo, G. The Reelin signaling pathway promotes dendritic spine development in hippocampal neurons. J. Neurosci. 2008, 28, 10339–10348. [Google Scholar] [CrossRef]
- Olson, E.C.; Kim, S.; Walsh, C.A. Impaired neuronal positioning and dendritogenesis in the neocortex after cell-autonomous Dab1 suppression. J. Neurosci. 2006, 26, 1767–1775. [Google Scholar] [CrossRef]
- Niu, S.; Renfro, A.; Quattrocchi, C.C.; Sheldon, M.; D’Arcangelo, G. Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron 2004, 41, 71–84. [Google Scholar] [CrossRef]
- Liu, W.S.; Pesold, C.; Rodriguez, M.A.; Carboni, G.; Auta, J.; Lacor, P.; Larson, J.; Condie, B.G.; Guidotti, A.; Costa, E. Down-regulation of dendritic spine and glutamic acid decarboxylase 67 expressions in the reelin haploinsufficient heterozygous reeler mouse. Proc. Natl. Acad. Sci. USA 2001, 98, 3477–3482. [Google Scholar] [CrossRef] [PubMed]
- Chameau, P.; Inta, D.; Vitalis, T.; Monyer, H.; Wadman, W.J.; van Hooft, J.A. The N-terminal region of reelin regulates postnatal dendritic maturation of cortical pyramidal neurons. Proc. Natl. Acad. Sci. USA 2009, 106, 7227–7232. [Google Scholar] [CrossRef] [PubMed]
- Manita, S.; Suzuki, T.; Homma, C.; Matsumoto, T.; Odagawa, M.; Yamada, K.; Ota, K.; Matsubara, C.; Inutsuka, A.; Sato, M.; et al. Top-Down Cortical Circuit for Accurate Sensory Perception. Neuron 2015, 86, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Larkum, M.E.; Petro, L.S.; Sachdev RN, S.; Muckli, L. A Perspective on cortical layering and layer-spanning neuronal elements. Front. Neuroanat. 2018, 12, 56. [Google Scholar] [CrossRef]
- Lörincz, A.; Notomi, T.; Tamás, G.; Shigemoto, R.; Nusser, Z. Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat. Neurosci. 2002, 5, 1185–1193. [Google Scholar] [CrossRef]
- Kupferman, J.V.; Basu, J.; Russo, M.J.; Guevarra, J.; Cheung, S.K.; Siegelbaum, S.A. Reelin signaling specifies the molecular identity of the pyramidal neuron distal dendritic compartment. Cell 2014, 158, 1335–1347. [Google Scholar] [CrossRef][Green Version]
- Meseke, M.; Neumuller, F.; Brunne, B.; Li, X.; Anstotz, M.; Pohlkamp, T.; Rogalla, M.M.; Herz, J.; Rune, G.M.; Bender, R.A. distal dendritic enrichment of HCN1 channels in hippocampal CA1 is promoted by estrogen, but does not require Reelin. eNeuro 2018, 5. [Google Scholar] [CrossRef]
- Bliss, T.V.; Chung, S.H. An electrophysiological study of the hippocampus of the ‘reeler’ mutant mouse. Nature 1974, 252, 153–155. [Google Scholar] [CrossRef]
- Silva, L.R.; Gutnick, M.J.; Connors, B.W. Laminar distribution of neuronal membrane properties in neocortex of normal and reeler mouse. J. Neurophysiol. 1991, 66, 2034–2040. [Google Scholar] [CrossRef]
- Shah, M.M.; Hammond, R.S.; Hoffman, D.A. Dendritic ion channel trafficking and plasticity. Trends Neurosci. 2010, 33, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Beffert, U.; Ertunc, M.; Tang, T.S.; Kavalali, E.T.; Bezprozvanny, I.; Herz, J. Reelin modulates NMDA receptor activity in cortical neurons. J. Neurosci. 2005, 25, 8209–8216. [Google Scholar] [CrossRef] [PubMed]
- Beffert, U.; Weeber, E.J.; Durudas, A.; Qiu, S.; Masiulis, I.; Sweatt, J.D.; Li, W.P.; Adelmann, G.; Frotscher, M.; Hammer, R.E.; et al. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron 2005, 47, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Ishida, A.; Shimazaki, K.; Terashima, T.; Kawai, N. An electrophysiological and immunohistochemical study of the hippocampus of the reeler mutant mouse. Brain Res. 1994, 662, 60–68. [Google Scholar] [CrossRef]
- Pujadas, L.; Gruart, A.; Bosch, C.; Delgado, L.; Teixeira, C.M.; Rossi, D.; De Lecea, L.; Martinez, A.; Delgado-Garcia, J.M.; Soriano, E. Reelin regulates postnatal neurogenesis and enhances spine hypertrophy and long-term potentiation. J. Neurosci. 2010, 30, 4636–4649. [Google Scholar] [CrossRef]
- Rogers, J.T.; Rusiana, I.; Trotter, J.; Zhao, L.; Donaldson, E.; Pak, D.T.; Babus, L.W.; Peters, M.; Banko, J.L.; Chavis, P.; et al. Reelin supplementation enhances cognitive ability, synaptic plasticity, and dendritic spine density. Learn. Mem. 2011, 18, 558–564. [Google Scholar] [CrossRef]
- Weeber, E.J.; Beffert, U.; Jones, C.; Christian, J.M.; Forster, E.; Sweatt, J.D.; Herz, J. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J. Biol. Chem. 2002, 277, 39944–39952. [Google Scholar] [CrossRef]
- Parnavelas, J.G. The origin and migration of cortical neurones: New vistas. Trends Neurosci. 2000, 23, 126–131. [Google Scholar] [CrossRef]
- D’Arcangelo, G. Reelin mouse mutants as models of cortical development disorders. Epilepsy Behav. 2006, 8, 81–90. [Google Scholar] [CrossRef]
- Caviness, V.S., Jr. Patterns of cell and fiber distribution in the neocortex of the reeler mutant mouse. J. Comp. Neurol 1976, 170, 435–447. [Google Scholar] [CrossRef]
- Terashima, T.; Takayama, C.; Ichikawa, R.; Inoue, Y. Dendritic arbolization of large pyramidal neurons in the motor cortex of normal and reeler mutant mouse. Okajimas Folia Anat. Jpn. 1992, 68, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Landrieu, P.; Goffinet, A. Inverted pyramidal neurons and their axons in the neocortex of reeler mutant mice. Cell Tissue Res. 1981, 218, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Terashima, T.; Inoue, K.; Inoue, Y.; Mikoshiba, K.; Tsukada, Y. Distribution and morphology of corticospinal tract neurons in reeler mouse cortex by the retrograde HRP method. J. Comp. Neurol. 1983, 218, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Markram, H.; Toledo-Rodriguez, M.; Wang, Y.; Gupta, A.; Silberberg, G.; Wu, C. Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci. 2004, 5, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Yabut, O.; Renfro, A.; Niu, S.; Swann, J.W.; Marin, O.; D’Arcangelo, G. Abnormal laminar position and dendrite development of interneurons in the reeler forebrain. Brain Res. 2007, 1140, 75–83. [Google Scholar] [CrossRef]
- Hevner, R.F.; Daza, R.A.; Englund, C.; Kohtz, J.; Fink, A. Postnatal shifts of interneuron position in the neocortex of normal and reeler mice: Evidence for inward radial migration. Neuroscience 2004, 124, 605–618. [Google Scholar] [CrossRef]
