Anti-ApoA-1 IgGs in Familial Hypercholesterolemia Display Paradoxical Associations with Lipid Profile and Promote Foam Cell Formation

 , , , and
, , , and 
Abstract
:1. Introduction
2. Experimental Section
2.1. Patients
2.2. Plasma Samples and Blood Analyses
2.3. Assessment of Anti-ApoA-1 IgG Levels
2.4. Reagents
2.5. Human Monocyte-Derived Macrophage Preparation
2.6. Protein Purification and Western Blot Analysis
2.7. Lipid Uptake by Oil Red O Staining
2.8. RNA Extraction
2.9. miRNA Quantitative PCR
2.10. Quantitative PCR
2.11. Anti-ApoA-1 IgG Modulation of Membrane Free Cholesterol. Assay of Cholesterol Oxidase
2.12. ACAT Activity Assessment
2.13. Measurement of Free Cholesterol Content in Cell Supernatant
2.14. Passive Diffusion Analysis
2.15. Statistics
3. Results
3.1. Anti-ApoA-1 IgG Associations with Lipid Profile in FH Children
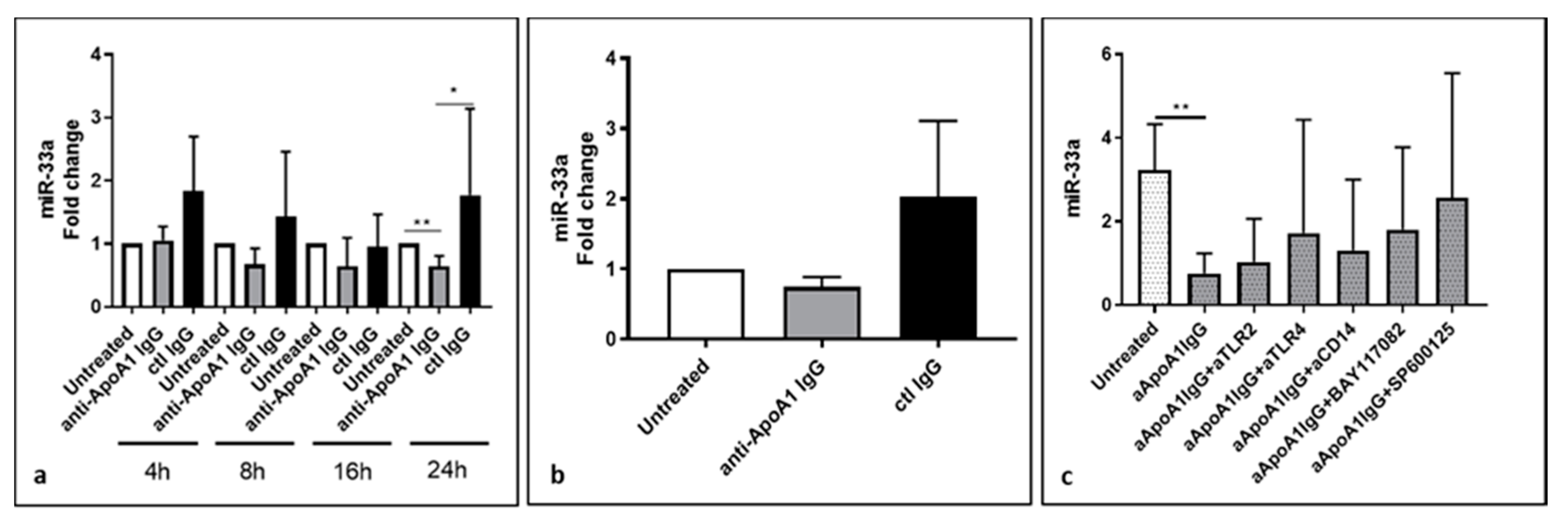
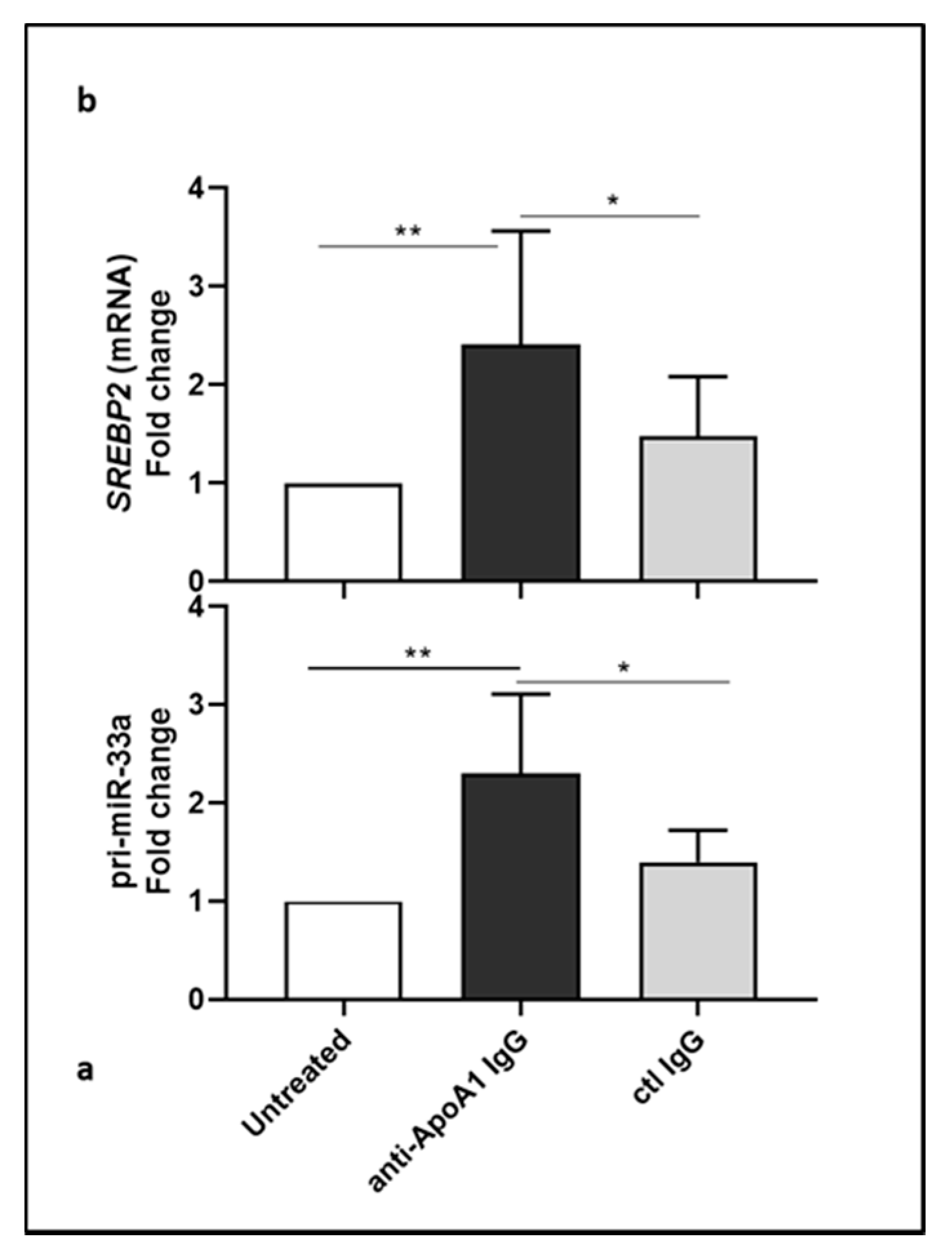
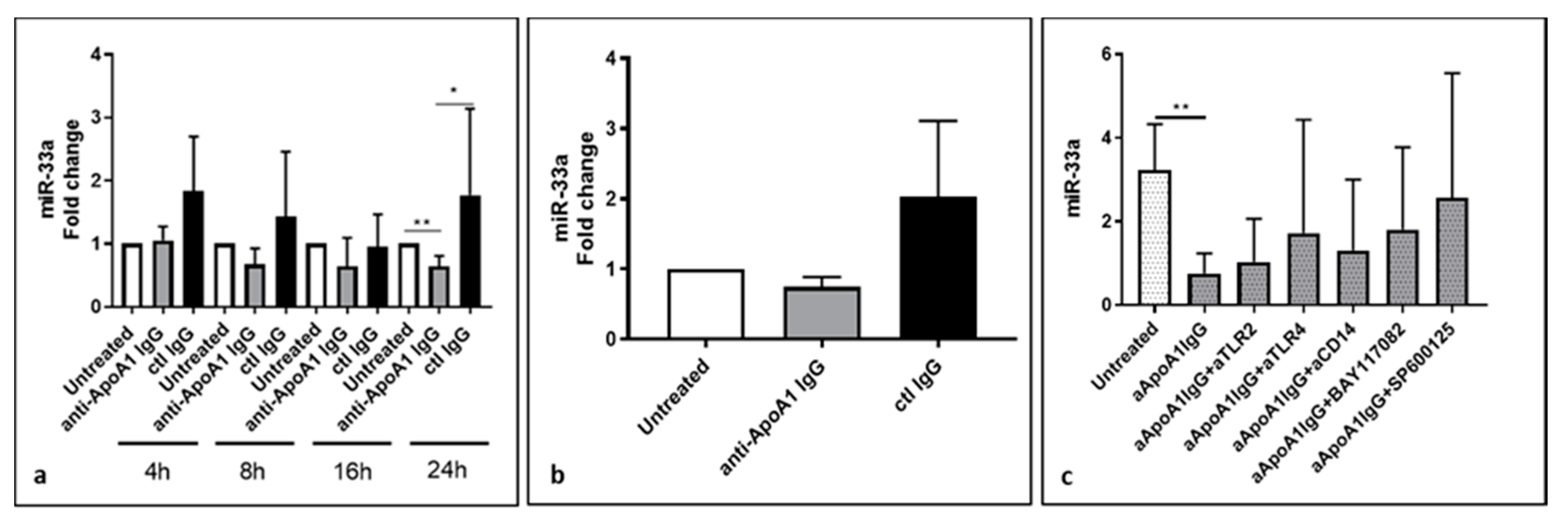
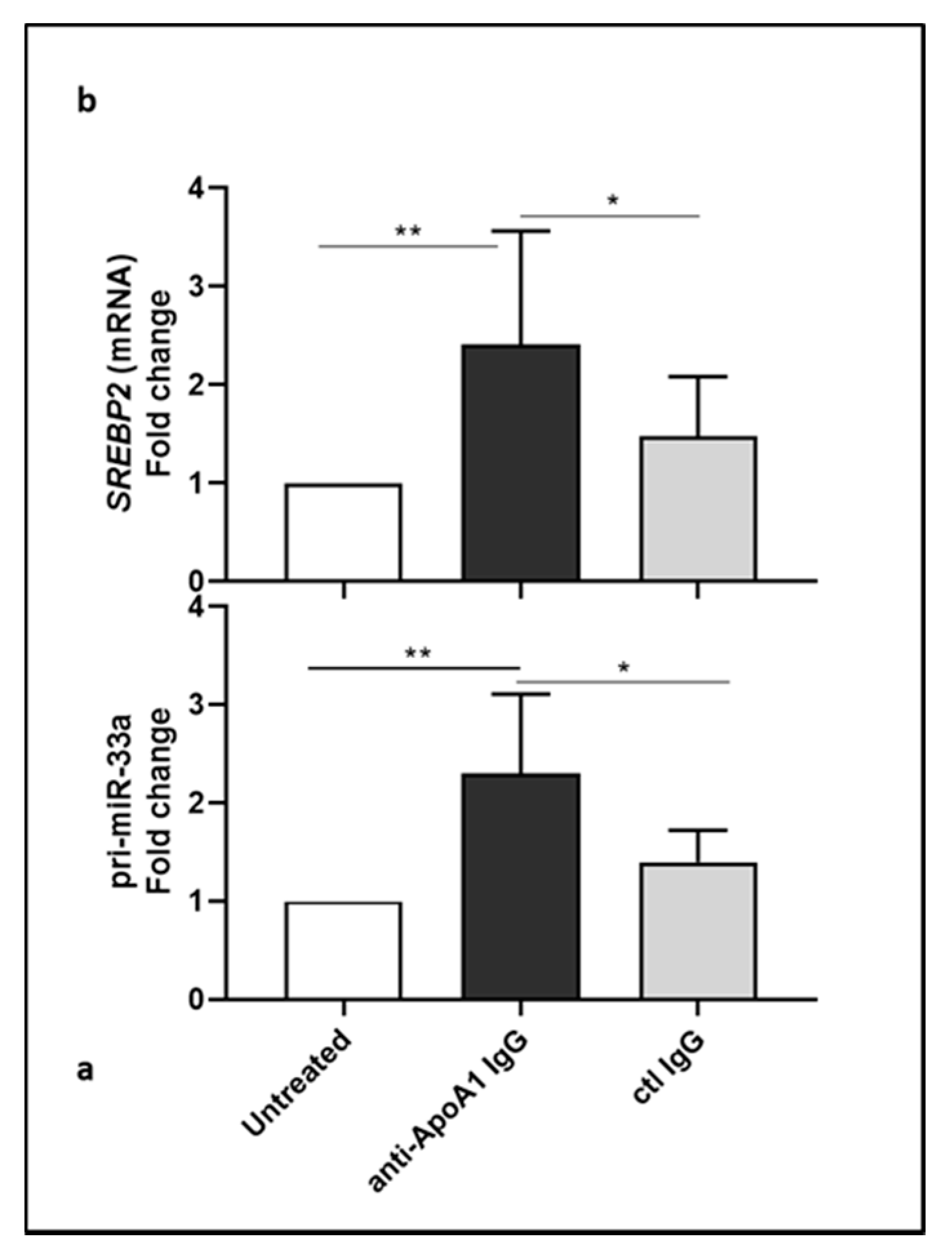
3.2. Lower Level of miR-33a in Anti-ApoA-1 IgG-Treated Human Macrophages
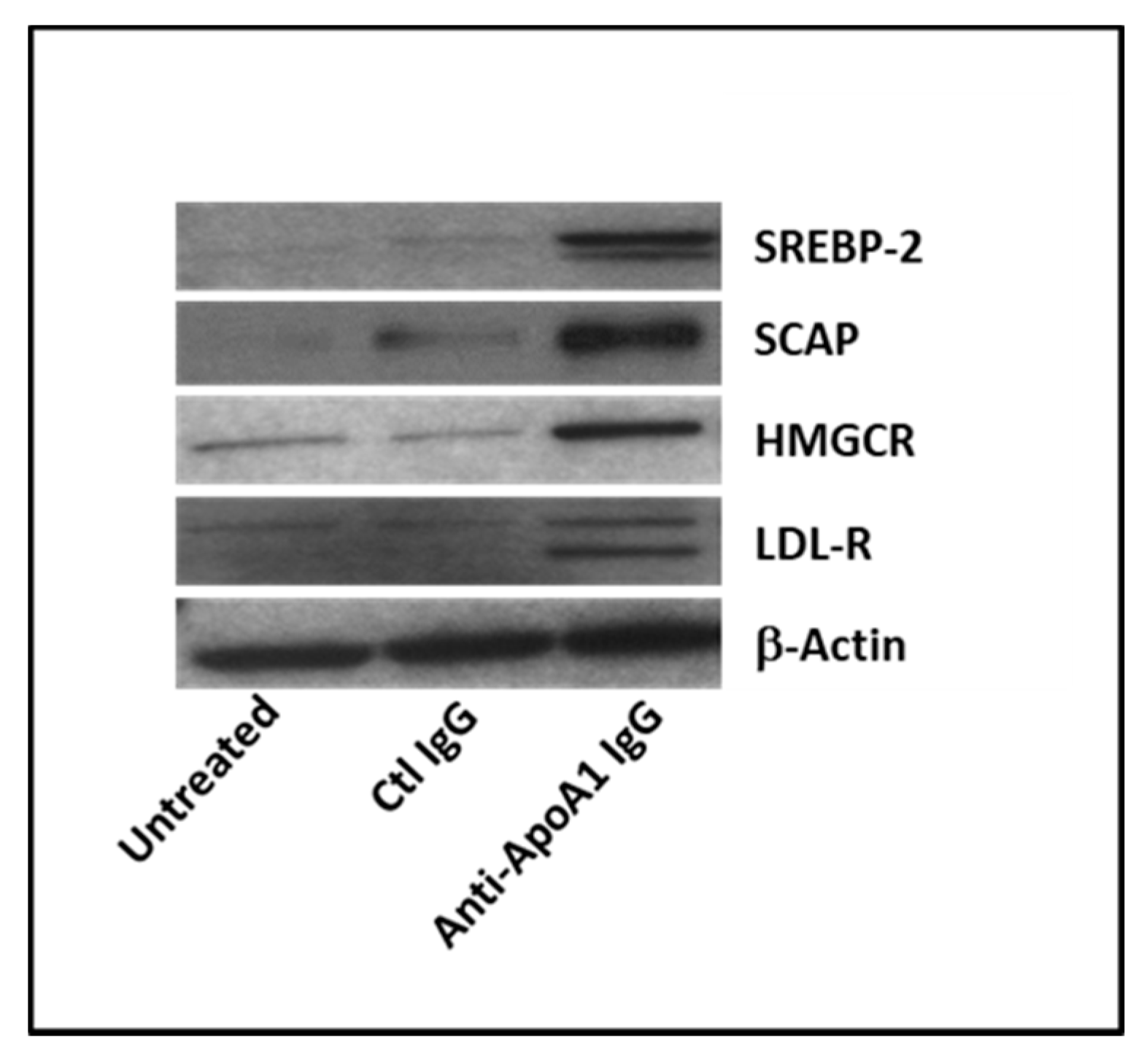
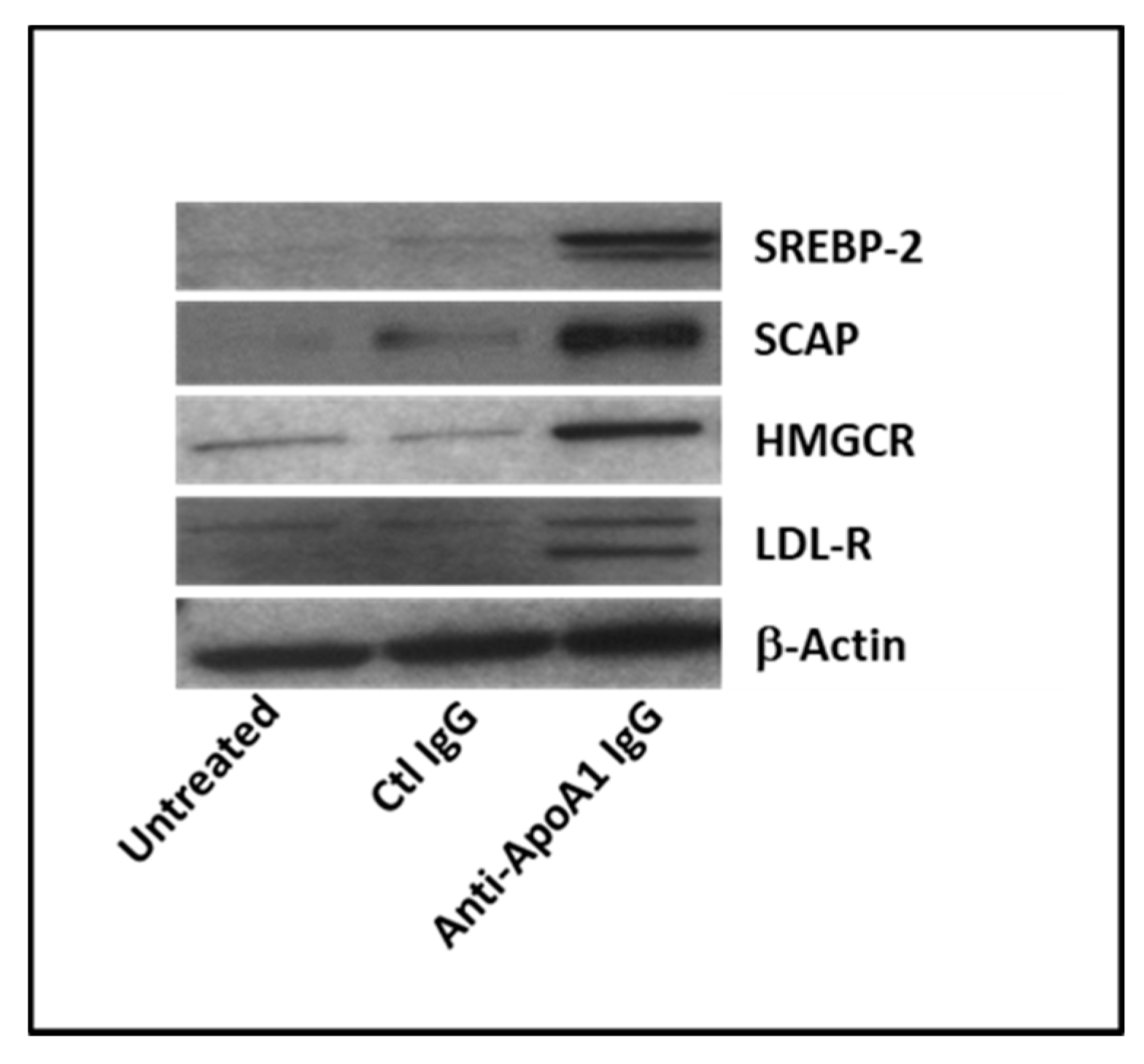
3.3. Anti-ApoA-1 IgGs Modulate Cholesterol-Regulating Proteins in Macrophages
