Ca2+ Flux: Searching for a Role in Efferocytosis of Apoptotic Cells in Atherosclerosis

 ,
,  and
and 
Abstract
1. Introduction

{kind=link}
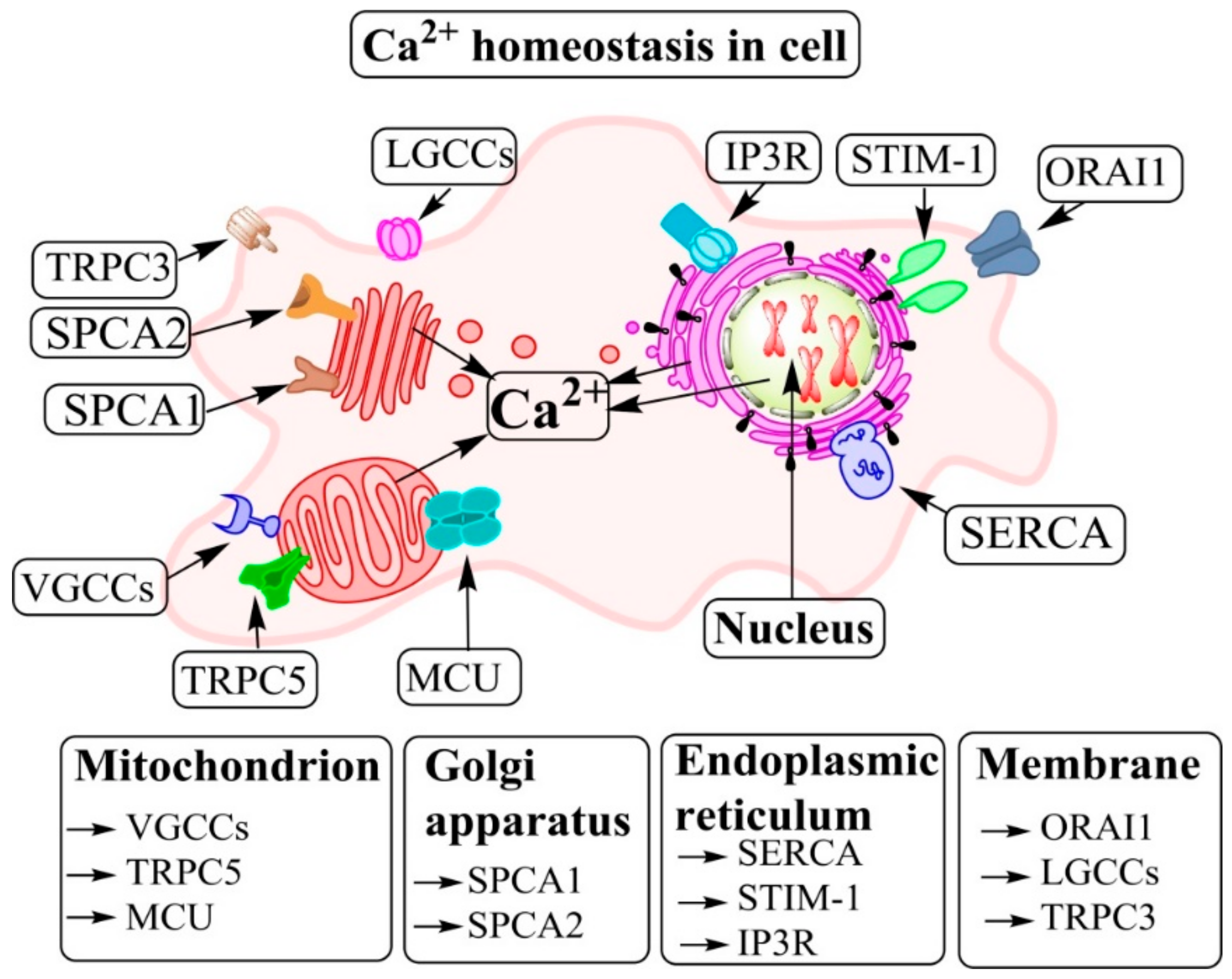{kind=link}
{kind=link}
{kind=link}
| Steps. | References | Molecules |
|---|---|---|
| Find-Me signal | [24,25,36] | LysoPC, ATP, P2Y2, ApoJ, ApoE4, Fractalkine (CX3CL1), S1P |
| Don’t Eat Me and Eat-Me signals | CD31, CD47, PtdSer, Caspase, MFG-E8, MerTK, Gas6, Protein S, | |
| Engulfment and processing | TRPC3, ABCA7, IRF8, IRF5, PPAR-δ/γ, p38 MAPK activities, CDKN2B, TLR3, TRAF6, UCP2, Cathepsin G, Rac | |
| Anti-inflammatory and tolerance responses | TGF-β, IL-10 |
2. Regulatory Roles of Ca2+ in Cellular Functions
3. The Role of Ca2+ in “Don’t Eat Me” Signalling
4. Scavenger Receptors and Efferocytosis in Atherosclerotic Plaques
5. The Vital Role of Ca2+ in the Efferocytic Engulfment Process
6. Role of Ca2+ Flux and Macrophage Metabolism in Efferocytosis of Apoptotic Cells in Atherosclerotic Plaques
7. Fundamental Role of Ca2+ Flux in Phagocytic Cells and in the Anti-Inflammatory Response to Engulfment of ACs in Efferocytosis and Atherosclerosis
8. Relations between ER, Ca2+, and Efferocytosis in Atherosclerosis
9. The Relation Between Ca2+/Calmodulin-Dependent Protein Kinase II Gamma (CaMKIIγ) and ER Stress in Atherosclerosis
10. The Relation Between Mitochondrial Ca2+ and Efferocytosis in Atherosclerosis
11. Roles of Mitochondria and AMP-Activated Protein Kinase (AMPK) in Efferocytosis in Atherosclerosis
12. Mitochondrial Fission and Related Factors in Efferocytosis and Atherosclerosis
13. Tumor Necrosis Factor Receptor-Associated Factor 6 (TRAF6) in Efferocytosis and Atherosclerosis
14. Interferon Regulatory Factor 8 (IRF8) and Factor 5 (IRF5) in Efferocytosis and Atherosclerosis
15. The Role of ORAI1 Store as a Part of the Operated Ca2+ Channel in Efferocytosis Related to Atherosclerosis
16. MicroRNAs and Their Effects on Efferocytosis Via Ca2+
17. Therapeutic Aspects
18. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; De Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Correction to: Heart Disease and Stroke Statistics-2017 Update: A Report from the American Heart Association. Circulation 2017, 135, e196. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, A.; Khorrami, M.S.; Hassanian, S.M.; Aghasizade, M.; Pasdar, A.; Maftouh, M.; Tabatabai, E.; Parizadeh, S.M.R.; Fazeli, M.; Ferns, G.A.; et al. The 9p21 Locus and its Potential Role in Atherosclerosis Susceptibility; Molecular Mechanisms and Clinical Implications. Curr. Pharm. Des. 2016, 22, 5730–5737. [Google Scholar] [CrossRef]
- Howson, J.M.M.; Zhao, W.; Barnes, D.R.; Ho, W.K.; Young, R.; Paul, D.S.; Waite, L.L.; Freitag, D.F.; Fauman, E.B.; Salfati, E.L.; et al. Fifteen New Risk Loci for Coronary Artery Disease Highlight Arterial-Wall-Specific Mechanisms. Nat. Genet. 2017, 49, 1113–1119. [Google Scholar] [CrossRef]
- Klarin, D.; Zhu, Q.M.; Emdin, C.A.; Chaffin, M.; Horner, S.; McMillan, B.J.; Leed, A.; Weale, M.E.; Spencer, C.C.A.; Aguet, F.; et al. Genetic Analysis in Uk Biobank Links Insulin Resistance and Transendothelial Migration Pathways to Coronary Artery Disease. Nat. Genet. 2017, 49, 1392–1397. [Google Scholar] [CrossRef]
- Polfus, L.M.; Smith, J.A.; Shimmin, L.C.; Bielak, L.F.; Morrison, A.C.; Kardia, S.L.; Peyser, P.A.; Hixson, J.E. Genome-Wide Association Study of Gene by Smoking Interactions in Coronary Artery Calcification. PLoS ONE 2013, 8, e74642. [Google Scholar] [CrossRef]
- Van Setten, J.; Isgum, I.; Smolonska, J.; Ripke, S.; De Jong, P.A.; Oudkerk, M.; De Koning, H.; Lammers, J.W.; Zanen, P.; Groen, H.J.; et al. Genome-Wide Association Study of Coronary and Aortic Calcification Implicates Risk Loci for Coronary Artery Disease and Myocardial Infarction. Atherosclerosis 2013, 228, 400–405. [Google Scholar] [CrossRef]
- O’Donnell, C.J.; Kavousi, M.; Smith, A.V.; Kardia, S.L.; Feitosa, M.F.; Hwang, S.J.; Sun, Y.V.; Province, M.A.; Aspelund, T.; Dehghan, A.; et al. Genome-Wide Association Study for Coronary Artery Calcification with Follow-Up in Myocardial Infarction. Circulation 2011, 124, 2855–2864. [Google Scholar] [CrossRef]
- Wojczynski, M.K.; Li, M.; Bielak, L.F.; Kerr, K.F.; Reiner, A.P.; Wong, N.D.; Yanek, L.R.; Qu, L.; White, C.C.; Lange, L.A.; et al. Genetics of Coronary Artery Calcification Among African Americans, a Meta-Analysis. BMC Med. Genet. 2013, 14, 75. [Google Scholar] [CrossRef] [PubMed]
- Divers, J.; Palmer, N.D.; Langefeld, C.D.; Brown, W.M.; Lu, L.; Hicks, P.J.; Smith, S.C.; Xu, J.; Terry, J.G.; Register, T.C.; et al. Genome-Wide Association Study of Coronary Artery Calcified Atherosclerotic Plaque in African Americans with Type 2 Diabetes. BMC Genet. 2017, 18, 105. [Google Scholar] [CrossRef] [PubMed]
- Kalampogias, A.; Siasos, G.; Oikonomou, E.; Tsalamandris, S.; Mourouzis, K.; Tsigkou, V.; Vavuranakis, M.; Zografos, T.; Deftereos, S.; Stefanadis, C.; et al. Basic Mechanisms in Atherosclerosis: The Role of Calcium. Med. Chem. 2016, 12, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y.; Ali, F. Atherosclerotic Cardiovascular Disease: A Review of Initiators and Protective Factors. Inflammopharmacology 2016, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Weissman, I.L.; Leeper, N.J. The Role of Efferocytosis in Atherosclerosis. Circulation 2017, 135, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, A.; Rezaee, M.; Kovanen, P.T.; Sahebkar, A. Efferocytosis in Atherosclerotic Lesions: Malfunctioning Regulatory Pathways and Control Mechanisms. Pharm. Ther. 2018, 188, 12–25. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Gheibi Hayat, S.M.; Butler, A.E.; Sahebkar, A. Effect of Soluble Cleavage Products of Important Receptors/Ligands on Efferocytosis: Their Role in Inflammatory, Autoimmune and Cardiovascular Disease. Ageing Res. Rev. 2019, 50, 43–57. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Bianconi, V.; Pirro, M.; Gheibi Hayat, S.M.; Johnston, T.P.; Sahebkar, A. Efferocytosis and Atherosclerosis: Regulation of Phagocyte Function by Micrornas. Trends Endocrinol. Metab. 2019, 30, 672–683. [Google Scholar] [CrossRef]
- Gonzalez, L.; Trigatti, B.L. Macrophage Apoptosis and Necrotic Core Development in Atherosclerosis: A Rapidly Advancing Field with Clinical Relevance to Imaging and Therapy. Can. J. Cardiol. 2017, 33, 303–312. [Google Scholar] [CrossRef]
- Wang, D.; Yang, Y.; Lei, Y.; Tzvetkov, N.T.; Liu, X.; Yeung, A.W.K.; Xu, S.; Atanasov, A.G. Targeting Foam Cell Formation in Atherosclerosis: Therapeutic Potential of Natural Products. Pharm. Rev. 2019, 71, 596–670. [Google Scholar] [CrossRef]
- Moore, K.J.; Tabas, I. Macrophages in the Pathogenesis of Atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef]