- Pla, R.; Borrell, V.; Flames, N.; Marin, O. Layer acquisition by cortical GABAergic interneurons is independent of Reelin signaling. J. Neurosci. 2006, 26, 6924–6934. [Google Scholar] [CrossRef]
- Hammond, V.; So, E.; Gunnersen, J.; Valcanis, H.; Kalloniatis, M.; Tan, S.S. Layer positioning of late-born cortical interneurons is dependent on Reelin but not p35 signaling. J. Neurosci. 2006, 26, 1646–1655. [Google Scholar] [CrossRef][Green Version]
- Lakoma, J.; Garcia-Alonso, L.; Luque, J.M. Reelin sets the pace of neocortical neurogenesis. Development 2011, 138, 5223–5234. [Google Scholar] [CrossRef]
- Nishikawa, S.; Goto, S.; Hamasaki, T.; Yamada, K.; Ushio, Y. Involvement of reelin and Cajal-Retzius cells in the developmental formation of vertical columnar structures in the cerebral cortex: Evidence from the study of mouse presubicular cortex. Cereb. Cortex 2002, 12, 1024–1030. [Google Scholar] [CrossRef][Green Version]
- Nishibe, M.; Katsuyama, Y.; Yamashita, T. Developmental abnormality contributes to cortex-dependent motor impairments and higher intracortical current requirement in the reeler homozygous mutants. Brain Struct. Funct. 2018, 223, 2575–2587. [Google Scholar] [CrossRef] [PubMed]
- Simmons, P.A.; Lemmon, V.; Pearlman, A.L. Afferent and efferent connections of the striate and extrastriate visual cortex of the normal and reeler mouse. J. Comp. Neurol. 1982, 211, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Wagener, R.J.; David, C.; Zhao, S.; Haas, C.A.; Staiger, J.F. The somatosensory cortex of reeler mutant mice shows absent layering but intact formation and behavioral activation of columnar somatotopic maps. J. Neurosci. 2010, 30, 15700–15709. [Google Scholar] [CrossRef] [PubMed]
- Pielecka-Fortuna, J.; Wagener, R.J.; Martens, A.K.; Goetze, B.; Schmidt, K.F.; Staiger, J.F.; Lowel, S. The disorganized visual cortex in reelin-deficient mice is functional and allows for enhanced plasticity. Brain Struct. Funct. 2015, 220, 3449–3467. [Google Scholar] [CrossRef]
- Guy, J.; Wagener, R.J.; Mock, M.; Staiger, J.F. persistence of functional sensory maps in the absence of cortical layers in the somsatosensory cortex of Reeler mice. Cereb. Cortex 2015, 25, 2517–2528. [Google Scholar] [CrossRef]
- Prume, M.; Rollenhagen, A.; Lubke, J.H.R. Structural and synaptic organization of the adult reeler mouse somatosensory neocortex: A comparative fine-scale electron microscopic study of Reeler with wild type mice. Front. Neuroanat. 2018, 12, 80. [Google Scholar] [CrossRef]
- Grossberg, S. Towards a unified theory of neocortex: Laminar cortical circuits for vision and cognition. Prog. Brain Res. 2007, 165, 79–104. [Google Scholar]
- D’Souza, R.D.; Burkhalter, A.A. Laminar Organization for Selective Cortico-Cortical Communication. Front. Neuroanat. 2017, 11, 71. [Google Scholar] [CrossRef]
- Guy, J.; Staiger, J.F. The Functioning of a Cortex without Layers. Front. Neuroanat. 2017, 11, 54. [Google Scholar] [CrossRef]
- Mikoshiba, K.; Nagaike, K.; Kohsaka, S.; Takamatsu, K.; Aoki, E.; Tsukada, Y. Developmental studies on the cerebellum from reeler mutant mouse in vivo and in vitro. Dev. Biol. 1980, 79, 64–80. [Google Scholar] [CrossRef]
- Altman, J.; Bayer, S.A. Development of the Cerebellar System in Relation to Its Evolution, Structure and Functions; CRC Press: Boca Raton, FL, USA, 1997. [Google Scholar]
- Cocito, C.; Merighi, A.; Giacobini, M.; Lossi, L. alterations of cell proliferation and apoptosis in the hypoplastic Reeler cerebellum. Front. Cell. Neurosci. 2016, 10, 141. [Google Scholar] [CrossRef] [PubMed]
- Castagna, C.; Merighi, A.; Lossi, L. Cell death and neurodegeneration in the postnatal development of cerebellar vermis in normal and Reeler mice. Ann. Anat. 2016, 207, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Mariani, J.; Crepel, F.; Mikoshiba, K.; Changeux, J.P.; Sotelo, C. Anatomical, physiological and biochemical studies of the cerebellum from Reeler mutant mouse. Philos. Trans. R. Soc. B 1977, 281, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Heckroth, J.A.; Goldowitz, D.; Eisenman, L.M. Purkinje cell reduction in the reeler mutant mouse: A quantitative immunohistochemical study. J. Comp. Neurol. 1989, 279, 546–555. [Google Scholar] [CrossRef]
- Yuasa, S.; Kitoh, J.; Oda, S.; Kawamura, K. Obstructed migration of Purkinje cells in the developing cerebellum of the reeler mutant mouse. Anat. Embryol. 1993, 188, 317–329. [Google Scholar] [CrossRef]
- Castagna, C.; Aimar, P.; Alasia, S.; Lossi, L. Post-natal development of the Reeler mouse cerebellum: An ultrastructural study. Ann. Anat. 2014, 196, 224–235. [Google Scholar] [CrossRef]
- Mariani, J. Extent of multiple innervation of Purkinje cells by climbing fibers in the olivocerebellar system of weaver, reeler, and staggerer mutant mice. J. Neurobiol. 1982, 13, 119–126. [Google Scholar] [CrossRef]
- Terashima, T.; Inoue, K.; Inoue, Y.; Mikoshiba, K.; Tsukada, Y. Observations on Golgi epithelial cells and granule cells in the cerebellum of the reeler mutant mouse. Brain Res. 1985, 350, 103–112. [Google Scholar] [CrossRef]
- Keays, D.A.; Tian, G.; Poirier, K.; Huang, G.J.; Siebold, C.; Cleak, J.; Oliver, P.L.; Fray, M.; Harvey, R.J.; Molnár, Z.; et al. Mutations in αtubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 2007, 128, 45–57. [Google Scholar] [CrossRef]
- Bahi-Buisson, N.; Poirier, K.; Fourniol, F.; Saillour, Y.; Valence, S.; Lebrun, N.; Hully, M.; Bianco, C.F.; Boddaert, N.; Elie, C.; et al. The wide spectrum of tubulinopathies: What are the key features for the diagnosis? Brain 2014, 137, 1676–1700. [Google Scholar] [CrossRef]
- Aiken, J.; Buscaglia, G.; Bates, E.A.; Moore, J.K. The α-tubulin gene TUBA1A in brain development: A key ingredient in the neuronal isotype blend. J. Dev. Biol. 2017, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, F.G.; Freddi, T.A.L.; Taranath, A.; Lakshmanan, R.; Goetti, R.; Feltrin, F.S.; Mankad, K.; Teixeira, S.R.; Hanagandi, P.B.; Arrigoni, F. Tubulinopathies. Top. Magn. Reson. Imaging 2018, 27, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Romaniello, R.; Arrigoni, F.; Fry, A.E.; Bassi, M.T.; Rees, M.I.; Borgatti, R.; Pilz, D.T.; Cushion, T.D. Tubulin genes and malformations of cortical development. Eur. J. Med. Genet. 2018, 61, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Dazzo, E.; Fanciulli, M.; Serioli, E.; Minervini, G.; Pulitano, P.; Binelli, S.; Di, B.C.; Luisi, C.; Pasini, E.; Striano, S.; et al. Heterozygous reelin mutations cause autosomal-dominant lateral temporal epilepsy. Am. J. Hum. Genet. 2015, 96, 992–1000. [Google Scholar] [CrossRef]