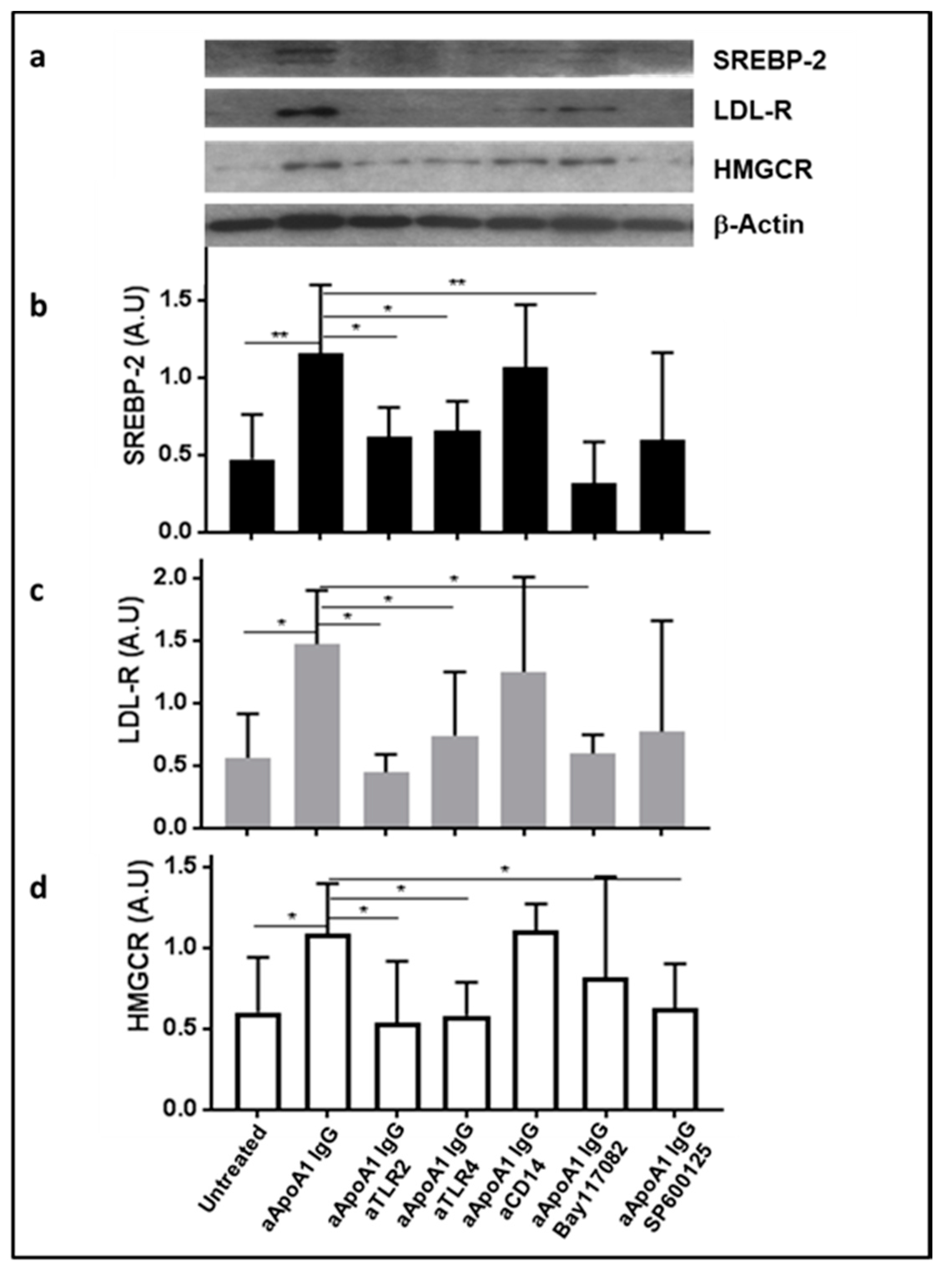
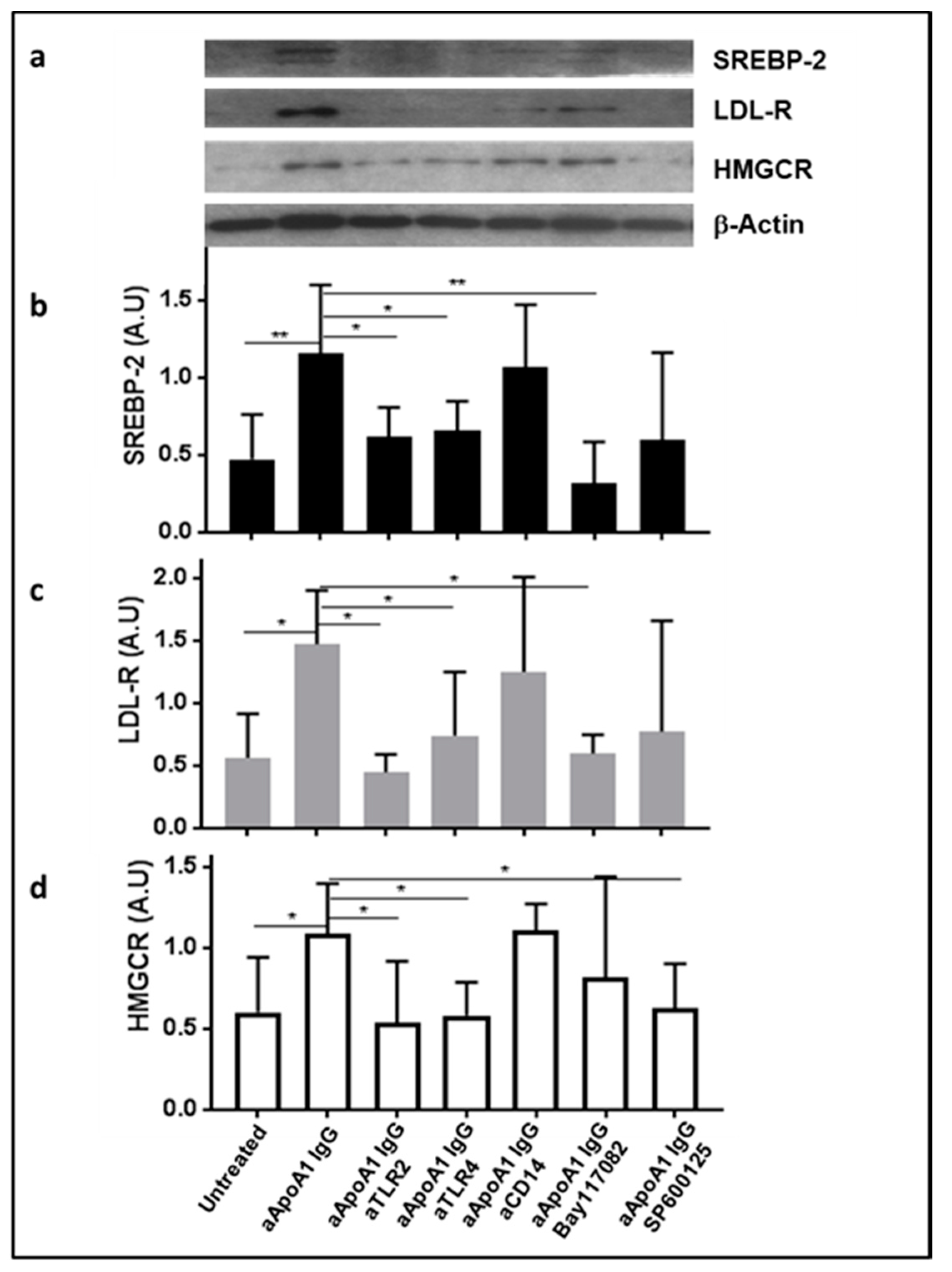
3.4. The Impact of Anti-ApoA-1 IgG on Cholesterol-Regulating Proteins Is Mediated by TLR2 and TLR4
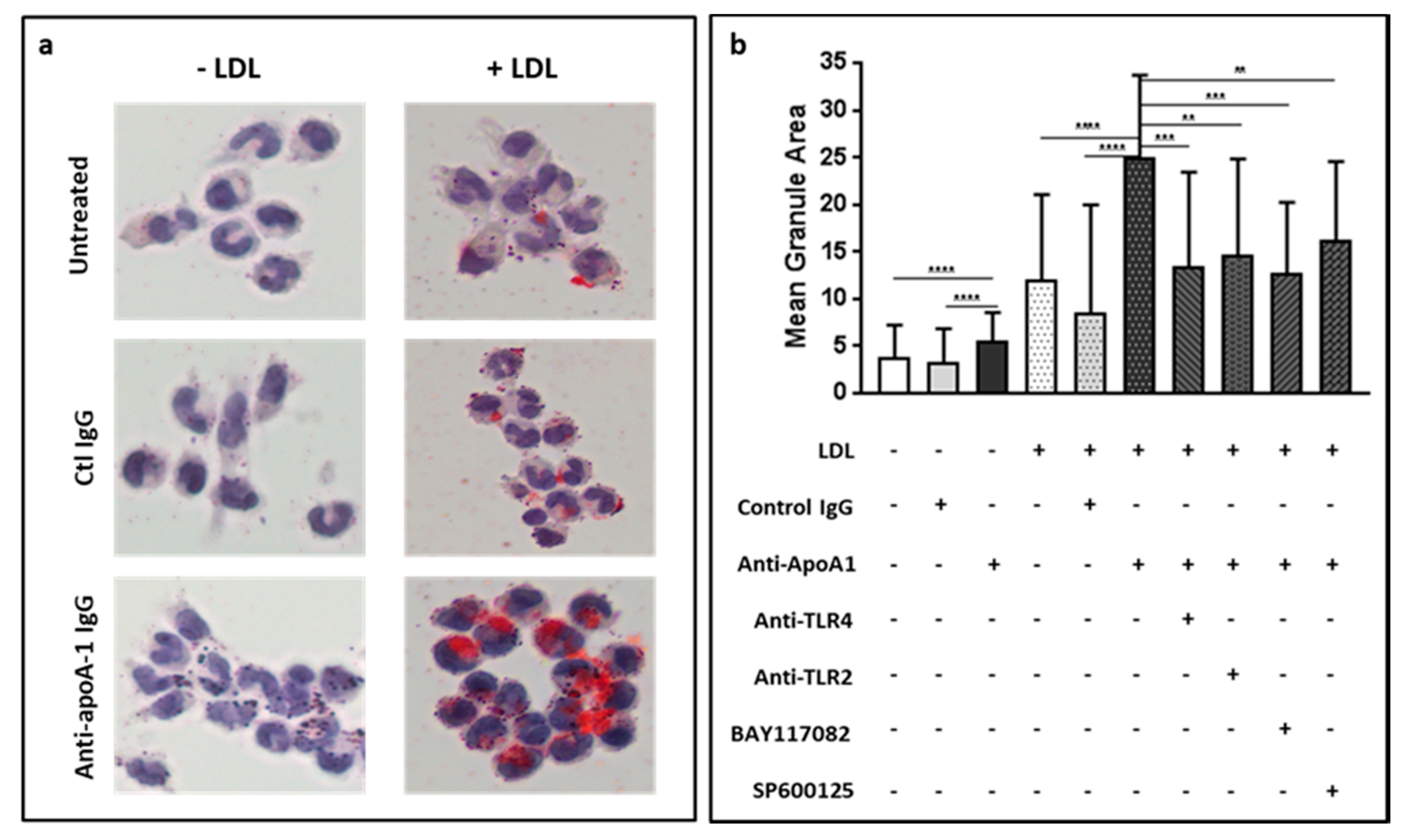
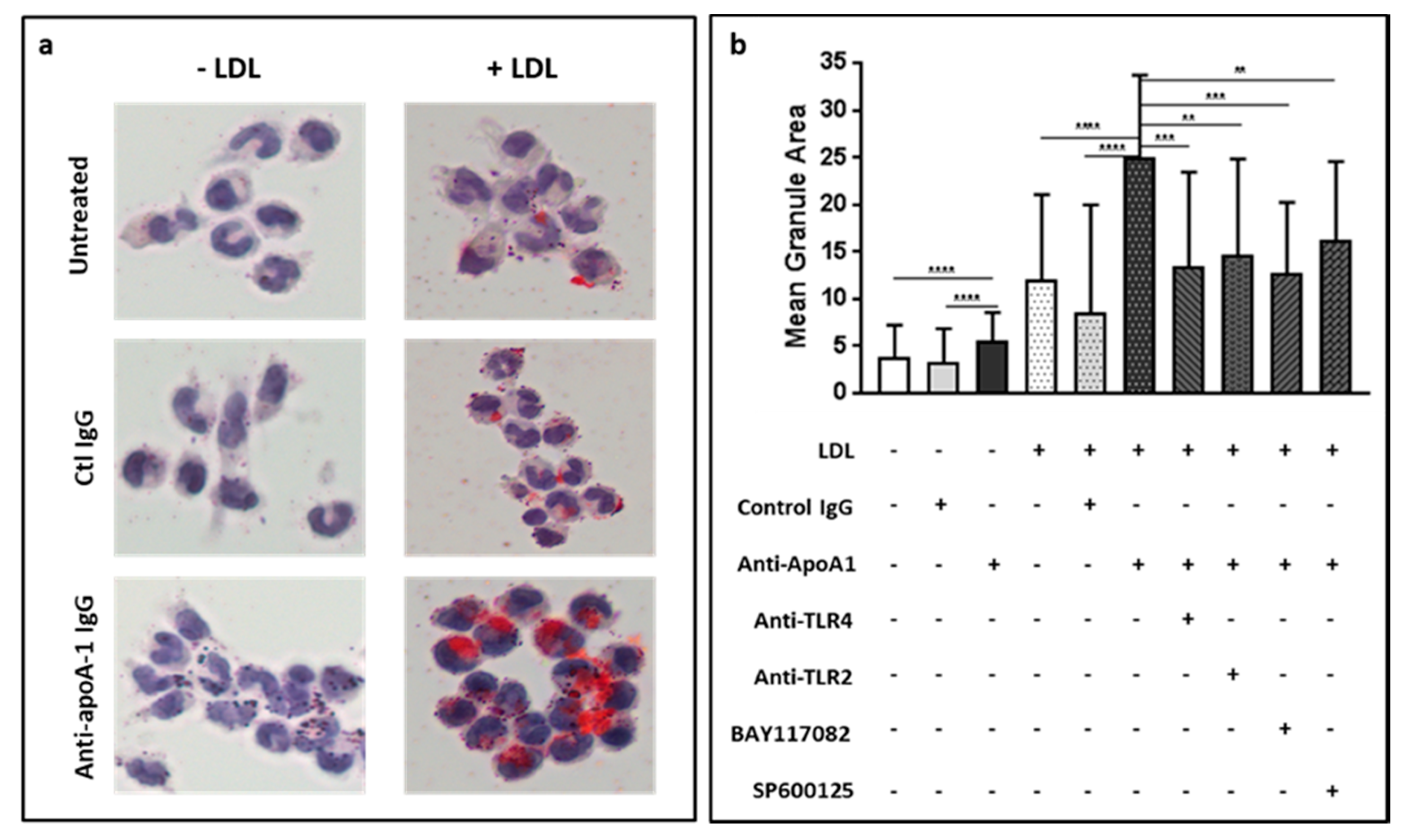
3.5. Anti-ApoA-1 IgGs Promote LDL Uptake and Foam Cell Formation
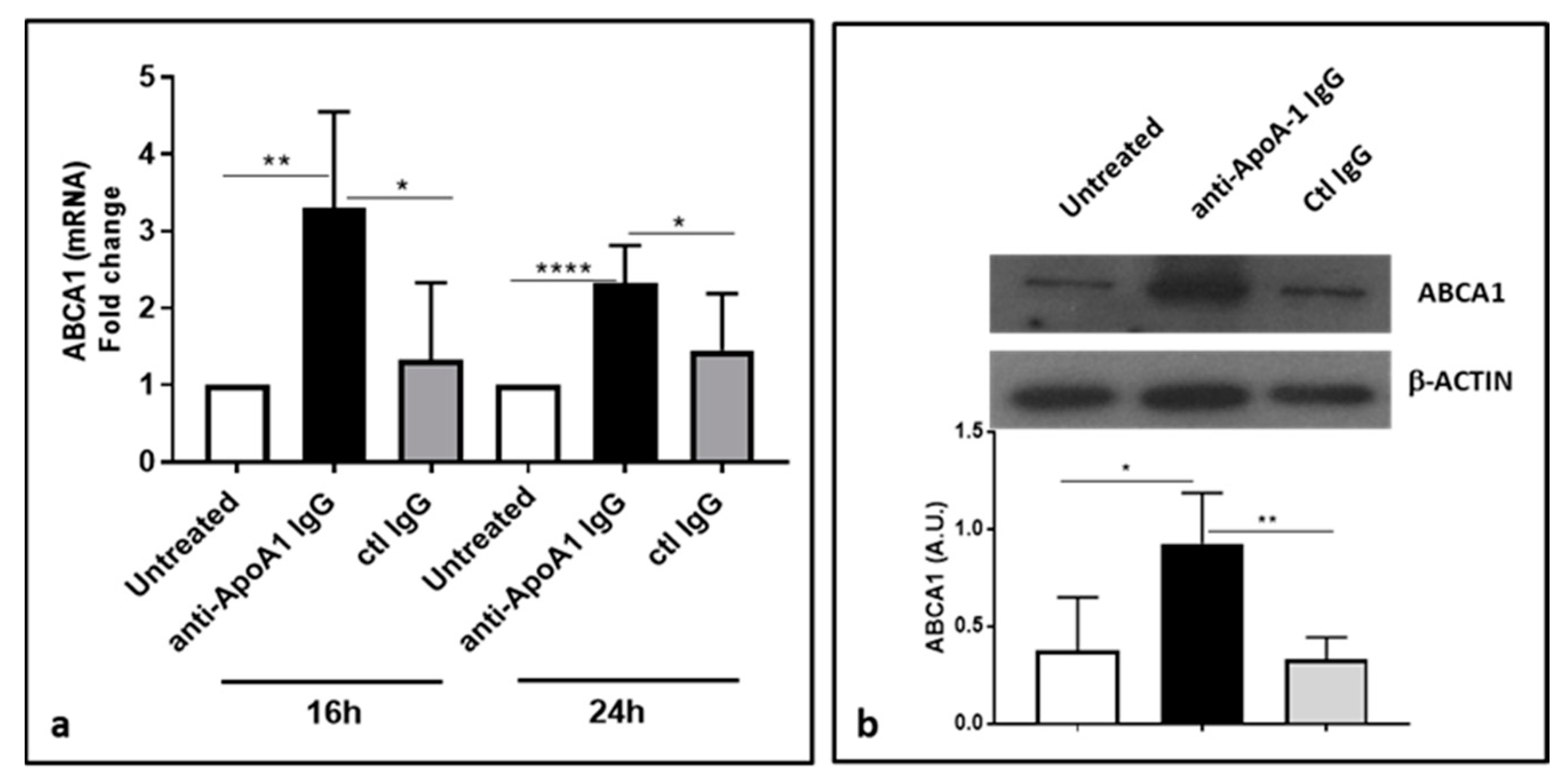
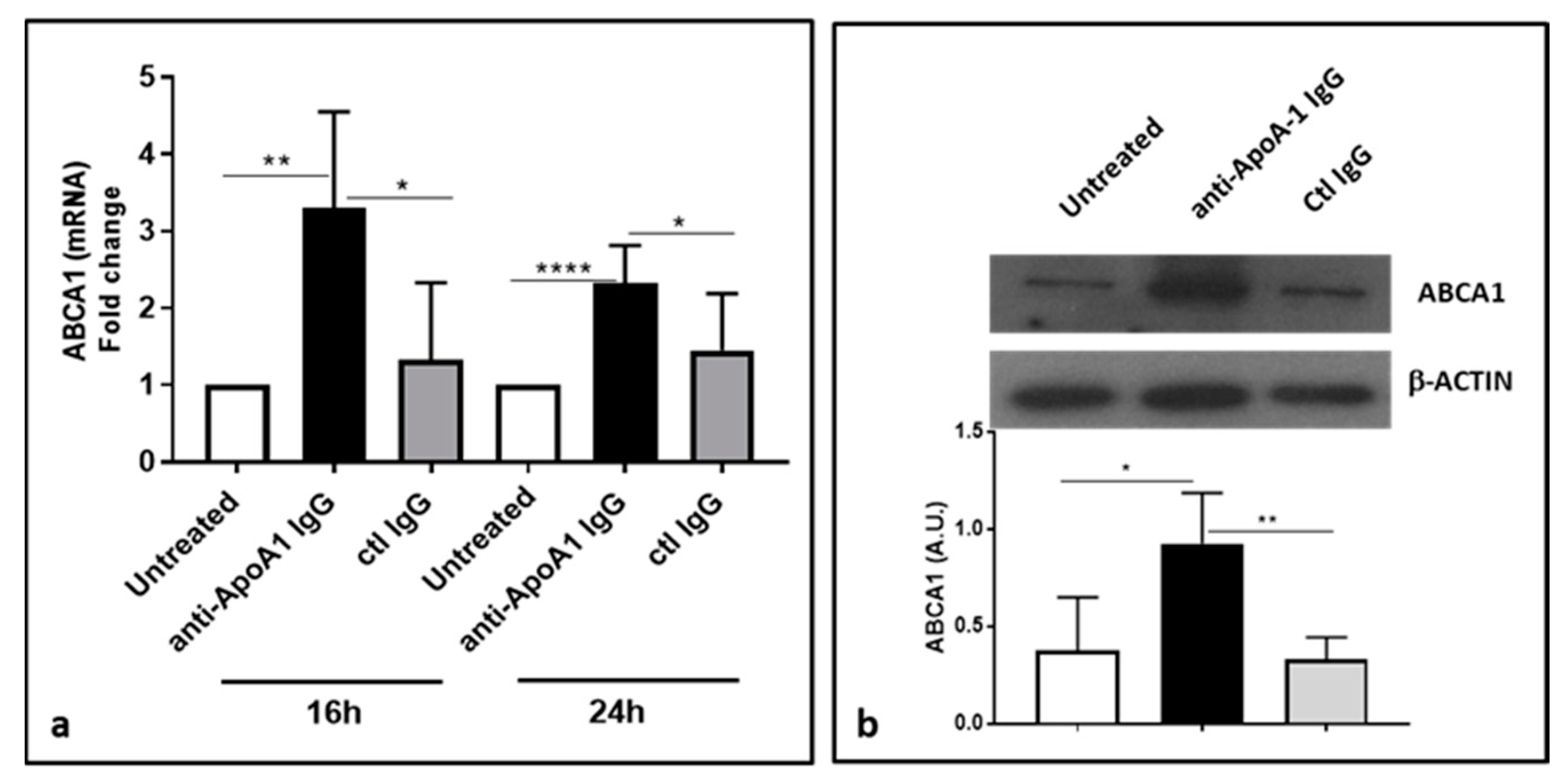