- Tano, J.Y.K.; Lee, R.H.; Vazquez, G. Macrophage Function in Atherosclerosis: Potential Roles of Trp Channels. Channels 2012, 6, 141–148. [Google Scholar] [CrossRef]
- Lewis, R.S. Calcium Signaling Mechanisms in t Lymphocytes. Annu. Rev. Immunol. 2001, 19, 497–521. [Google Scholar] [CrossRef] [PubMed]
- Gronski, M.A.; Kinchen, J.M.; Juncadella, I.J.; Franc, N.C.; Ravichandran, K.S. An Essential Role for Calcium Flux in Phagocytes for Apoptotic Cell Engulfment and the Anti-Inflammatory Response. Cell Death Differ. 2009, 16, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Tosello Trampont, A.C.; Elliott, M.R.; Lu, M.; Haney, L.B.; Ma, Z.; Klibanov, A.L.; Mandell, J.W.; Ravichandran, K.S. Bai1 is an Engulfment Receptor for Apoptotic Cells Upstream of the Elmo/Dock180/Rac Module. Nature 2007, 450, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, I.S. Engulfment Signals and the Phagocytic Machinery for Apoptotic Cell Clearance. Exp. Mol. Med. 2017, 49, e331. [Google Scholar] [CrossRef]
- Gregory, C. Sent by the Scent of Death. Nature 2009, 461, 181. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Pasdar, A.; Rezaee, M.; Fazeli, M.; Soleimanpour, S.; Hassanian, S.M.; FarshchiyanYazdi, Z.; Younesi Rad, T.; Ferns, G.A.; Avan, A. The Current Status and Perspectives Regarding the Clinical Implication of Intracellular Calcium in Breast Cancer. J. Cell Physiol. 2018, 233, 5623–5641. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium Signalling: Dynamics, Homeostasis and Remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Parkash, J.; Chaudhry, M.A.; Rhoten, W.B. Ca2+Sensing Receptor Activation by cacl2 Increases [ca2+] i Resulting in Enhanced Spatial Interactions with Calbindin-D28k Protein. Int. J. Mol. Med. 2004, 13, 3–11. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The Mitochondrial Calcium Uniporter is a Highly Selective ion Channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef]
- Hajnoczky, G.; Csordas, G.; Das, S.; Garcia Perez, C.; Saotome, M.; Sinha Roy, S.; Yi, M. Mitochondrial Calcium Signalling and Cell Death: Approaches for Assessing the Role of Mitochondrial ca2+Uptake in Apoptosis. Cell Calcium 2006, 40, 553–560. [Google Scholar] [CrossRef]
- Zhou, A.X.; Tabas, I. The Upr in Atherosclerosis. Semin. Immunopathol. 2013, 35, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lhotak, S.; Hilditch, B.A.; Austin, R.C. Activation of the Unfolded Protein Response Occurs at All Stages of Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice. Circulation 2005, 111, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Erbay, E.; Babaev, V.R.; Mayers, J.R.; Makowski, L.; Charles, K.N.; Snitow, M.E.; Fazio, S.; Wiest, M.M.; Watkins, S.M.; Linton, M.F.; et al. Reducing Endoplasmic Reticulum Stress Through a Macrophage Lipid Chaperone Alleviates Atherosclerosis. Nat. Med. 2009, 15, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Naik, V.; Leaf, E.M.; Hu, J.H.; Yang, H.Y.; Nguyen, N.B.; Giachelli, C.M.; Speer, M.Y. Sources of Cells that Contribute to Atherosclerotic Intimal Calcification: An in Vivo Genetic Fate Mapping Study. Cardiovasc. Res. 2012, 94, 545–554. [Google Scholar] [CrossRef]
- Cuttell, L.; Vaughan, A.; Silva, E.; Escaron, C.J.; Lavine, M.; Van Goethem, E.; Eid, J.P.; Quirin, M.; Franc, N.C. Undertaker, a Drosophila Junctophilin, Links Draper-Mediated Phagocytosis and Calcium Homeostasis. Cell 2008, 135, 524–534. [Google Scholar] [CrossRef]
- Elliott, M.R.; Ravichandran, K.S. Clearance of Apoptotic Cells: Implications in Health and Disease. J. Cell Biol. 2010, 189, 1059–1070. [Google Scholar] [CrossRef]
- Pettit, E.J.; Fay, F.S. Cytosolic Free Calcium and the Cytoskeleton in the Control of Leukocyte Chemotaxis. Physiol. Rev. 1998, 78, 949–967. [Google Scholar] [CrossRef]
- Yang, S.; Huang, X.Y. Ca2+Influx Through l-Type ca2+Channels Controls the Trailing Tail Contraction in Growth Factor-Induced Fibroblast Cell Migration. J. Biol. Chem. 2005, 280, 27130–27137. [Google Scholar] [CrossRef]
- Lee, J.; Ishihara, A.; Oxford, G.; Johnson, B.; Jacobson, K. Regulation of cell movement is mediated by stretch-activated calcium channels. Nature 1999, 400, 382–386. [Google Scholar] [CrossRef]
- Ozcan, L.; Tabas, I. Calcium Signalling and er Stress in Insulin Resistance and Atherosclerosis. J. Intern. Med. 2016, 280, 457–464. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan Barmatz, V.; Herrmann, S.; et al. Nclx is an Essential Component of Mitochondrial na+/ca2+Exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Shennan, D.B. Calcium Transport by Mammary Secretory Cells: Mechanisms Underlying Transepithelial Movement. Cell Mol. Biol. Lett. 2008, 13, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Tartakoff, A.M. The Confined Function Model of the Golgi Complex: Center for Ordered Processing of Biosynthetic Products of the Rough Endoplasmic Reticulum. Int. Rev. Cytol. 1983, 85, 221–252. [Google Scholar] [PubMed]
- Phair, R.D. Cellular Calcium and Atherosclerosis: A Brief Review. Cell Calcium 1988, 9, 275–284. [Google Scholar] [CrossRef]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-Surface Calreticulin Initiates Clearance of Viable or Apoptotic Cells Through Trans-Activation of Lrp on the Phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef]
- Isenberg, J.S.; Hyodo, F.; Pappan, L.K.; Abu Asab, M.; Tsokos, M.; Krishna, M.C.; Frazier, W.A.; Roberts, D.D. Blocking Thrombospondin-1/cd47 Signaling Alleviates Deleterious Effects of Aging on Tissue Responses to Ischemia. Arter. Thromb. Vasc. Biol. 2007, 27, 2582–2588. [Google Scholar] [CrossRef]
- Zhang, S.; Yeap, X.Y.; DeBerge, M.; Naresh, N.K.; Wang, K.; Jiang, Z.; Wilcox, J.E.; White, S.M.; Morrow, J.P.; Burridge, P.W.; et al. Acute cd47 Blockade During Ischemic Myocardial Reperfusion Enhances Phagocytosis-Associated Cardiac Repair. JACC Basic. Transl. Sci. 2017, 2, 386–397. [Google Scholar] [CrossRef]
- Soto Pantoja, D.R.; Kaur, S.; Roberts, D.D. Cd47 Signaling Pathways Controlling Cellular Differentiation and Responses to Stress. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 212–230. [Google Scholar] [CrossRef]
- Rogers, N.M.; Sharifi Sanjani, M.; Csanyi, G.; Pagano, P.J.; Isenberg, J.S. Thrombospondin-1 and cd47 Regulation of Cardiac, Pulmonary and Vascular Responses in Health and Disease. Matrix. Biol. 2014, 37, 92–101. [Google Scholar] [CrossRef]
- Schwartz, M.A.; Brown, E.J.; Fazeli, B. A 50-Kda Integrin-Associated Protein is Required for Integrin-Regulated Calcium Entry in Endothelial Cells. J. Biol. Chem. 1993, 268, 19931–19934. [Google Scholar]
- Martinelli, R.; Newton, G.; Carman, C.V.; Greenwood, J.; Luscinskas, F.W. Novel Role of cd47 in Rat Microvascular Endothelium: Signaling and Regulation of t-Cell Transendothelial Migration. Arter. Thromb. Vasc. Biol. 2013, 33, 2566–2576. [Google Scholar] [CrossRef] [PubMed]