- Michelucci, R.; Pulitano, P.; Di Bonaventura, C.; Binelli, S.; Luisi, C.; Pasini, E.; Striano, S.; Striano, P.; Coppola, G.; La Neve, A.; et al. The clinical phenotype of autosomal dominant lateral temporal lobe epilepsy related to reelin mutations. Epilepsy Behav. 2017, 68, 103–107. [Google Scholar] [CrossRef]
- Ceská, K.; Aulická, Š.; Horák, O.; Danhofer, P.; Ríha, P.; Marecek, R.; Šenkyrík, J.; Rektor, I.; Brázdil, M.; Oslejsková, H. Autosomal dominant temporal lobe epilepsy associated with heterozygous reelin mutation: 3 T brain MRI study with advanced neuroimaging methods. Epilepsy Behav. Case Rep. 2019, 11, 39–42. [Google Scholar] [CrossRef]
- Hwang, E.; Brown, R.E.; Kocsis, B.; Kim, T.; McKenna, J.T.; McNally, J.M.; Han, H.B.; Choi, J.H. Optogenetic stimulation of basal forebrain parvalbumin neurons modulates the cortical topography of auditory steady-state responses. Brain Struct. Funct. 2019, 224, 1505–1518. [Google Scholar] [CrossRef]
- Ammassari-Teule, M.; Sgobio, C.; Biamonte, F.; Marrone, C.; Mercuri, N.B.; Keller, F. Reelin haploinsufficiency reduces the density of PV+neurons in circumscribed regions of the striatum and selectively alters striatal-based behaviors. Psychopharmacology 2009, 204, 511–521. [Google Scholar] [CrossRef]
- Nullmeier, S.; Panther, P.; Dobrowolny, H.; Frotscher, M.; Zhao, S.; Schwegler, H.; Wolf, R. Region-specific alteration of GABAergic markers in the brain of heterozygous reeler mice. Eur. J. Neurosci. 2011, 33, 689–698. [Google Scholar] [CrossRef]
- Ding, L.; Satish, S.; Zhou, C.; Gallagher, M.J. Cortical activation in generalized seizures. Epilepsia 2019, 60, 1932–1941. [Google Scholar] [CrossRef]
- Boycott, K.M.; Bonnemann, C.; Herz, J.; Neuert, S.; Beaulieu, C.; Scott, J.N.; Venkatasubramanian, A.; Parboosingh, J.S. Mutations in VLDLR as a cause for autosomal recessive cerebellar ataxia with mental retardation (dysequilibrium syndrome). J. Child Neurol. 2009, 24, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Trommsdorff, M.; Gotthardt, M.; Hiesberger, T.; Shelton, J.; Stockinger, W.; Nimpf, J.; Hammer, R.E.; Richardson, J.A.; Herz, J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 1999, 97, 689–701. [Google Scholar] [CrossRef]
- Korwek, K.M.; Trotter, J.H.; LaDu, M.J.; Sullivan, P.M.; Weeber, E.J. ApoE isoform-dependent changes in hippocampal synaptic function. Mol. Neurodegener. 2009, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Matilla-Dueñas, A.; Volpini, V. Spinocerebellar ataxia type 37. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Serrano-Munuera, C.; Corral-Juan, M.; Stevanin, G.; San Nicolás, H.; Roig, C.; Corral, J.; Campos, B.; De Jorge, L.; Morcillo-Suárez, C.; Navarro, A.; et al. New subtype of spinocerebellar ataxia with altered vertical eye movements mapping to chromosome 1p32 subtype of SCA with altered vertical eye movements. JAMA Neurol. 2013, 70, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Seixas, A.I.; Loureiro, J.R.; Costa, C.; Ordóñez-Ugalde, A.S.; Marcelino, H.; Oliveira, C.L.; Loureiro, J.L.; Dhingra, A.; Brandão, E.; Cruz, V.T.; et al. A Pentanucleotide ATTTC Repeat Insertion in the Non-coding Region of DAB1, Mapping to SCA37, Causes Spinocerebellar Ataxia. Am. J. Hum. Genet. 2017, 101, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Ware, M.L.; Fox, J.W.; González, J.L.; Davis, N.M.; De Rouvroit, C.L.; Russo, C.J.; Chua, S.C.; Goffinet, A.M.; Walsh, C.A. Aberrant splicing of a mouse disabled homolog, mdab1, in the scrambler Mouse. Neuron 1997, 19, 239–249. [Google Scholar] [CrossRef]
- Dobyns, W.B.; Das, S. PAFAH1B1-Associated Lissencephaly/Subcortical Band Heterotopia. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2014. [Google Scholar]
- Katayama, K.I.; Hayashi, K.; Inoue, S.; Sakaguchi, K.; Nakajima, K. Enhanced expression of Pafah1b1 causes over-migration of cerebral cortical neurons into the marginal zone. Brain Struct. Funct. 2017, 222, 4283–4291. [Google Scholar] [CrossRef]
- Hunt, R.F.; Dinday, M.T.; Hindle-Katel, W.; Baraban, S.C. LIS1 Deficiency Promotes Dysfunctional synaptic integration of granule cells generated in the developing and adult dentate gyrus. J. Neurosci. 2012, 32, 12862–12875. [Google Scholar] [CrossRef]
- Zhang, G.; Assadi, A.H.; McNeil, R.S.; Beffert, U.; Wynshaw-Boris, A.; Herz, J.; Clark, G.D.; D’Arcangelo, G. The Pafah1b complex interacts with the Reelin receptor VLDLR. PLoS ONE 2007, 2, e252. [Google Scholar] [CrossRef]
- Enevoldson, T.P.; Sanders, M.D.; Harding, A.E. Autosomal dominant cerebellar ataxia with pigmentary macular dystrophy. A clinical and genetic study of eight families. Brain 1994, 117, 445–460. [Google Scholar] [CrossRef]
- McCullough, S.D.; Xu, X.; Dent, S.Y.; Bekiranov, S.; Roeder, R.G.; Grant, P.A. Reelin is a target of polyglutamine expanded ataxin-7 in human spinocerebellar ataxia type 7 (SCA7) astrocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 21319–21324. [Google Scholar] [CrossRef] [PubMed]
- Lammert, D.B.; Howell, B.W. RELN Mutations in Autism Spectrum Disorder. Front. Cell Neurosci. 2016, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Persico, A.M.; D’Agruma, L.; Maiorano, N.; Totaro, A.; Militerni, R.; Bravaccio, C.; Wassink, T.H.; Schneider, C.; Melmed, R.; Trillo, S.; et al. Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol. Psychiatry 2001, 6, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, X.; Zhang, C.; Mundo, E.; Macciardi, F.; Grayson, D.R.; Guidotti, A.R.; Holden, J.J.A. Reelin gene alleles and susceptibility to autism spectrum disorders. Mol. Psychiatry 2002, 7, 1012–1017. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Holt, R.; Barnby, G.; Maestrini, E.; Bacchelli, E.; Brocklebank, D.; Sousa, I.S.; Mulder, E.J.; Kantojärvi, K.; Järvelä, I.; Klauck, S.M.; et al. Linkage and candidate gene studies of autism spectrum disorders in European populations. Eur. J. Hum. Genet. 2010, 18, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Devlin, B.; Bennett, P.; Dawson, G.; Figlewicz, D.A.; Grigorenko, E.L.; McMahon, W.; Minshew, N.; Pauls, D.; Smith, M.; Spence, M.A.; et al. Alleles of a reelin CGG repeat do not convey liability to autism in a sample from the CPEA network. Am. J. Med. Genet. 2004, 126, 46–50. [Google Scholar] [CrossRef]