3.6. Anti-ApoA-1 IgGs Upregulate ABCA1
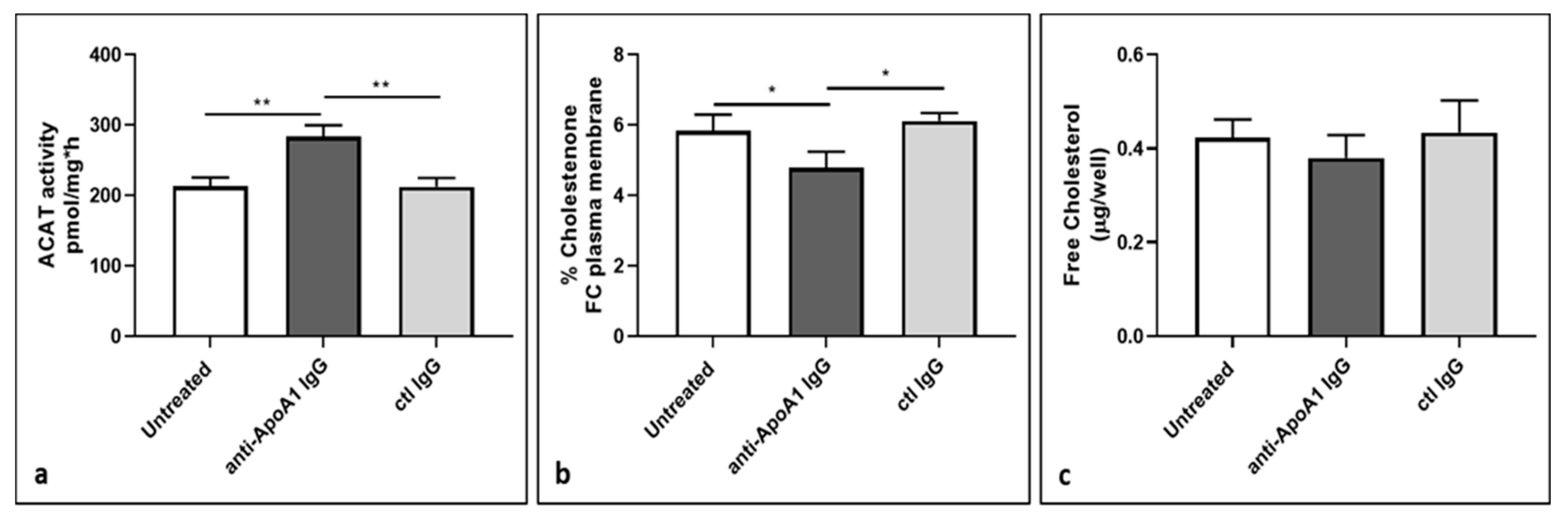
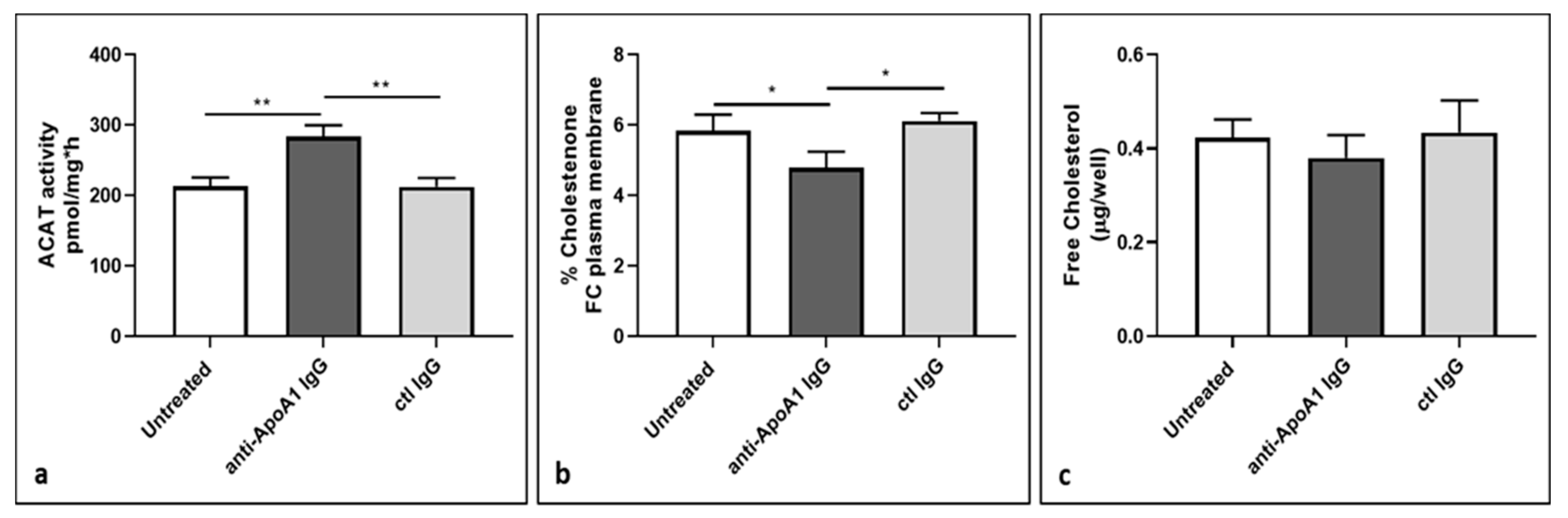
3.7. ACAT Activity and Cellular Cholesterol Distribution in Macrophages Following Anti-ApoA1 IgG Treatment
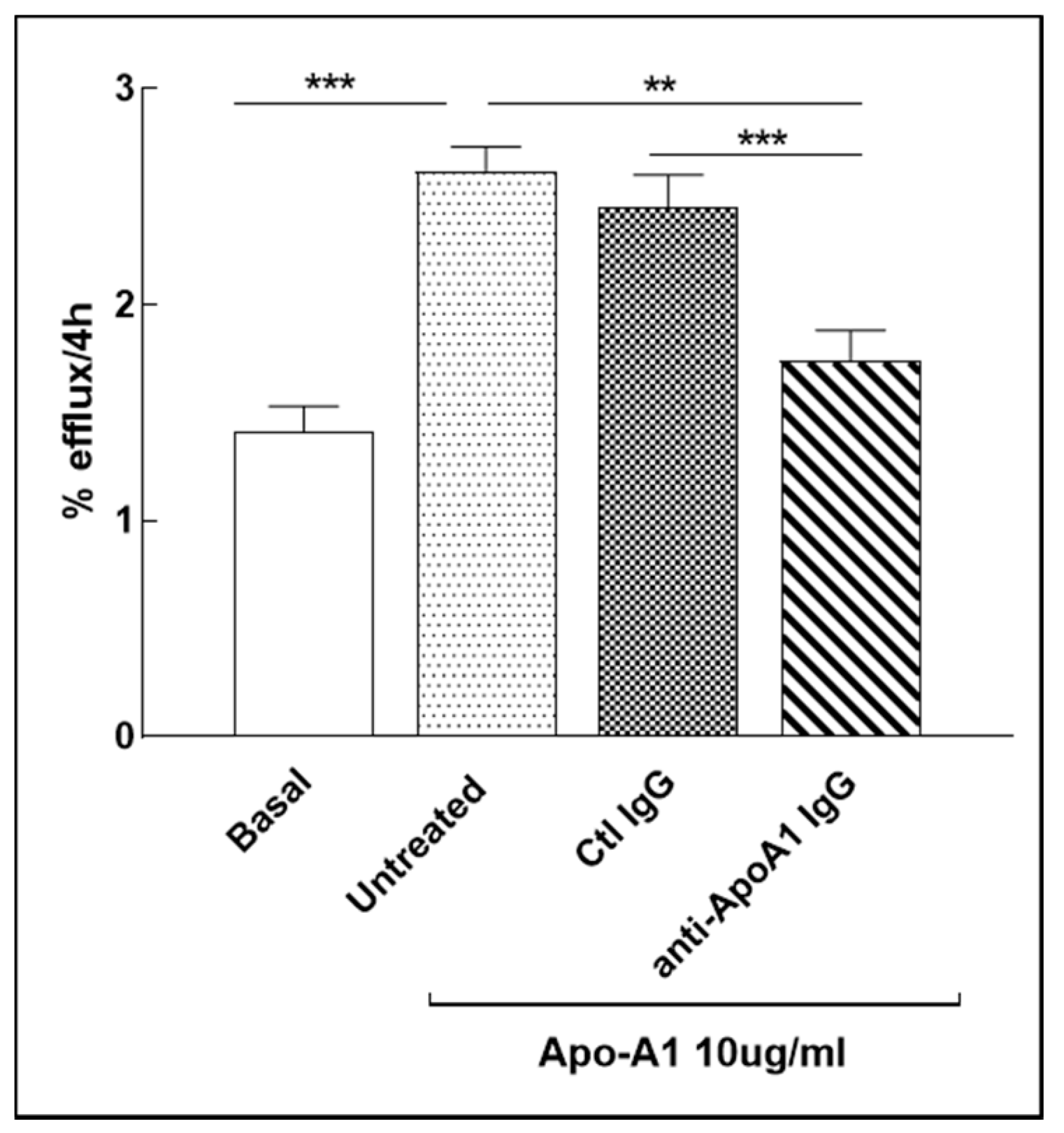
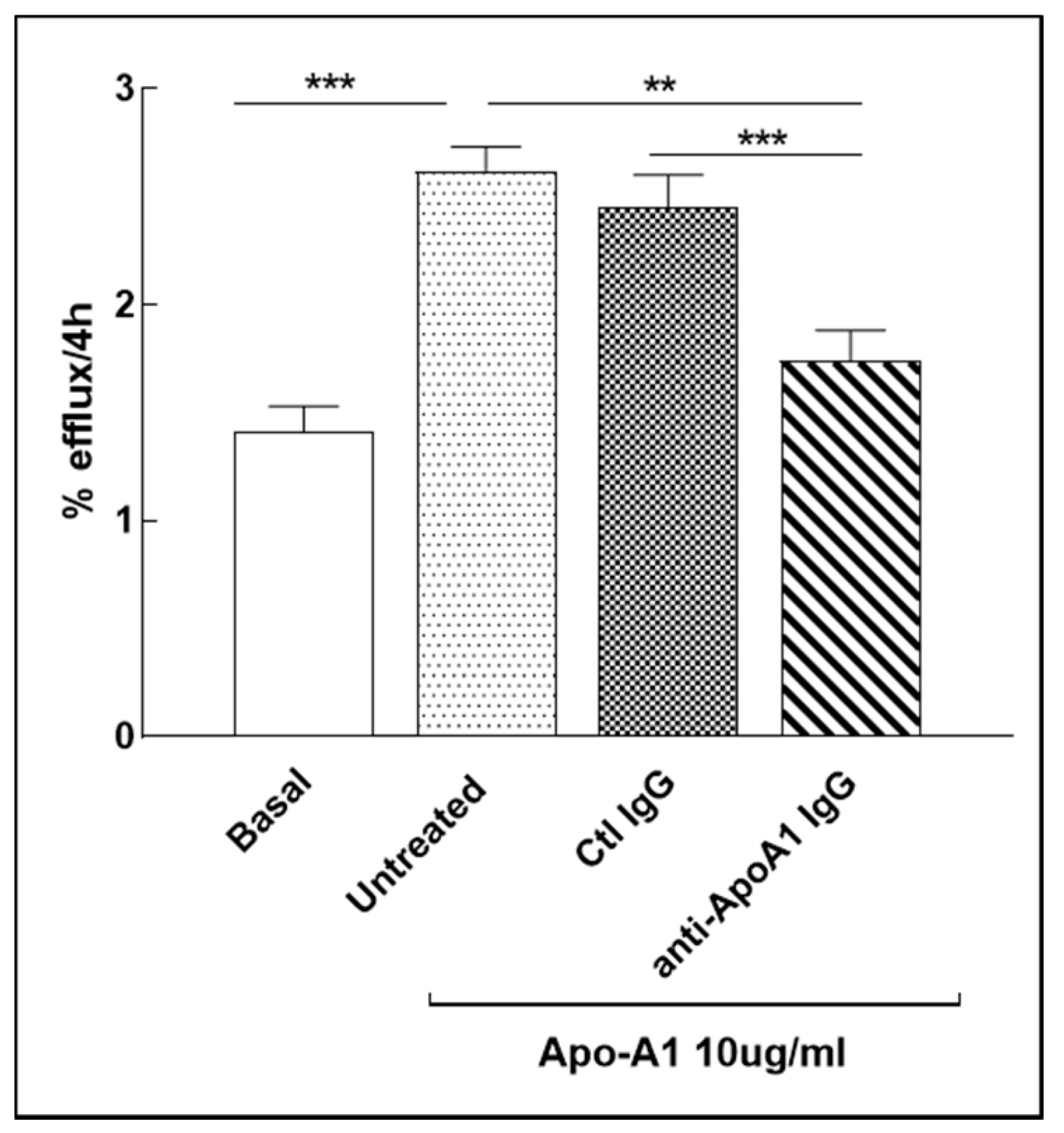
3.8. Anti-ApoA-1 IgG Impact on Cholesterol Passive