- Ii, M.; Takenaka, H.; Asai, J.; Ibusuki, K.; Mizukami, Y.; Maruyama, K.; Yoon, Y.S.; Wecker, A.; Luedemann, C.; Eaton, E.; et al. Endothelial Progenitor Thrombospondin-1 Mediates Diabetes-Induced Delay in Reendothelialization Following Arterial Injury. Circ. Res. 2006, 98, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J. Nitric Oxide as a Unique Signaling Molecule in the Vascular System: A Historical Overview. J. Physiol. Pharmacol. 2002, 53, 503–514. [Google Scholar] [PubMed]
- Lincoln, T.M.; Dey, N.; Sellak, H. Invited Review: Cgmp-Dependent Protein Kinase Signaling Mechanisms in Smooth Muscle: From the Regulation of Tone to Gene Expression. J. Appl. Physiol. (1985) 2001, 91, 1421–1430. [Google Scholar] [CrossRef]
- Kojima, Y.; Volkmer, J.P.; McKenna, K.; Civelek, M.; Lusis, A.J.; Miller, C.L.; Direnzo, D.; Nanda, V.; Ye, J.; Connolly, A.J.; et al. Cd47-Blocking Antibodies Restore Phagocytosis and Prevent Atherosclerosis. Nature 2016, 536, 86–90. [Google Scholar] [CrossRef]
- Ryan, J.J. Cd47-Blocking Antibodies and Atherosclerosis. JACC Basic. Transl. Sci. 2016, 1, 413–415. [Google Scholar] [CrossRef][Green Version]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger Receptors in Homeostasis and Immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef]
- Kzhyshkowska, J.; Neyen, C.; Gordon, S. Role of Macrophage Scavenger Receptors in Atherosclerosis. Immunobiology 2012, 217, 492–502. [Google Scholar] [CrossRef]
- Savill, J.; Hogg, N.; Ren, Y.; Haslett, C. Thrombospondin Cooperates with cd36 and the Vitronectin Receptor in Macrophage Recognition of Neutrophils Undergoing Apoptosis. J. Clin. Investig. 1992, 90, 1513–1522. [Google Scholar] [CrossRef]
- Beppu, M.; Hora, M.; Watanabe, T.; Watanabe, M.; Kawachi, H.; Mishima, E.; Makino, M.; Kikugawa, K. Substrate-Bound Fibronectin Enhances Scavenger Receptor Activity of Macrophages by Calcium Signaling. Arch. Biochem. Biophys. 2001, 390, 243–252. [Google Scholar] [CrossRef]
- Deng, T.L.; Yu, L.; Ge, Y.K.; Zhang, L.; Zheng, X.X. Intracellular-Free Calcium Dynamics and f-Actin Alteration in the Formation of Macrophage Foam Cells. Biochem. Biophys. Res. Commun. 2005, 338, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, S.O.; Zhou, G.; Silverstein, R.L. Vav Protein Guanine Nucleotide Exchange Factor Regulates cd36 Protein-Mediated Macrophage Foam Cell Formation Via Calcium and Dynamin-Dependent Processes. J. Biol. Chem. 2011, 286, 36011–36019. [Google Scholar] [CrossRef] [PubMed]
- Penberthy, K.K.; Ravichandran, K.S. Apoptotic Cell Recognition Receptors and Scavenger Receptors. Immunol. Rev. 2016, 269, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ogura, S.; Chen, J.; Little, P.J.; Moss, J.; Liu, P. Lox-1 in Atherosclerosis: Biological Functions and Pharmacological Modifiers. Cell Mol. Life Sci. 2013, 70, 2859–2872. [Google Scholar] [CrossRef]
- Murphy, J.E.; Tacon, D.; Tedbury, P.R.; Hadden, J.M.; Knowling, S.; Sawamura, T.; Peckham, M.; Phillips, S.E.; Walker, J.H.; Ponnambalam, S. Lox-1 Scavenger Receptor Mediates Calcium-Dependent Recognition of Phosphatidylserine and Apoptotic Cells. Biochem. J. 2006, 393, 107–115. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Li, W.; Park, Y.M.; Rahaman, S.O. Mechanisms of Cell Signaling by the Scavenger Receptor cd36: Implications in Atherosclerosis and Thrombosis. Trans. Am. Clin. Climatol. Assoc. 2010, 121, 206–220. [Google Scholar]
- Henry, P.D. Atherogenesis, Calcium and Calcium Antagonists. Am. J. Cardiol. 1990, 66, 3–6. [Google Scholar] [CrossRef]
- Sugano, M.; Tsuchida, K.; Makino, N. Nifedipine Prevents Apoptosis of Endothelial Cells Induced by Oxidized Low-Density Lipoproteins. J. Cardiovasc. Pharm. 2002, 40, 146–152. [Google Scholar] [CrossRef]
- Pasterkamp, G.; Den Ruijter, H.M.; Libby, P. Temporal Shifts in Clinical Presentation and Underlying Mechanisms of Atherosclerotic Disease. Nat. Rev. Cardiol. 2017, 14, 21–29. [Google Scholar] [CrossRef]
- Heikkila, H.M.; Latti, S.; Leskinen, M.J.; Hakala, J.K.; Kovanen, P.T.; Lindstedt, K.A. Activated Mast Cells Induce Endothelial Cell Apoptosis by a Combined Action of Chymase and Tumor Necrosis Factor-Alpha. Arter. Thromb. Vasc. Biol. 2008, 28, 309–314. [Google Scholar] [CrossRef]
- Timmins, J.M.; Ozcan, L.; Seimon, T.A.; Li, G.; Malagelada, C.; Backs, J.; Backs, T.; Bassel Duby, R.; Olson, E.N.; Anderson, M.E.; et al. Calcium/Calmodulin-Dependent Protein Kinase ii Links er Stress with Fas and Mitochondrial Apoptosis Pathways. J. Clin. Investig. 2009, 119, 2925–2941. [Google Scholar] [CrossRef] [PubMed]
- Tano, J.Y.; Smedlund, K.; Lee, R.; Abramowitz, J.; Birnbaumer, L.; Vazquez, G. Impairment of Survival Signaling and Efferocytosis in Trpc3-Deficient Macrophages. Biochem. Biophys. Res. Commun. 2011, 410, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, R.; Henkels, K.M.; Wrenshall, L.E.; Kanaho, Y.; Di Paolo, G.; Frohman, M.A.; Gomez Cambronero, J. Oxidized Ldl Phagocytosis During Foam Cell Formation in Atherosclerotic Plaques Relies on a pld2-cd36 Functional Interdependence. J. Leukoc. Biol. 2018, 103, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Tsujimura, H.; Nagamura Inoue, T.; Tamura, T.; Ozato, K. Ifn Consensus Sequence Binding Protein/Ifn Regulatory Factor-8 Guides Bone Marrow Progenitor Cells Toward the Macrophage Lineage. J. Immunol. 2002, 169, 1261–1269. [Google Scholar] [CrossRef]
- Miyanishi, M.; Tada, K.; Koike, M.; Uchiyama, Y.; Kitamura, T.; Nagata, S. Identification of Tim4 as a Phosphatidylserine Receptor. Nature 2007, 450, 435–439. [Google Scholar] [CrossRef]
- Kaplan, G. Differences in the Mode of Phagocytosis with fc and c3 Receptors in Macrophages. Scand. J. Immunol. 1977, 6, 797–807. [Google Scholar] [CrossRef]
- Wennerberg, K.; Der, C.J. Rho Family Gtpases: It’s Not Only Rac and Rho (and i Like it). J. Cell Sci. 2004, 117, 1301–1312. [Google Scholar] [CrossRef]
- Mao, Y.; Finnemann, S.C. Regulation of Phagocytosis by Rho Gtpases. Small GTPases 2015, 6, 89–99. [Google Scholar] [CrossRef]
- Caron, E.; Hall, A. Identification of Two Distinct Mechanisms of Phagocytosis Controlled by Different Rho Gtpases. Science 1998, 282, 1717–1721. [Google Scholar] [CrossRef]
- Kinchen, J.M.; Ravichandran, K.S. Identification of Two Evolutionarily Conserved Genes Regulating Processing of Engulfed Apoptotic Cells. Nature 2010, 464, 778–782. [Google Scholar] [CrossRef]
- Grimsley, C.M.; Kinchen, J.M.; Tosello Trampont, A.C.; Brugnera, E.; Haney, L.B.; Lu, M.; Chen, Q.; Klingele, D.; Hengartner, M.O.; Ravichandran, K.S. Dock180 and Elmo1 Proteins Cooperate to Promote Evolutionarily Conserved Rac-Dependent Cell Migration. J. Biol. Chem. 2004, 279, 6087–6097. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, C.; De La Rosa, G.; Anderson, C.S.; Murphy, P.S.; Capece, T.; Kim, M.; Elliott, M.R. Essential Role of Elmo1 in Dock2-Dependent Lymphocyte Migration. J. Immunol. 2014, 192, 6062–6070. [Google Scholar] [CrossRef] [PubMed]