- Bonora, E.; Beyer, K.S.; Lamb, J.A.; Parr, J.R.; Klauck, S.M.; Benner, A.; Paolucci, M.; Abbott, A.; Ragoussis, I.; Poustka, A.; et al. Analysis of reelin as a candidate gene for autism. Mol. Psychiatry 2003, 8, 885–892. [Google Scholar] [CrossRef][Green Version]
- Wang, Z.; Hong, Y.; Zou, L.; Zhong, R.; Zhu, B.; Shen, N.; Chen, W.; Lou, J.; Ke, J.; Zhang, T.; et al. Reelin gene variants and risk of autism spectrum disorders: An integrated meta-analysis. Am. J. Med. Genet. 2014, 165, 192–200. [Google Scholar] [CrossRef]
- Parihar, R.; Ganesh, S. Autism genes: The continuum that connects us all. J. Genet. 2016, 95, 481–483. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Ercument Cicek, A.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.F.; Stevens, C.; Wang, L.S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H. Reelin mutations in mouse and man: From reeler mouse to schizophrenia, mood disorders, autism and lissencephaly. Mol. Psychiatry 2001, 6, 129–133. [Google Scholar] [CrossRef]
- Ecker, C. The neuroanatomy of autism spectrum disorder: An overview of structural neuroimaging findings and their translatability to the clinical setting. Autism 2017, 21, 18–28. [Google Scholar] [CrossRef]
- Lange, N.; Travers, B.G.; Bigler, E.D.; Prigge, M.B.; Froehlich, A.L.; Nielsen, J.A.; Cariello, A.N.; Zielinski, B.A.; Anderson, J.S.; Fletcher, P.T.; et al. Longitudinal volumetric brain changes in autism spectrum disorder ages 6–35 years. Autism Res. 2015, 8, 82–93. [Google Scholar] [CrossRef]
- Shen, M.D.; Nordahl, C.W.; Young, G.S.; Wootton-Gorges, S.L.; Lee, A.; Liston, S.E.; Harrington, K.R.; Ozonoff, S.; Amaral, D.G. Early brain enlargement and elevated extra-axial fluid in infants who develop autism spectrum disorder. Brain 2013, 136, 2825–2835. [Google Scholar] [CrossRef]
- Schumann, C.M.; Bloss, C.S.; Barnes, C.C.; Wideman, G.M.; Carper, R.A.; Akshoomoff, N.; Pierce, K.; Hagler, D.; Schork, N.; Lord, C.; et al. Longitudinal magnetic resonance imaging study of cortical development through early childhood in autism. J. Neurosci. 2010, 30, 4419–4427. [Google Scholar] [CrossRef]
- Hazlett, H.C.; Poe, M.D.; Gerig, G.; Styner, M.; Chappell, C.; Smith, R.G.; Vachet, C.; Piven, J. Early brain overgrowth in autism associated with an increase in cortical surface area before age 2 years. Arch. Gen. Psychiatry 2011, 68, 467–476. [Google Scholar] [CrossRef]
- Minshew, N.J.; Sweeney, J.A.; Bauman, M.L.; Webb, S.J. Neurologic aspects of autism. In Handbook of Autism and Pervasive Developmental Disorders; Volkmar, F.R., Paul, R., Klin, A., Cohen, D., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005; pp. 473–514. [Google Scholar] [CrossRef]
- Courchesne, E.; Yeung-Courchesne, R.; Hesselink, J.R.; Jernigan, T.L. Hypoplasia of cerebellar vermal lobules VI and VII in autism. N. Engl. J. Med. 1988, 318, 1349–1354. [Google Scholar] [CrossRef]
- Courchesne, E.; Saitoh, O.; Townsend, J.; Yeung-Courchesne, R.; Press, G.; Lincoln, A.; Haas, R.; Schreibman, L. Cerebellar hypoplasia and hyperplasia in infantile autism. Lancet 1994, 343, 63–64. [Google Scholar] [CrossRef]
- Scott, J.A.; Schumann, C.M.; Goodlin-Jones, B.L.; Amaral, D.G. A comprehensive volumetric analysis of the cerebellum in children and adolescents with autism spectrum disorder. Autism Res. 2009, 2, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Piven, J.; Saliba, K.; Bailey, J.; Arndt, S. An MRI study of autism: The cerebellum revisited. Neurology 1997, 49, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Lucibello, S.; Verdolotti, T.; Giordano, F.M.; Lapenta, L.; Infante, A.; Piludu, F.; Tartaglione, T.; Chieffo, D.; Colosimo, C.; Mercuri, E.; et al. Brain morphometry of preschool age children affected by autism spectrum disorder: Correlation with clinical findings. Clin. Anat. 2019, 32, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Oguro-Ando, A.; Zuko, A.; Kleijer, K.T.E.; Burbach, J.P.H. A current view on contactin-4,-5, and-6: Implications in neurodevelopmental disorders. Mol. Cell. Neurosci. 2017, 81, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Kemper, T.L.; Bauman, M.L. The contribution of neuropathologic studies to the understanding of autism. Neurol. Clin. 1993, 11, 175–187. [Google Scholar] [CrossRef]
- Amaral, D.G.; Schumann, C.M.; Nordahl, C.W. Neuroanatomy of autism. Trends Neurosci. 2008, 31, 137–145. [Google Scholar] [CrossRef]
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.D.; et al. Autism spectrum disorder: Neuropathology and animal models. Acta Neuropathol. 2017, 134, 537–566. [Google Scholar] [CrossRef]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Imaki, H.; Wegiel, J.; Marchi, E.; Ma, S.Y.; Chauhan, A.; Chauhan, V.; Bobrowicz, T.W.; et al. The neuropathology of autism: Defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010, 119, 755–770. [Google Scholar] [CrossRef]
- Wegiel, J.; Flory, M.; Kuchna, I.; Nowicki, K.; Ma, S.Y.; Imaki, H.; Wegiel, J.; Frackowiak, J.; Kolecka, B.M.; Wierzba-Bobrowicz, T.; et al. Neuronal nucleus and cytoplasm volume deficit in children with autism and volume increase in adolescents and adults. Acta Neuropathol. Commun. 2015, 3, 2. [Google Scholar] [CrossRef]
- Casanova, M.F. Neuropathological and genetic findings in autism: The significance of a putative minicolumnopathy. Neuroscientist 2006, 12, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Mukaetova-Ladinska, E.B.; Arnold, H.; Jaros, E.; Perry, R.; Perry, E. Depletion of MAP2 expression and laminar cytoarchitectonic changes in dorsolateral prefrontal cortex in adult autistic individuals. Neuropathol. Appl. Neurobiol. 2004, 30, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Raymond, G.V.; Bauman, M.L.; Kemper, T.L. Hippocampus in autism: A Golgi analysis. Acta Neuropathol. 1996, 91, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Hutsler, J.J.; Zhang, H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010, 1309, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Camacho, J.; Ejaz, E.; Ariza, J.; Noctor, S.C.; Martínez-Cerdeño, V.N. RELN-expressing neuron density in layer I of the superior temporal lobe is similar in human brains with autism and in age-matched controls. Neurosci. Lett. 2014, 579, 163–167. [Google Scholar] [CrossRef]