- Tosello Trampont, A.C.; Nakada Tsukui, K.; Ravichandran, K.S. Engulfment of Apoptotic Cells is Negatively Regulated by Rho-Mediated Signaling. J. Biol. Chem. 2003, 278, 49911–49919. [Google Scholar] [CrossRef] [PubMed]
- Erwig, L.P.; McPhilips, K.A.; Wynes, M.W.; Ivetic, A.; Ridley, A.J.; Henson, P.M. Differential Regulation of Phagosome Maturation in Macrophages and Dendritic Cells Mediated by Rho Gtpases and Ezrin-Radixin-Moesin (Erm) Proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 12825–12830. [Google Scholar] [CrossRef] [PubMed]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional Kinases in Cell Behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Ravichandran, K.S. The Dynamics of Apoptotic Cell Clearance. Dev. Cell 2016, 38, 147–160. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, Rac, and cdc42 Gtpases Regulate the Assembly of Multimolecular Focal Complexes Associated with Actin Stress Fibers, Lamellipodia, and Filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Maatta, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized Phospholipids are Proinflammatory and Proatherogenic in Hypercholesterolaemic Mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef]
- Schwartz, C.J.; Valente, A.J.; Sprague, E.A.; Kelley, J.L.; Cayatte, A.J.; Mowery, J. Atherosclerosis. Potential Targets for Stabilization and Regression. Circulation 1992, 86, 117–123. [Google Scholar]
- Fisher, M. Atherosclerosis: Cellular Aspects and Potential Interventions. Cereb. Brain. Metab. Rev. 1991, 3, 114–133. [Google Scholar]
- Matsumura, T.; Sakai, M.; Kobori, S.; Biwa, T.; Takemura, T.; Matsuda, H.; Hakamata, H.; Horiuchi, S.; Shichiri, M. Two Intracellular Signaling Pathways for Activation of Protein Kinase c are Involved in Oxidized Low-Density Lipoprotein-Induced Macrophage Growth. Arter. Thromb. Vasc. Biol. 1997, 17, 3013–3020. [Google Scholar] [CrossRef] [PubMed]
- Maziere, C.; Morliere, P.; Massy, Z.; Kamel, S.; Louandre, C.; Conte, M.A.; Maziere, J.C. Oxidized Low-Density Lipoprotein Elicits An Intracellular Calcium Rise and Increases the Binding Activity of the Transcription Factor Nfat. Free Radic. Biol. Med. 2005, 38, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Vindis, C.; Elbaz, M.; Escargueil Blanc, I.; Auge, N.; Heniquez, A.; Thiers, J.C.; Negre Salvayre, A.; Salvayre, R. Two Distinct Calcium-Dependent Mitochondrial Pathways are Involved in Oxidized Ldl-Induced Apoptosis. Arter. Thromb. Vasc. Biol. 2005, 25, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Colles, S.M.; Maxson, J.M.; Carlson, S.G.; Chisolm, G.M. Oxidized Ldl-Induced Injury and Apoptosis in Atherosclerosis. Potential Roles for Oxysterols. Trends Cardiovasc. Med. 2001, 11, 131–138. [Google Scholar] [CrossRef]
- Napoli, C. Oxidation of Ldl, Atherogenesis, and Apoptosis. Ann. N. Y. Acad. Sci. 2003, 1010, 698–709. [Google Scholar] [CrossRef]
- Graham, S.; Yuan, J.P.; Ma, R. Canonical Transient Receptor Potential Channels in Diabetes. Exp. Biol. Med. 2012, 237, 111–118. [Google Scholar] [CrossRef]
- Wuensch, T.; Thilo, F.; Krueger, K.; Scholze, A.; Ristow, M.; Tepel, M. High glucose-induced oxidative stress increases transient receptor potential channel expression in human monocytes. Diabetes 2010, 59, 844–849. [Google Scholar] [CrossRef]
- Tano, J.Y.; Vazquez, G. Requirement for Non-Regulated, Constitutive Calcium Influx in Macrophage Survival Signaling. Biochem. Biophys. Res. Commun. 2011, 407, 432–437. [Google Scholar] [CrossRef]
- Morimoto, K.; Yamashita, Y.; Takaki, M.; Tanaka, T. Cigarette Smoke Extract Induced Endoplasmic Reticulum Stress Suppresses Efferocytosis by Macrophages Through Rhoa Activation. Am. J. Respir. Crit. Care Med. 2015, 191, A2736. [Google Scholar]
- Cash, J.G.; Kuhel, D.G.; Basford, J.E.; Jaeschke, A.; Chatterjee, T.K.; Weintraub, N.L.; Hui, D.Y. Apolipoprotein e4 Impairs Macrophage Efferocytosis and Potentiates Apoptosis by Accelerating Endoplasmic Reticulum Stress. J. Biol. Chem. 2012, 287, 27876–27884. [Google Scholar] [CrossRef]
- Mallat, Z.; Tedgui, A. Apoptosis in the Vasculature: Mechanisms and Functional Importance. Br. J. Pharmacol. 2000, 130, 947–962. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.J.; Libby, P. Progression of Atheroma: A Struggle Between Death and Procreation. Arter. Thromb. Vasc. Biol. 2002, 22, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of Ero1-Alpha-Mediated Stimulation of Inositol 1,4,5-Triphosphate Receptor Activity in Endoplasmic Reticulum Stress-Induced Apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Scull, C.; Ozcan, L.; Tabas, I. Nadph Oxidase Links Endoplasmic Reticulum Stress, Oxidative Stress, and Pkr Activation to Induce Apoptosis. J. Cell Biol. 2010, 191, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Merched, A.J.; Ko, K.; Gotlinger, K.H.; Serhan, C.N.; Chan, L. Atherosclerosis: Evidence for Impairment of Resolution of Vascular Inflammation Governed by Specific Lipid Mediators. FASEB J. 2008, 22, 3595–3606. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Ozcan, L.; Spolitu, S.; Hellmann, J.; Spite, M.; Backs, J.; Tabas, I. Resolvin d1 Limits 5-Lipoxygenase Nuclear Localization and Leukotriene b4 Synthesis by Inhibiting a Calcium-Activated Kinase Pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 14530–14535. [Google Scholar] [CrossRef]
- Ricci, R.; Sumara, G.; Sumara, I.; Rozenberg, I.; Kurrer, M.; Akhmedov, A.; Hersberger, M.; Eriksson, U.; Eberli, F.R.; Becher, B.; et al. Requirement of jnk2 for Scavenger Receptor a-Mediated Foam Cell Formation in Atherogenesis. Science 2004, 306, 1558–1561. [Google Scholar] [CrossRef]
- Liang, S.J.; Zeng, D.Y.; Mai, X.Y.; Shang, J.Y.; Wu, Q.Q.; Yuan, J.N.; Yu, B.X.; Zhou, P.; Zhang, F.R.; Liu, Y.Y.; et al. Inhibition of Orai1 Store-Operated Calcium Channel Prevents Foam Cell Formation and Atherosclerosis. Arter. Thromb. Vasc. Biol. 2016, 36, 618–628. [Google Scholar] [CrossRef]