- Courchesne, E.; Mouton, P.R.; Calhoun, M.E.; Semendeferi, K.; Ahrens-Barbeau, C.; Hallet, M.J.; Barnes, C.C.; Pierce, K. Neuron number and size in prefrontal cortex of children with autism. JAMA 2011, 306, 2001–2010. [Google Scholar] [CrossRef]
- Hashemi, E.; Ariza, J.; Rogers, H.; Noctor, S.C.; Martínez-Cerdeño, V.N. The number of parvalbumin-expressing interneurons is decreased in the prefrontal cortex in autism. Cereb. Cortex 2017, 27, 1931–1943. [Google Scholar]
- Jacot-Descombes, S.; Uppal, N.; Wicinski, B.; Santos, M.; Schmeidler, J.; Giannakopoulos, P.; Heinsein, H.; Schmitz, C.; Hof, P.R. Decreased pyramidal neuron size in Brodmann areas 44 and 45 in patients with autism. Acta Neuropathol. 2012, 124, 67–79. [Google Scholar] [CrossRef]
- Zhubi, A.; Chen, Y.; Guidotti, A.; Grayson, D.R. Epigenetic regulation of RELN and GAD1 in the frontal cortex (FC) of autism spectrum disorder (ASD) subjects. Int. J. Dev. Neurosci. 2017, 62, 63–72. [Google Scholar] [CrossRef]
- Van Kooten, I.A.J.; Palmen, S.J.M.C.; Von Cappeln, P.; Steinbusch, H.W.M.; Korr, H.; Heinsen, H.; Hof, P.R.; Van Engeland, H.; Schmitz, C. Neurons in the fusiform gyrus are fewer and smaller in autism. Brain 2008, 131, 987–999. [Google Scholar] [CrossRef]
- Oblak, A.L.; Rosene, D.L.; Kemper, T.L.; Bauman, M.L.; Blatt, G.J. Altered posterior cingulate cortical cyctoarchitecture, but normal density of neurons and interneurons in the posterior cingulate cortex and fusiform gyrus in autism. Autism Res. 2011, 4, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.P.; Semendeferi, K.; Courchesne, E. No reduction of spindle neuron number in frontoinsular cortex in autism. Brain Cogn. 2007, 64, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Allman, J.M.; Watson, K.K.; Tetreault, N.A.; Hakeem, A.Y. Intuition and autism: A possible role for Von Economo neurons. Trends Cogn. Sci. 2005, 9, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.; Uppal, N.; Butti, C.; Wicinski, B.; Schmeidler, J.; Giannakopoulos, P.; Heinsen, H.; Schmitz, C.; Hof, P.R. Von Economo neurons in autism: A stereologic study of the frontoinsular cortex in children. Brain Res. 2011, 1380, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Simms, M.L.; Kemper, T.L.; Timbie, C.M.; Bauman, M.L.; Blatt, G.J. The anterior cingulate cortex in autism: Heterogeneity of qualitative and quantitative cytoarchitectonic features suggests possible subgroups. Acta Neuropathol. 2009, 118, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Uppal, N.; Wicinski, B.; Buxbaum, J.D.; Heinsen, H.; Schmitz, C.; Hof, P.R. Neuropathology of the Anterior Midcingulate Cortex in Young Children with Autism. J. Neuropathol. Exp. Neurol. 2014, 73, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Weidenheim, K.M.; Goodman, L.; Dickson, D.W.; Gillberg, C.; Råstam, M.; Rapin, I. Etiology and Pathophysiology of Autistic Behavior: Clues from Two Cases with an Unusual Variant of Neuroaxonal Dystrophy. J. Child Neurol. 2001, 16, 809–819. [Google Scholar] [CrossRef]
- Hussman, J.P.; Chung, R.H.; Griswold, A.J.; Jaworski, J.M.; Salyakina, D.; Ma, D.; Konidari, I.; Whitehead, P.L.; Vance, J.M.; Martin, E.R.; et al. A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Mol. Autism 2011, 2, 1. [Google Scholar] [CrossRef]
- Ichinohe, N.; Knight, A.; Ogawa, M.; Ohshima, T.; Mikoshiba, K.; Yoshihara, Y.; Terashima, T.; Rockland, K.S. Unusual Patch–Matrix Organization in the Retrosplenial Cortex of the Reeler Mouse and Shaking Rat Kawasaki. Cereb. Cortex 2007, 18, 1125–1138. [Google Scholar] [CrossRef]
- Ichinohe, N. Small-Scale Module of the Rat Granular retrosplenial cortex: An example of the minicolumn-like structure of the cerebral cortex. Front. Neuroanat. 2012, 5, 69. [Google Scholar] [CrossRef]
- Lui, J.; Hansen, D.; Kriegstein, A. Development and Evolution of the Human Neocortex. Cell 2011, 146, 18–36. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Fuso, A.; Laviola, G. Nicotine restores Wt-like levels of reelin and GAD67 gene expression in brain of heterozygous reeler mice. Neurotox. Res. 2013, 24, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.; Luthert, P.; Dean, A.; Harding, B.; Janota, I.; Montgomery, M.; Rutter, M.; Lantos, P. A clinicopathological study of autism. Brain 1998, 121, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, Y.A.; Kemper, T.L.; Bauman, M.L.; Blatt, G.J. Parvalbumin-, calbindin-, and calretinin-immunoreactive hippocampal interneuron density in autism. Acta Neurol. Scand. 2010, 121, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Zhao, L.; Trotter, J.H.; Rusiana, I.; Peters, M.M.; Li, Q.; Donaldson, E.; Banko, J.L.; Keenoy, K.E.; Rebeck, G.W.; et al. Reelin supplementation recovers sensorimotor gating, synaptic plasticity and associative learning deficits in the heterozygous reeler mouse. J. Psychopharmacol. 2013, 27, 386–395. [Google Scholar] [CrossRef]
- Qiu, S.; Zhao, L.F.; Korwek, K.M.; Weeber, E.J. Differential Reelin-Induced Enhancement of NMDA and AMPA Receptor Activity in the Adult Hippocampus. J. Neurosci. 2006, 26, 12943. [Google Scholar] [CrossRef]
- Qiu, S.; Korwek, K.M.; Pratt-Davis, A.R.; Peters, M.; Bergman, M.Y.; Weeber, E.J. Cognitive disruption and altered hippocampus synaptic function in Reelin haploinsufficient mice. Neurobiol. Learn. Mem. 2006, 85, 228–242. [Google Scholar] [CrossRef]
- Hellwig, S.; Hack, I.; Kowalski, J.; Brunne, B.; Jarowyj, J.; Unger, A.; Bock, H.H.; Junghans, D.; Frotscher, M. Role for Reelin in neurotransmitter release. J. Neurosci. 2011, 31, 2352–2360. [Google Scholar] [CrossRef]
- Bauman, M.; Kemper, T.L. Histoanatomic observations of the brain in early infantile autism. Neurology 1985, 35, 866–874. [Google Scholar] [CrossRef]
- Schumann, C.M.; Amaral, D.G. Stereological Analysis of Amygdala Neuron Number in Autism. J. Neurosci. 2006, 26, 7674–7679. [Google Scholar] [CrossRef]