- Doran, A.C.; Ozcan, L.; Cai, B.; Zheng, Z.; Fredman, G.; Rymond, C.C.; Dorweiler, B.; Sluimer, J.C.; Hsieh, J.; Kuriakose, G.; et al. Camkiigamma Suppresses an Efferocytosis Pathway in Macrophages and Promotes Atherosclerotic Plaque Necrosis. J. Clin. Investig. 2017, 127, 4075–4089. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the Mechanisms of Apoptosis Induced by Endoplasmic Reticulum Stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liang, C.P.; DeVries Seimon, T.; Ranalletta, M.; Welch, C.L.; Collins Fletcher, K.; Accili, D.; Tabas, I.; Tall, A.R. Macrophage Insulin Receptor Deficiency Increases Er Stress-Induced Apoptosis and Necrotic Core Formation in Advanced Atherosclerotic Lesions. Cell Metab. 2006, 3, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.P.; Han, S.; Li, G.; Tabas, I.; Tall, A.R. Impaired Mek Signaling and Serca Expression Promote er Stress and Apoptosis in Insulin-Resistant Macrophages and are Reversed by Exenatide Treatment. Diabetes 2012, 61, 2609–2620. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, M.; Ciani, L.; Kuriakose, G.; Westover, E.J.; Dura, M.; Covey, D.F.; Freed, J.H.; Maxfield, F.R.; Lytton, J.; et al. Enrichment of Endoplasmic Reticulum with Cholesterol Inhibits Sarcoplasmic-Endoplasmic Reticulum Calcium Atpase-2b Activity in Parallel with Increased Order of Membrane Lipids: Implications for Depletion of Endoplasmic Reticulum Calcium Stores and Apoptosis in Cholesterol-Loaded Macrophages. J. Biol. Chem. 2004, 279, 37030–37039. [Google Scholar]
- Anderson, H.A.; Maylock, C.A.; Williams, J.A.; Paweletz, C.P.; Shu, H.; Shacter, E. Serum-Derived Protein s Binds to Phosphatidylserine and Stimulates the Phagocytosis of Apoptotic Cells. Nat. Immunol. 2003, 4, 87–91. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Ohashi, K.; Mizuno, K.; Nakano, T. Promotion of the Uptake of Ps Liposomes and Apoptotic Cells by a Product of Growth Arrest-Specific Gene, Gas6. J. Biochem. 2000, 127, 411–417. [Google Scholar] [CrossRef]
- Uehara, H.; Shacter, E. Auto-Oxidation and Oligomerization of Protein s on the Apoptotic Cell Surface is Required for Mer Tyrosine Kinase-Mediated Phagocytosis of Apoptotic Cells. J. Immunol. 2008, 180, 2522–2530. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as Sensors and Regulators of Calcium Signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. The ca2+Concentration of the Endoplasmic Reticulum is a Key Determinant of Ceramide-Induced Apoptosis: Significance for the Molecular Mechanism of Bcl-2 Action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef]
- Curry, M.C.; Peters, A.A.; Kenny, P.A.; Roberts Thomson, S.J.; Monteith, G.R. Mitochondrial Calcium Uniporter Silencing Potentiates Caspase-Independent Cell Death in Mda-Mb-231 Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2013, 434, 695–700. [Google Scholar] [CrossRef]
- Park, D.; Han, C.Z.; Elliott, M.R.; Kinchen, J.M.; Trampont, P.C.; Das, S.; Collins, S.; Lysiak, J.J.; Hoehn, K.L.; Ravichandran, K.S. Continued Clearance of Apoptotic Cells Critically Depends on the Phagocyte Ucp2 Protein. Nature 2011, 477, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Lee, H.N.; Kim, W.; Surh, Y.J. Docosahexaenoic Acid Induces m2 Macrophage Polarization Through Peroxisome Proliferator-Activated Receptor Gamma Activation. Life Sci. 2015, 120, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Park, D.W.; Stigler, W.S.; Creighton, J.; Ravi, S.; Darley Usmar, V.; Zmijewski, J.W. Mitochondria and Amp-Activated Protein Kinase-Dependent Mechanism of Efferocytosis. J. Biol. Chem. 2013, 288, 26013–26026. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R. The Role of Mitochondria in Ischemic Heart Disease. J. Cardiovasc. Pharmacol. 1996, 28, 1–10. [Google Scholar]
- Yu, W.L.; Sun, T.W.; Qi, C.; Zhao, H.K.; Ding, Z.Y.; Zhang, Z.W.; Sun, B.B.; Shen, J.; Chen, F.; Zhu, Y.J.; et al. Enhanced Osteogenesis and Angiogenesis by Mesoporous Hydroxyapatite Microspheres-Derived Simvastatin Sustained Release System for Superior Bone Regeneration. Sci. Rep. 2017, 7, 44129. [Google Scholar] [CrossRef] [PubMed]
- Ricquier, D.; Bouillaud, F. Mitochondrial Uncoupling Proteins: From Mitochondria to the Regulation of Energy Balance. J. Physiol. 2000, 529, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Blanc, J.; Alves Guerra, M.; Esposito, B.; Rousset, S.; Gourdy, P.; Ricquier, D.; Tedgui, A.; Miroux, B.; Mallat, Z. Protective Role of Uncoupling Protein 2 in Atherosclerosis. Circulation 2003, 107, 388–390. [Google Scholar] [CrossRef]
- Frank, S.; Gaume, B.; Bergmann Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The Role of Dynamin-Related Protein 1, a Mediator of Mitochondrial Fission, in Apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef]
- Savchenko, A.S.; Borissoff, J.I.; Martinod, K.; De Meyer, S.F.; Gallant, M.; Erpenbeck, L.; Brill, A.; Wang, Y.; Wagner, D.D. Vwf-Mediated Leukocyte Recruitment with Chromatin Decondensation by pad4 Increases Myocardial Ischemia/Reperfusion Injury in Mice. Blood 2014, 123, 141–148. [Google Scholar] [CrossRef]
- Di Bartolo, B.A.; Schoppet, M.; Mattar, M.Z.; Rachner, T.D.; Shanahan, C.M.; Kavurma, M.M. Calcium and Osteoprotegerin Regulate Igf1r Expression to Inhibit Vascular Calcification. Cardiovasc. Res. 2011, 91, 537–545. [Google Scholar] [CrossRef]
- Ishida, T.; Mizushima, S.; Azuma, S.; Kobayashi, N.; Tojo, T.; Suzuki, K.; Aizawa, S.; Watanabe, T.; Mosialos, G.; Kieff, E.; et al. Identification of Traf6, a Novel Tumor Necrosis Factor Receptor-Associated Factor Protein that Mediates Signaling from An Amino-Terminal Domain of the cd40 Cytoplasmic Region. J. Biol. Chem. 1996, 271, 28745–28748. [Google Scholar] [CrossRef] [PubMed]
- Seibold, K.; Ehrenschwender, M. P62 Regulates cd40-Mediated Nfκb Activation in Macrophages Through Interaction with traf6. Biochem. Biophys. Res. Commun. 2015, 464, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Polykratis, A.; Van Loo, G.; Xanthoulea, S.; Hellmich, M.; Pasparakis, M. Conditional Targeting of Tumor Necrosis Factor Receptor-Associated Factor 6 Reveals Opposing Functions of Toll-Like Receptor Signaling in Endothelial and Myeloid Cells in a Mouse Model of Atherosclerosis. Circulation 2012, 126, 1739–1751. [Google Scholar] [CrossRef]
- Lutgens, E.; Lievens, D.; Beckers, L.; Wijnands, E.; Soehnlein, O.; Zernecke, A.; Seijkens, T.; Engel, D.; Cleutjens, J.; Keller, A.M.; et al. Deficient cd40-Traf6 Signaling in Leukocytes Prevents Atherosclerosis by Skewing the Immune Response Toward an Antiinflammatory Profile. J. Exp. Med. 2010, 207, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Scheller, M.; Foerster, J.; Heyworth, C.M.; Waring, J.F.; Lohler, J.; Gilmore, G.L.; Shadduck, R.K.; Dexter, T.M.; Horak, I. Altered Development ad Cytokine Responses of Myeloid Progenitors in the Absence of Transcription Factor, Interferon Consensus Sequence Binding Protein. Blood 1999, 94, 3764–3771. [Google Scholar] [CrossRef]
- Doring, Y.; Soehnlein, O.; Drechsler, M.; Shagdarsuren, E.; Chaudhari, S.M.; Meiler, S.; Hartwig, H.; Hristov, M.; Koenen, R.R.; Hieronymus, T.; et al. Hematopoietic Interferon Regulatory Factor 8-Deficiency Accelerates Atherosclerosis in Mice. Arter. Thromb. Vasc. Biol. 2012, 32, 1613–1623. [Google Scholar] [CrossRef]