- Wegiel, J.; Flory, M.; Kuchna, I.; Nowicki, K.; Ma, S.Y.; Imaki, H.; Wegiel, J.; Cohen, I.L.; London, E.; Wisniewski, T.; et al. Stereological study of the neuronal number and volume of 38 brain subdivisions of subjects diagnosed with autism reveals significant alterations restricted to the striatum, amygdala and cerebellum. Acta Neuropathol. Commun. 2014, 2, 141. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.T.; Barger, N.; Amaral, D.G.; Schumann, C.M. Stereological study of amygdala glial populations in adolescents and adults with autism spectrum disorder. PLoS ONE 2014, 9, e110356. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.P.; Bernard, A.; Thompson, C.L.; Ng, L.; Boe, A.; Mortrud, M.; Hawrylycz, M.J.; Jones, A.R.; Hevner, R.F.; Lein, E.S. Cell-type-specific consequences of reelin deficiency in the mouse neocortex, hippocampus, and amygdala. J. Comp. Neurol. 2011, 519, 2061–2089. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Aldinger, K.A.; Ashwood, P.; Bauman, M.L.; Blaha, C.D.; Blatt, G.J.; Chauhan, A.; Chauhan, V.; Dager, S.R.; Dickson, P.E.; et al. Consensus paper: Pathological role of the cerebellum in autism. Cerebellum 2012, 11, 777–807. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, A.M.; Crocetti, D.; Mostofsky, S.H.; Stoodley, C.J. Cerebellar gray matter and lobular volumes correlate with core autism symptoms. Neuroimage Clin. 2015, 7, 631–639. [Google Scholar] [CrossRef]
- Hampson, D.R.; Blatt, G.J. Autism spectrum disorders and neuropathology of the cerebellum. Front. Neurosci. 2015, 9, 420. [Google Scholar] [CrossRef] [PubMed]
- Skefos, J.; Cummings, C.; Enzer, K.; Holiday, J.; Weed, K.; Levy, E.; Yuce, T.; Kemper, T.; Bauman, M. Regional alterations in purkinje cell density in patients with autism. PLoS ONE 2014, 9, e81255. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Halt, A.R.; Realmuto, G.; Earle, J.; Kist, D.A.; Thuras, P.; Merz, A. Purkinje cell size is reduced in cerebellum of patients with autism. Cell. Mol. Neurobiol. 2002, 22, 171–175. [Google Scholar] [CrossRef]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Imaki, H.; Wegiel, J.; Ma, S.Y.; Azmitia, E.C.; Banerjee, P.; Flory, M.; Cohen, I.L.; et al. Contribution of olivofloccular circuitry developmental defects to atypical gaze in autism. Brain Res. 2013, 1512, 106–122. [Google Scholar] [CrossRef]
- Whitney, E.R.; Kemper, T.L.; Rosene, D.L.; Bauman, M.L.; Blatt, G.J. Density of cerebellar basket and stellate cells in autism: Evidence for a late developmental loss of Purkinje cells. J. Neurosci. Res. 2009, 87, 2245–2254. [Google Scholar] [CrossRef]
- Maloku, E.; Covelo, I.R.; Hanbauer, I.; Guidotti, A.; Kadriu, B.; Hu, Q.; Davis, J.M.; Costa, E. Lower number of cerebellar Purkinje neurons in psychosis is associated with reduced reelin expression. Proc. Natl. Acad. Sci. USA 2010, 107, 4407–4411. [Google Scholar] [CrossRef] [PubMed]
- Magliaro, C.; Cocito, C.; Bagatella, S.; Merighi, A.; Ahluwalia, A.; Lossi, L. The number of Purkinje neurons and their topology in the cerebellar vermis of normal and reln haplodeficient mouse. Ann. Anat. 2016, 207, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Keller, R.; Basta, R.; Salerno, L.; Elia, M. Autism, epilepsy, and synaptopathies: A not rare association. Neurol. Sci. 2017, 38, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Mercati, O.; Huguet, G.; Danckaert, A.; André-Leroux, G.; Maruani, A.; Bellinzoni, M.; Rolland, T.; Gouder, L.; Mathieu, A.; Buratti, J.; et al. CNTN6 mutations are risk factors for abnormal auditory sensory perception in autism spectrum disorders. Mol. Psychiatry 2016, 22, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Castagna, C.; Merighi, A.; Lossi, L. Decreased expression of synaptophysin 1 (SYP1 major synaptic vesicle protein p38) and contactin 6 (CNTN6/NB3) in the cerebellar vermis of reln haplodeficient mice. Cell. Mol. Neurobiol. 2019, 39, 833–856. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, M.; Tochitsky, I.; Buchholz, D.E.; Winden, K.; Kujala, V.; Kapur, K.; Cataltepe, D.; Turner, D.; Han, M.J.; Woolf, C.J.; et al. Purkinje cells derived from TSC patients display hypoexcitability and synaptic deficits associated with reduced FMRP levels and reversed by rapamycin. Mol. Psychiatry 2018, 23, 2167–2183. [Google Scholar] [CrossRef]
- Tassano, E.; Uccella, S.; Giacomini, T.; Severino, M.; Fiorio, P.; Gimelli, G.; Ronchetto, P. Clinical and molecular characterization of two patients with CNTN6 copy number variations. Cytogenet. Genome Res. 2018, 156, 144–149. [Google Scholar] [CrossRef]
- Garcia-Ortiz, J.E.; Zarazúa-Niño, A.I.; Hernández-Orozco, A.A.; Reyes-Oliva, E.A.; Pérez-Ávila, C.E.; Becerra-Solano, L.E.; Galán-Huerta, K.A.; Rivas-Estilla, A.M.; Córdova-Fletes, C. Case Report: Whole exome sequencing unveils an inherited truncating variant in CNTN6 (p. Ser189Ter) in a mexican child with autism spectrum disorder. J. Autism Dev. Disord. 2019. [Google Scholar] [CrossRef]
- Hu, J.; Liao, J.; Sathanoori, M.; Kochmar, S.; Sebastian, J.; Yatsenko, S.A.; Surti, U. CNTN6 copy number variations in 14 patients: A possible candidate gene for neurodevelopmental and neuropsychiatric disorders. J. Neurodev. Disord. 2015, 7, 26. [Google Scholar] [CrossRef]
- Stoodley, C.J.; Schmahmann, J.D. Evidence for topographic organization in the cerebellum of motor control versus cognitive and affective processing. Cortex 2010, 46, 831–844. [Google Scholar] [CrossRef]
- Schmahmann, J.D.; Weilburg, J.B.; Sherman, J.C. The neuropsychiatry of the cerebellum—insights from the clinic. Cerebellum 2007, 6, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Catani, M.; Jones, D.K.; Daly, E.; Embiricos, N.; Deeley, Q.; Pugliese, L.; Curran, S.; Robertson, D.; Murphy, D.G. Altered cerebellar feedback projections in Asperger syndrome. Neuroimage 2008, 41, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Catani, M.; Dell’Acqua, F.; de Schotten, M.T. A revised limbic system model for memory, emotion and behaviour. Neurosci. Biobehav. Rev. 2013, 37, 1724–1737. [Google Scholar] [CrossRef] [PubMed]
- Bauman, M.L. Microscopic neuroanatomic abnormalities in autism. Pediatrics 1991, 87, 791–796. [Google Scholar]