- Crosslin, D.R.; McDavid, A.; Weston, N.; Zheng, X.; Hart, E.; De Andrade, M.; Kullo, I.J.; McCarty, C.A.; Doheny, K.F.; Pugh, E.; et al. Genetic Variation Associated with Circulating Monocyte Count in the Emerge Network. Hum. Mol. Genet. 2013, 22, 2119–2127. [Google Scholar] [CrossRef]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. Irf5 Promotes Inflammatory Macrophage Polarization and th1-Th17 Responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef]
- Seneviratne, A.N.; Edsfeldt, A.; Cole, J.E.; Kassiteridi, C.; Swart, M.; Park, I.; Green, P.; Khoyratty, T.; Saliba, D.; Goddard, M.E.; et al. Interferon Regulatory Factor 5 Controls Necrotic Core Formation in Atherosclerotic Lesions by Impairing Efferocytosis. Circulation 2017, 136, 1140–1154. [Google Scholar] [CrossRef]
- Fan, J.H.; Gao, L.B.; Pan, X.M.; Li, C.; Liang, W.B.; Liu, J.; Li, Y.; Zhang, L. Association Between irf-5 Polymorphisms and Risk of Acute Coronary Syndrome. DNA Cell Biol. 2010, 29, 19–23. [Google Scholar] [CrossRef]
- Hall, J.L.; Wei, L.N. Could Silencing irf5 Improve Healing of a Myocardial Infarct Through the Reprogramming of the Macrophage Population? J. Am. Coll. Cardiol. 2014, 63, 1567–1568. [Google Scholar] [CrossRef][Green Version]
- Galdiero, M.R.; Garlanda, C.; Jaillon, S.; Marone, G.; Mantovani, A. Tumor Associated Macrophages and Neutrophils in Tumor Progression. J. Cell Physiol. 2013, 228, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Natoli, G. Transcriptional Regulation of Macrophage Polarization: Enabling Diversity with Identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W. Forms and Functions of Store-Operated Calcium Entry Mediators, Stim and Orai. Adv. Biol. Regul. 2018, 68, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. Stim1 is a ca2+Sensor that Activates Crac Channels and Migrates from the ca2+store to the Plasma Membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef] [PubMed]
- Mignen, O.; Thompson, J.L.; Shuttleworth, T.J. Orai1 Subunit Stoichiometry of the Mammalian Crac Channel Pore. J. Physiol. 2008, 586, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.H.; Michalak, M.; Verkhratsky, A. Calcium Signalling: Past, Present and Future. Cell Calcium 2005, 38, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, H.; Wang, Y.; Hu, J.; Sun, Z.; Xu, S. Down-Regulation of Hsa-Mir-148b Inhibits Vascular Smooth Muscle Cells Proliferation and Migration by Directly Targeting hsp90 in Atherosclerosis. Am. J. Transl. Res. 2017, 9, 629–637. [Google Scholar]
- Madrigal Matute, J.; Lopez Franco, O.; Blanco Colio, L.M.; Munoz Garcia, B.; Ramos Mozo, P.; Ortega, L.; Egido, J.; Martin Ventura, J.L. Heat Shock Protein 90 Inhibitors Attenuate Inflammatory Responses in Atherosclerosis. Cardiovasc. Res. 2010, 86, 330–337. [Google Scholar] [CrossRef]
- Cimino, D.; De Pitta, C.; Orso, F.; Zampini, M.; Casara, S.; Penna, E.; Quaglino, E.; Forni, M.; Damasco, C.; Pinatel, E.; et al. Mir148b is a Major Coordinator of Breast Cancer Progression in a Relapse-Associated Microrna Signature by Targeting Itga5, Rock1, pik3ca, Nras, and csf1. FASEB J. 2013, 27, 1223–1235. [Google Scholar] [CrossRef]
- Hodge, S.; Hodge, G.; Jersmann, H.; Matthews, G.; Ahern, J.; Holmes, M.; Reynolds, P.N. Azithromycin Improves Macrophage Phagocytic Function and Expression of Mannose Receptor in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2008, 178, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Janssen, W.J.; Fessler, M.B.; McPhillips, K.A.; Borges, V.M.; Bowler, R.P.; Xiao, Y.Q.; Kench, J.A.; Henson, P.M.; Vandivier, R.W. Lovastatin Enhances Clearance of Apoptotic Cells (Efferocytosis) with Implications for Chronic Obstructive Pulmonary Disease. J. Immunol. 2006, 176, 7657–7665. [Google Scholar] [CrossRef] [PubMed]
- Bruining, N.; De Winter, S.; Roelandt, J.R.; Rodriguez Granillo, G.A.; Heller, I.; Van Domburg, R.T.; Hamers, R.; De Feijter, P.J. Coronary Calcium Significantly Affects Quantitative Analysis of Coronary Ultrasound: Importance for Atherosclerosis Progression/Regression Studies. Coron. Artery Dis. 2009, 20, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Magnani, B.; Dal Palu, C.; Zanchetti, A. Preliminary Clinical Experience with Calcium Antagonists in Atherosclerosis. Verapamil in Hypertension Atherosclerosis Study Investigators. Drugs 1992, 44, 128–133. [Google Scholar] [CrossRef]
- Zanchetti, A.; Bond, M.G.; Hennig, M.; Neiss, A.; Mancia, G.; Dal Palu, C.; Hansson, L.; Magnani, B.; Rahn, K.H.; Reid, J.L.; et al. Calcium Antagonist Lacidipine Slows Down Progression of Asymptomatic Carotid Atherosclerosis: Principal Results of the European Lacidipine Study on Atherosclerosis (Elsa), a Randomized, Double-Blind, Long-Term Trial. Circulation 2002, 106, 2422–2427. [Google Scholar] [CrossRef]
- Kwon, O.; Kang, S.J.; Kang, S.H.; Lee, P.H.; Yun, S.C.; Ahn, J.M.; Park, D.W.; Lee, S.W.; Kim, Y.H.; Lee, C.W.; et al. Relationship Between Serum Inflammatory Marker Levels and the Dynamic Changes in Coronary Plaque Characteristics after Statin Therapy. Circ. Cardiovasc. Imaging 2017, 10, e005934. [Google Scholar] [CrossRef]
- Lee, D.H.; Chun, E.J.; Hur, J.H.; Min, S.H.; Lee, J.E.; Oh, T.J.; Kim, K.M.; Jang, H.C.; Han, S.J.; Kang, D.K.; et al. Effect of Sarpogrelate, a Selective 5-Ht2a Receptor Antagonist, on Characteristics of Coronary Artery Disease in Patients with Type 2 Diabetes. Atherosclerosis 2017, 257, 47–54. [Google Scholar] [CrossRef]
- Matsumoto, S.; Ibrahim, R.; Gregoire, J.C.; L’Allier, P.L.; Pressacco, J.; Tardif, J.C.; Budoff, M.J. Effect of Treatment with 5-Lipoxygenase Inhibitor via-2291 (Atreleuton) on Coronary Plaque Progression: A Serial ct Angiography Study. Clin. Cardiol. 2017, 40, 210–215. [Google Scholar] [CrossRef]
- Park, S.J.; Kang, S.J.; Ahn, J.M.; Chang, M.; Yun, S.C.; Roh, J.H.; Lee, P.H.; Park, H.W.; Yoon, S.H.; Park, D.W.; et al. Effect of Statin Treatment on Modifying Plaque Composition: A Double-Blind, Randomized Study. J. Am. Coll. Cardiol. 2016, 67, 1772–1783. [Google Scholar] [CrossRef]
- Koskinas, K.C.; Zaugg, S.; Yamaji, K.; Garcia Garcia, H.M.; Taniwaki, M.; Klingenberg, R.; Moschovitis, A.; Luscher, T.F.; Van Tits, L.J.; Matter, C.M.; et al. Changes of Coronary Plaque Composition Correlate with c-Reactive Protein Levels in Patients with st-Elevation Myocardial Infarction Following High-Intensity Statin Therapy. Atherosclerosis 2016, 247, 154–160. [Google Scholar] [CrossRef]
- Matsumoto, S.; Nakanishi, R.; Li, D.; Alani, A.; Rezaeian, P.; Prabhu, S.; Abraham, J.; Fahmy, M.A.; Dailing, C.; Flores, F.; et al. Aged Garlic Extract Reduces Low Attenuation Plaque in Coronary Arteries of Patients with Metabolic Syndrome in a Prospective Randomized Double-Blind Study. J. Nutr. 2016, 146, 427S–432S. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.; Libby, P.; Nissen, S.E.; Wolski, K.; Ballantyne, C.M.; Barter, P.J.; Chapman, M.J.; Erbel, R.; Raichlen, J.S.; Uno, K.; et al. Long-Term Effects of Maximally Intensive Statin Therapy on Changes in Coronary Atheroma Composition: Insights from Saturn. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Eshtehardi, P.; McDaniel, M.C.; Dhawan, S.S.; Binongo, J.N.; Krishnan, S.K.; Golub, L.; Corban, M.T.; Raggi, P.; Quyyumi, A.A.; Samady, H. Effect of Intensive Atorvastatin Therapy on Coronary Atherosclerosis Progression, Composition, Arterial Remodeling, and Microvascular Function. J. Invasive Cardiol. 2012, 24, 522–529. [Google Scholar] [PubMed]