- Stanfield, A.C.; McIntosh, A.M.; Spencer, M.D.; Philip, R.; Gaur, S.; Lawrie, S.M. Towards a neuroanatomy of autism: A systematic review and meta-analysis of structural magnetic resonance imaging studies. Eur. Psychiatry 2008, 23, 289–299. [Google Scholar] [CrossRef]
- Limperopoulos, C.; Bassan, H.; Gauvreau, K.; Robertson, R.L., Jr.; Sullivan, N.R.; Benson, C.B.; Avery, L.; Stewart, J.; Soul, J.S.; Ringer, S.A.; et al. Does cerebellar injury in premature infants contribute to the high prevalence of long-term cognitive, learning, and behavioral disability in survivors? Pediatrics 2007, 120, 584–593. [Google Scholar] [CrossRef]
- D’Mello, A.M.; Stoodley, C.J. Cerebro-cerebellar circuits in autism spectrum disorder. Front. Neurosci. 2015, 9, 408. [Google Scholar] [CrossRef]
- Stoodley, C.J. Distinct regions of the cerebellum show gray matter decreases in autism, ADHD, and developmental dyslexia. Front. Syst. Neurosci. 2014, 8, 92. [Google Scholar] [CrossRef]
- Insel, T.R. Rethinking schizophrenia. Nature 2010, 468, 187–193. [Google Scholar] [CrossRef]
- Van, L.; Boot, E.; Bassett, A.S. Update on the 22q11.2 deletion syndrome and its relevance to schizophrenia. Curr. Opin. Psychiatry 2017, 30, 191–196. [Google Scholar] [CrossRef]
- Ishii, K.; Kubo, K.I.; Nakajima, K. Reelin and Neuropsychiatric Disorders. Front. Cell. Neurosci. 2016, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Chen, S.; Xue, L.; Chen, J.H.; Shi, Y.W.; Zhao, H. SNP variation of RELN gene and schizophrenia in a chinese population: A hospital-based case-control study. Front. Genet. 2019, 10, 175. [Google Scholar] [CrossRef] [PubMed]
- Tost, H.; Weinberger, D.R. RELN rs7341475 and schizophrenia risk: Confusing, yet somehow intriguing. Biol. Psychiatry 2011, 69, e19. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, D.R. Future of Days Past: Neurodevelopment and Schizophrenia. Schizophr. Bull. 2017, 43, 1164–1168. [Google Scholar] [CrossRef]
- Dietsche, B.; Kircher, T.; Falkenberg, I. Structural brain changes in schizophrenia at different stages of the illness: A selective review of longitudinal magnetic resonance imaging studies. Aust. N. Z. J. Psychiatry 2017, 51, 500–508. [Google Scholar] [CrossRef]
- Kahn, R.S.; Sommer, I.E.; Murray, R.M.; Meyer-Lindenberg, A.; Weinberger, D.R.; Cannon, T.D.; O’Donovan, M.; Correll, C.U.; Kane, J.M.; Van Os, J.; et al. Schizophrenia. Nat. Rev. Dis. Primers 2015, 1, 15067. [Google Scholar] [CrossRef]
- Bakhshi, K.; Chance, S.A. The neuropathology of schizophrenia: A selective review of past studies and emerging themes in brain structure and cytoarchitecture. Neuroscience 2015, 303, 82–102. [Google Scholar] [CrossRef]
- Hoistad, M.; Heinsen, H.; Wicinski, B.; Schmitz, C.; Hof, P.R. Stereological assessment of the dorsal anterior cingulate cortex in schizophrenia: Absence of changes in neuronal and glial densities. Neuropathol. Appl. Neurobiol. 2013, 39, 348–361. [Google Scholar] [CrossRef]
- Guidotti, A.; Auta, J.; Davis, J.M.; Di-Giorgi-Gerevini, V.; Dwivedi, Y.; Grayson, D.R.; Impagnatiello, F.; Pandey, G.; Pesold, C.; Sharma, R.; et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: A postmortem brain study. Arch. Gen. Psychiatry 2000, 57, 1061–1069. [Google Scholar] [CrossRef]
- Dienel, S.J.; Lewis, D.A. Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol. Dis. 2019, 131, 104208. [Google Scholar] [CrossRef]
- Curley, A.A.; Lewis, D.A. Cortical basket cell dysfunction in schizophrenia. J. Physiol. 2012, 590, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Curley, A.A.; Glausier, J.R.; Volk, D.W. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012, 35, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Ruzicka, W.B.; Zhubi, A.; Veldic, M.; Grayson, D.R.; Costa, E.; Guidotti, A. Selective epigenetic alteration of layer I GABAergic neurons isolated from prefrontal cortex of schizophrenia patients using laser-assisted microdissection. Mol. Psychiatry 2007, 12, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A. The chandelier neuron in schizophrenia. Dev. Neurobiol. 2011, 71, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Glausier, J.R.; Lewis, D.A. Dendritic spine pathology in schizophrenia. Neuroscience 2013, 251, 90–107. [Google Scholar] [CrossRef]
- MacDonald, M.L.; Alhassan, J.; Newman, J.T.; Richard, M.; Gu, H.; Kelly, R.M.; Sampson, A.R.; Fish, K.N.; Penzes, P.; Wills, Z.P.; et al. Selective loss of smaller spines in schizophrenia. AJP 2017, 174, 586–594. [Google Scholar] [CrossRef]
- Matrisciano, F.; Tueting, P.; Dalal, I.; Kadriu, B.; Grayson, D.R.; Davis, J.M.; Nicoletti, F.; Guidotti, A. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology 2013, 68, 184–194. [Google Scholar] [CrossRef]
- Guidotti, A.; Grayson, D.R.; Caruncho, H.J. Epigenetic RELN dysfunction in schizophrenia and related neuropsychiatric disorders. Front. Cell. Neurosci. 2016, 10, 89. [Google Scholar] [CrossRef]
- Gothelf, D.; Soreni, N.; Nachman, R.P.; Tyano, S.; Hiss, Y.; Reiner, O.; Weizman, A. Evidence for the involvement of the hippocampus in the pathophysiology of schizophrenia. Eur. Neuropsychopharmacol. 2000, 10, 389–395. [Google Scholar] [CrossRef]
- Konradi, C.; Yang, C.K.; Zimmerman, E.I.; Lohmann, K.M.; Gresch, P.; Pantazopoulos, H.; Berretta, S.; Heckers, S. Hippocampal interneurons are abnormal in schizophrenia. Schizophr. Res. 2011, 131, 165–173. [Google Scholar] [CrossRef]
- Mavroudis, I.A.; Petrides, F.; Manani, M.; Chatzinikolaou, F.; Ciobica, A.S.; Padurariu, M.; Kazis, D.; Njau, S.N.; Costa, V.G.; Baloyannis, S.J. Purkinje cells pathology in schizophrenia. A morphometric approach. Rom. J. Morphol. Embryol. 2017, 58, 419–424. [Google Scholar] [PubMed]
- Eastwood, S.L.; Cotter, D.; Harrison, P.J. Cerebellar synaptic protein expression in schizophrenia. Neuroscience 2001, 105, 219–229. [Google Scholar] [CrossRef]
- Bey, A.L.; Jiang, Y.H. Overview of mouse models of autism spectrum disorders. Curr. Protoc. Pharmacol. 2014, 66, 5–26. [Google Scholar] [PubMed]
- Tueting, P.; Costa, E.; Dwivedi, Y.; Guidotti, A.; Impagnatiello, F.; Manev, R.; Pesold, C. The phenotypic characteristics of heterozygous reeler mouse. Neuroreport 1999, 10, 1329–1334. [Google Scholar] [CrossRef]