- Kojima, T.; Miyauchi, K.; Yokoyama, T.; Yokoyama, K.; Kurata, T.; Suwa, S.; Kawamura, M.; Tamura, H.; Okazaki, S.; Inoue, K.; et al. Azelnidipine and Amlodipine Anti-Coronary Atherosclerosis Trial in Hypertensive Patients Undergoing Coronary Intervention by Serial Volumetric Intravascular Ultrasound Analysis in Juntendo University (Alps-J). Circ. J. 2011, 75, 1071–1079. [Google Scholar] [CrossRef]
- Luscher, T.F.; Pieper, M.; Tendera, M.; Vrolix, M.; Rutsch, W.; Van Den Branden, F.; Gil, R.; Bischoff, K.O.; Haude, M.; Fischer, D.; et al. A Randomized Placebo-Controlled Study on the Effect of Nifedipine on Coronary Endothelial Function and Plaque Formation in Patients with Coronary Artery Disease: The Encore ii Study. Eur. Heart J. 2009, 30, 1590–1597. [Google Scholar] [CrossRef]
- Serhan, C.N.; Dalli, J.; Karamnov, S.; Choi, A.; Park, C.K.; Xu, Z.Z.; Ji, R.R.; Zhu, M.; Petasis, N.A. Macrophage Proresolving Mediator Maresin 1 Stimulates Tissue Regeneration and Controls Pain. FASEB J. 2012, 26, 1755–1765. [Google Scholar] [CrossRef]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: Novel Macrophage Mediators with Potent Antiinflammatory and Proresolving Actions. J. Exp. Med. 2009, 206, 15–23. [Google Scholar] [CrossRef]
- Quan, H.; Kim, J.M.; Lee, H.J.; Lee, S.H.; Choi, J.I.; Bae, H.B. Aicar Enhances the Phagocytic Ability of Macrophages Towards Apoptotic Cells Through p38 Mitogen Activated Protein Kinase Activation Independent of Amp-Activated Protein Kinase. PLoS ONE 2015, 10, e0127885. [Google Scholar] [CrossRef]
- Schutters, K.; Kusters, D.H.; Chatrou, M.L.; Montero Melendez, T.; Donners, M.; Deckers, N.M.; Krysko, D.V.; Vandenabeele, P.; Perretti, M.; Schurgers, L.J.; et al. Cell Surface-Expressed Phosphatidylserine as Therapeutic Target to Enhance Phagocytosis of Apoptotic Cells. Cell Death Differ. 2013, 20, 49–56. [Google Scholar] [CrossRef]
- Kamaly, N.; Fredman, G.; Subramanian, M.; Gadde, S.; Pesic, A.; Cheung, L.; Fayad, Z.A.; Langer, R.; Tabas, I.; Farokhzad, O.C. Development and in Vivo Efficacy of Targeted Polymeric Inflammation-Resolving Nanoparticles. Proc. Natl. Acad. Sci. USA 2013, 110, 6506–6511. [Google Scholar] [CrossRef]
- Bagalkot, V.; Deiuliis, J.A.; Rajagopalan, S.; Maiseyeu, A. “Eat Me” Imaging and Therapy. Adv. Drug Deliv. Rev. 2016, 99, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Beer, L.; Zimmermann, M.; Mitterbauer, A.; Ellinger, A.; Gruber, F.; Narzt, M.S.; Zellner, M.; Gyongyosi, M.; Madlener, S.; Simader, E.; et al. Analysis of the Secretome of Apoptotic Peripheral Blood Mononuclear Cells: Impact of Released Proteins and Exosomes for Tissue Regeneration. Sci. Rep. 2015, 5, 16662. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, H.; Li, Y.; Kanellakis, P.; Tay, C.; Cao, A.; Tipping, P.; Bobik, A.; Toh, B.H.; Kyaw, T. Phosphatidylserine Liposomes Mimic Apoptotic Cells to Attenuate Atherosclerosis by Expanding Polyreactive igm Producing b1a Lymphocytes. Cardiovasc. Res. 2015, 106, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Mevorach, D.; Zuckerman, T.; Reiner, I.; Shimoni, A.; Samuel, S.; Nagler, A.; Rowe, J.M.; Or, R. Single Infusion of Donor Mononuclear Early Apoptotic Cells as Prophylaxis for Graft-Versus-Host Disease in Myeloablative Hla-Matched Allogeneic Bone Marrow Transplantation: A Phase i/iia Clinical Trial. Biol. Blood Marrow Transplant. 2014, 20, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Parmley, W.W.; Blumlein, S.; Sievers, R. Modification of Experimental Atherosclerosis by Calcium-Channel Blockers. Am. J. Cardiol. 1985, 55, B165–B171. [Google Scholar] [CrossRef]
- Endo, A. A Historical Perspective on the Discovery of Statins. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Kovanen, P.T.; Goldstein, J.L. Regulation of Plasma Cholesterol by Lipoprotein Receptors. Science 1981, 212, 628–635. [Google Scholar] [CrossRef]
- Rodriguez, F.; Maron, D.J.; Knowles, J.W.; Virani, S.S.; Lin, S.; Heidenreich, P.A. Association Between Intensity of Statin Therapy and Mortality in Patients with Atherosclerotic Cardiovascular Disease. JAMA Cardiol. 2017, 2, 47–54. [Google Scholar] [CrossRef]
- Robson, J. Lipid Modification: Cardiovascular Risk Assessment and the Modification of Blood Lipids for the Primary and Secondary Prevention of Cardiovascular Disease. Heart 2008, 94, 1331–1332. [Google Scholar] [CrossRef]
- Son, B.K.; Kozaki, K.; Iijima, K.; Eto, M.; Kojima, T.; Ota, H.; Senda, Y.; Maemura, K.; Nakano, T.; Akishita, M.; et al. Statins Protect Human Aortic Smooth Muscle Cells from Inorganic Phosphate-Induced Calcification by Restoring gas6-Axl Survival Pathway. Circ. Res. 2006, 98, 1024–1031. [Google Scholar] [CrossRef]
- Son, B.K.; Kozaki, K.; Iijima, K.; Eto, M.; Nakano, T.; Akishita, M.; Ouchi, Y. Gas6/axl-Pi3k/akt Pathway Plays a Central Role in the Effect of Statins on Inorganic Phosphate-Induced Calcification of Vascular Smooth Muscle Cells. Eur. J. Pharmacol. 2007, 556, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zou, B.; Hou, Y.; Yan, W.; Chen, T.; Qu, S. Extracellular Vesicles in Vascular Calcification. Clin. Chim. Acta 2019, 499, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Joo, H.J.; Jung, H.W.; Lim, D.S. Investigating Potential Mediator Between Statin and Coronary Artery Calcification. PLoS ONE 2018, 13, e0203702. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, Y.; Yu, C.M.; Ji, Q.W.; Cai, M.; Zhao, Y.X.; Zhou, Y.J. Current Understanding of Coronary Artery Calcification. J. Geriatr. Cardiol. 2015, 12, 668–675. [Google Scholar]
- Lehmann, N.; Mohlenkamp, S.; Mahabadi, A.A.; Schmermund, A.; Roggenbuck, U.; Seibel, R.; Gronemeyer, D.; Budde, T.; Dragano, N.; Stang, A.; et al. Effect of Smoking and Other Traditional Risk Factors on the Onset of Coronary Artery Calcification: Results of the Heinz Nixdorf Recall Study. Atherosclerosis 2014, 232, 339–345. [Google Scholar] [CrossRef]
- Mohlenkamp, S.; Schmermund, A.; Lehmann, N.; Roggenbuck, U.; Dragano, N.; Stang, A.; Moebus, S.; Beck, E.M.; Schluter, C.; Sack, S.; et al. Subclinical Coronary Atherosclerosis and Resting Ecg Abnormalities in an Unselected General Population. Atherosclerosis 2008, 196, 786–794. [Google Scholar] [CrossRef]
- Ikegami, Y.; Inoue, I.; Inoue, K.; Shinoda, Y.; Iida, S.; Goto, S.; Nakano, T.; Shimada, A.; Noda, M. The Annual Rate of Coronary Artery Calcification with Combination Therapy with a pcsk9 Inhibitor and a Statin is Lower Than That with Statin Monotherapy. NPJ Aging Mech. Dis. 2018, 4, 7. [Google Scholar] [CrossRef]
- Baigent, C.; Keech, A.; Kearney, P.M.; Blackwell, L.; Buck, G.; Pollicino, C.; Kirby, A.; Sourjina, T.; Peto, R.; Collins, R.; et al. Efficacy and Safety of Cholesterol-Lowering Treatment: Prospective Meta-Analysis of Data from 90,056 Participants in 14 Randomised Trials of Statins. Lancet 2005, 366, 1267–1278. [Google Scholar]
- Han, X.; Zhang, Y.; Yin, L.; Zhang, L.; Wang, Y.; Zhang, H.; Li, B. Statin in the Treatment of Patients with Myocardial Infarction: A Meta-Analysis. Medicine 2018, 97, e0167. [Google Scholar] [CrossRef]
- Puri, R.; Nicholls, S.J.; Shao, M.; Kataoka, Y.; Uno, K.; Kapadia, S.R.; Tuzcu, E.M.; Nissen, S.E. Impact of Statins on Serial Coronary Calcification During Atheroma Progression and Regression. J. Am. Coll. Cardiol. 2015, 65, 1273–1282. [Google Scholar] [CrossRef]
- Nissen, S.E.; Tuzcu, E.M.; Schoenhagen, P.; Crowe, T.; Sasiela, W.J.; Tsai, J.; Orazem, J.; Magorien, R.D.; O’Shaughnessy, C.; Ganz, P. Statin Therapy, Ldl Cholesterol, C-Reactive Protein, and Coronary Artery Disease. N. Engl. J. Med. 2005, 352, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Henein, M.; Granasen, G.; Wiklund, U.; Schmermund, A.; Guerci, A.; Erbel, R.; Raggi, P. High dose and Long-Term Statin Therapy Accelerate Coronary Artery Calcification. Int. J. Cardiol. 2015, 184, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Nakazato, R.; Gransar, H.; Berman, D.S.; Cheng, V.Y.; Lin, F.Y.; Achenbach, S.; Al Mallah, M.; Budoff, M.J.; Cademartiri, F.; Callister, T.Q.; et al. Statins Use and Coronary Artery Plaque Composition: Results from the International Multicenter Confirm Registry. Atherosclerosis 2012, 225, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Rivner, H.; Denis, R.; Tang, F.; Marzouka, G.R. Abstract 17248: Statin Therapy Increases Coronary Artery Calcification Scores. Circulation 2018, 138, A17248. [Google Scholar]
- Achenbach, S.; Ropers, D.; Pohle, K.; Leber, A.; Thilo, C.; Knez, A.; Menendez, T.; Maeffert, R.; Kusus, M.; Regenfus, M.; et al. Influence of Lipid-Lowering Therapy on the Progression of Coronary Artery Calcification: A Prospective Evaluation. Circulation 2002, 106, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Budoff, M.J.; Lane, K.L.; Bakhsheshi, H.; Mao, S.; Grassmann, B.O.; Friedman, B.C.; Brundage, B.H. Rates of Progression of Coronary Calcium by Electron Beam Tomography. Am. J. Cardiol. 2000, 86, 8–11. [Google Scholar] [CrossRef]
- Cheng, X.W.; Huang, Z.; Kuzuya, M.; Okumura, K.; Murohara, T. Cysteine Protease Cathepsins in Atherosclerosis-Based Vascular Disease and its Complications. Hypertension 2011, 58, 978–986. [Google Scholar] [CrossRef]
- Mirjanic Azaric, B.; Vekic, J.; Zeljkovic, A.; Jelic Ivanovic, Z.; Djeric, M.; Milivojac, T.; Pecar Fonovic, U.; Marc, J.; Kos, J.; Cerne, D. Interrelated Cathepsin s-Lowering and Ldl Subclass Profile Improvements Induced by Atorvastatin in the Plasma of Stable Angina Patients. J. Atheroscler. Thromb. 2014, 21, 868–877. [Google Scholar] [CrossRef]
- Kalia, N.K.; Miller, L.G.; Nasir, K.; Blumenthal, R.S.; Agrawal, N.; Budoff, M.J. Visualizing Coronary Calcium is Associated with Improvements in Adherence to Statin Therapy. Atherosclerosis 2006, 185, 394–399. [Google Scholar] [CrossRef]
- Raggi, P.; Callister, T.Q.; Shaw, L.J. Progression of Coronary Artery Calcium and Risk of First Myocardial Infarction in Patients Receiving Cholesterol-Lowering Therapy. Arter. Thromb. Vasc. Biol. 2004, 24, 1272–1277. [Google Scholar] [CrossRef]
- Martinet, W.; Coornaert, I.; Puylaert, P.; De Meyer, G.R.Y. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 2019, 10, 306. [Google Scholar] [CrossRef] [PubMed]




| Author and Year | Disorders | Intervention | Numbers of Patients | Effect on Plaque Calcification | Effect on Plaque Volume |
|---|---|---|---|---|---|
| Bruining et al., 2009 [153] | Coronary artery disease | Perindopril (angiotensin-converting enzyme inhibitors) | 118 patients | ↓ | ↓ |
| Magnani et al., 1992 [154] | Atherosclerosis and plaque evolution, with hypertension | Verapamil, dihydropyridines and diphenylalkylamines (calcium antagonists) | 550 patients | ↓ | ↓ |
| Zanchetti et al., 2002 [155] | Asymptomatic carotid atherosclerosis | Lacidipine (calcium antagonist) | 2334 patients | ↓ | ↓ |
| Kwon et al., 2017 [156] | Coronary artery disease | Rosuvastatin (HMG-CoA reductase inhibitor) | 218 patients | ↑ | ↓ |
| Lee et al., 2017 [157] | Diabetic patients with 10–75% coronary artery stenosis | Sarpogrelate (5-HT2A receptor antagonist) | 40 patients | ↓ | ↓ |
| Matsumoto et al., 2017 [158] | Patients with recent acute coronary syndrome | Atreleuton (5-lipoxygenase inhibitor VIA-2291) | 54 patients | ↓ | ↓ |
| Park et al., 2016 [159] | Statin and atheroma vulnerability evaluation study | Rosuvastatin (HMG-CoA reductase inhibitor) | 225 patients | Unchanged | ↓ |
| Koskinas et al., 2016 [160] | ST-elevation myocardial infarction | Rosuvastatin (HMG-CoA reductase inhibitor | 44 patients | ↑ | ↓ |
| Matsumoto et al., 2016 [161] | Metabolic syndrome | Aged garlic extract | 27 patients | ↓ | ↓ |
| Puri et al., 2014 [162] | Coronary atheroma | Rosuvastatin vs. atorvastatin (HMG-CoA reductase inhibitors) | 71 patients | ↑ | ↓ |
| Eshtehardi et al., 2012 [163] | Moderate coronary artery disease | Atorvastatin (HMG-CoA reductase) | 20 patients | ↑ | ↓ |
| Kojima et al., 2011 [164] | Hypertension | Azelnidipine and amlodipine (calcium channel blockers) | 199 patients | ↓ | ↓ |
| Lüscher et al., 2009 [165] | Coronary artery disease | Nifedipine (calcium channel blocker) | 454 patients | ↓ | ↓ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tajbakhsh, A.; Kovanen, P.T.; Rezaee, M.; Banach, M.; Sahebkar, A. Ca2+ Flux: Searching for a Role in Efferocytosis of Apoptotic Cells in Atherosclerosis. J. Clin. Med. 2019, 8, 2047. https://doi.org/10.3390/jcm8122047
Tajbakhsh A, Kovanen PT, Rezaee M, Banach M, Sahebkar A. Ca2+ Flux: Searching for a Role in Efferocytosis of Apoptotic Cells in Atherosclerosis. Journal of Clinical Medicine. 2019; 8(12):2047. https://doi.org/10.3390/jcm8122047
Chicago/Turabian StyleTajbakhsh, Amir, Petri T. Kovanen, Mahdi Rezaee, Maciej Banach, and Amirhossein Sahebkar. 2019. "Ca2+ Flux: Searching for a Role in Efferocytosis of Apoptotic Cells in Atherosclerosis" Journal of Clinical Medicine 8, no. 12: 2047. https://doi.org/10.3390/jcm8122047
APA StyleTajbakhsh, A., Kovanen, P. T., Rezaee, M., Banach, M., & Sahebkar, A. (2019). Ca2+ Flux: Searching for a Role in Efferocytosis of Apoptotic Cells in Atherosclerosis. Journal of Clinical Medicine, 8(12), 2047. https://doi.org/10.3390/jcm8122047