- Brigman, J.L.; Padukiewicz, K.E.; Sutherland, M.L.; Rothblat, L.A. Executive functions in the heterozygous reeler mouse model of schizophrenia. Behav. Neurosci. 2006, 120, 984–988. [Google Scholar] [CrossRef]
- Ognibene, E.; Adriani, W.; Macri, S.; Laviola, G. Neurobehavioural disorders in the infant reeler mouse model: Interaction of genetic vulnerability and consequences of maternal separation. Behav. Brain Res. 2007, 177, 142–149. [Google Scholar] [CrossRef]
- Ognibene, E.; Adriani, W.; Granstrem, O.; Pieretti, S.; Laviola, G. Impulsivity–anxiety-related behavior and profiles of morphine-induced analgesia in heterozygous reeler mice. Brain Res. 2007, 1131, 173–180. [Google Scholar] [CrossRef]
- Barr, A.M.; Fish, K.N.; Markou, A.; Honer, W.G. Heterozygous reeler mice exhibit alterations in sensorimotor gating but not presynaptic proteins. Eur. J. Neurosci. 2008, 27, 2568–2574. [Google Scholar] [CrossRef]
- Salinger, W.L.; Ladrow, P.; Wheeler, C. Behavioral phenotype of the reeler mutant mouse: Effects of RELN gene dosage and social isolation. Behav. Neurosci. 2003, 117, 1257–1275. [Google Scholar] [CrossRef]
- Podhorna, J.; Didriksen, M. The heterozygous reeler mouse: Behavioural phenotype. Behav. Brain Res. 2004, 153, 43–54. [Google Scholar] [CrossRef]
- Krueger, D.D.; Howell, J.L.; Hebert, B.F.; Olausson, P.; Taylor, J.R.; Nairn, A.C. Assessment of cognitive function in the heterozygous reeler mouse. Psychopharmacology 2006, 189, 95–104. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Michetti, C.; Romano, E.; Altabella, L.; Caruso, A.; Castelluccio, P.; Bedse, G.; Gaetani, S.; Canese, R.; Laviola, G.; Scattoni, M.L. Mapping pathological phenotypes in reelin mutant mice. Front. Pediatr. 2014, 2, 95. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Teixeira, C.M.; Martín, E.D.; Sahún, I.; Masachs, N.; Pujadas, L.; Corvelo, A.; Bosch, C.; Rossi, D.; Martinez, A.; Maldonado, R.; et al. Overexpression of Reelin Prevents the Manifestation of Behavioral Phenotypes Related to Schizophrenia and Bipolar Disorder. Neuropsychopharmacology 2011, 36, 2395–2405. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Michetti, C.; Caruso, A.; Laviola, G.; Scattoni, M.L. Characterization of neonatal vocal and motor repertoire of reelin mutant mice. PLoS ONE 2013, 8, e64407. [Google Scholar] [CrossRef] [PubMed]
- Ottman, R.; Risch, N.; Hauser, W.A.; Pedley, T.A.; Lee, J.H.; Barker-Cummings, C.; Lustenberger, A.; Nagle, K.J.; Lee, K.S.; Scheuer, M.L.; et al. Localization of a gene for partial epilepsy to chromosome 10q. Nat. Genet. 1995, 10, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, R.; Pasini, E.; Nobile, C. Lateral temporal lobe epilepsies: Clinical and genetic features. Epilepsia 2009, 50, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.M.; Flavelle, S.; Bureau, A.; Glass, H.C.; Fujiwara, T.M.; Wirrell, E.; Davey, K.; Chudley, A.E.; Scott, J.N.; McLeod, D.R.; et al. Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am. J. Hum. Genet. 2005, 77, 477–483. [Google Scholar] [CrossRef]
- Garden, G. Spinocerebellar Ataxia Type 7. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Marino, M.J. The use and misuse of statistical methodologies in pharmacology research. Biochem. Pharmacol. 2014, 87, 78–92. [Google Scholar] [CrossRef]
- Motulsky, H.J. Common misconceptions about data analysis and statistics. Br. J. Pharmacol. 2015, 172, 2126–2132. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Transmission | Causative Gene(s) | Reeler Mutants of Translational Interest | Other Mouse Models |
|---|---|---|---|---|
| LIS 2 | Autosomal recessive | RELN | Homozygous | see text |
| ADLTE | Autosomal dominant | RELN (in 17.5% of cases) | Heterozygous | LG11-mutated |
| VLDLR-associated cerebellar hypoplasia | Autosomal recessive | VLDLR | Homozygous | VLDLR knock-out |
| SCA37 | Autosomal dominant | DAB1 | Homozygous | DAB1 knock-out apoER2 knock-out |
| PAFAH1B1-associated lissencephaly | Autosomal dominant | PAFAH1B1 | Homozygous | Lis1+/− |
| SCA7 | Autosomal dominant | ATXN7 | Homozygous | SCA7 knock-in |
| Autism | Isolated cases Multifactorial | see https://omim.org # 209850 | Heterozygous | see text |
| Schizophrenia | Autosomal dominant | see https://omim.org # 181500 | Heterozygous | see text |
| Division of CNS | Region/Division | Subdivision/Nucleus | Type(s) of Alteration | Ref |
|---|---|---|---|---|
| Forebrain | Olfactory bulb |
| [33,34] | |
| Striatum |
| [35] | ||
| Diencephalon |
| [36] | ||
| Mammilary bodies |
| [37] | ||
| Midbrain | Rostral colliculus |
| [38] | |
| Mesencephalic nucleus of V |
| [39] | ||
| Substantia nigra |
| [40] | ||
| Medulla oblongata and pons | Dorsal cochlear nucleus |
| [41] | |
| Inferior olivary nucleus |
| [42] | ||
| Somatic motorneurons (Nucleus ambiguous, facial and trigeminal) |
| [6,43] | ||
| Pontine nuclei |
| [44] | ||
| Spinal cord | Dorsal horn (laminae I-II) | Nociceptive |
| [45] |
| Lateral horn | Preganglionic sympathetic and parasympathetic neurons |
| [46,47] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lossi, L.; Castagna, C.; Granato, A.; Merighi, A. The Reeler Mouse: A Translational Model of Human Neurological Conditions, or Simply a Good Tool for Better Understanding Neurodevelopment? J. Clin. Med. 2019, 8, 2088. https://doi.org/10.3390/jcm8122088
Lossi L, Castagna C, Granato A, Merighi A. The Reeler Mouse: A Translational Model of Human Neurological Conditions, or Simply a Good Tool for Better Understanding Neurodevelopment? Journal of Clinical Medicine. 2019; 8(12):2088. https://doi.org/10.3390/jcm8122088
Chicago/Turabian StyleLossi, Laura, Claudia Castagna, Alberto Granato, and Adalberto Merighi. 2019. "The Reeler Mouse: A Translational Model of Human Neurological Conditions, or Simply a Good Tool for Better Understanding Neurodevelopment?" Journal of Clinical Medicine 8, no. 12: 2088. https://doi.org/10.3390/jcm8122088
APA StyleLossi, L., Castagna, C., Granato, A., & Merighi, A. (2019). The Reeler Mouse: A Translational Model of Human Neurological Conditions, or Simply a Good Tool for Better Understanding Neurodevelopment? Journal of Clinical Medicine, 8(12), 2088. https://doi.org/10.3390/jcm8122088