Diffusion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Libby, P.; Tabas, I.; Fredman, G.; Fisher, E.A. Inflammation and Its Resolution as Determinants of Acute Coronary Syndromes. Circ. Res. 2014, 114, 1867–1879. [Google Scholar] [CrossRef]
- Satta, N.; Vuilleumier, N. Auto-Antibodies as Possible Markers and Mediators of Ischemic, Dilated, and Rhythmic Cardiopathies. Curr. Drug Targets 2015, 16, 342–360. [Google Scholar] [CrossRef]
- Antiochos, P.; Marques-Vidal, P.; Virzi, J.; Pagano, S.; Satta, N.; Bastardot, F.; Hartley, O.; Montecucco, F.; Mach, F.; Waeber, G.; et al. Association between Anti-Apolipoprotein a-1 Antibodies and Cardiovascular Disease in the General Population. Results from the Colaus Study. Thromb. Haemost. 2016, 116, 764–771. [Google Scholar] [CrossRef]
- Antiochos, P.; Marques-Vidal, P.; Virzi, J.; Pagano, S.; Satta, N.; Hartley, O.; Montecucco, F.; Mach, F.; Kutalik, Z.; Waeber, G.; et al. Impact of Cd14 Polymorphisms on Anti-Apolipoprotein a-1 Igg-Related Coronary Artery Disease Prediction in the General Population. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2342–2349. [Google Scholar] [CrossRef]
- Antiochos, P.; Marques-Vidal, P.; Virzi, J.; Pagano, S.; Satta, N.; Hartley, O.; Montecucco, F.; Mach, F.; Kutalik, Z.; Waeber, G.; et al. Anti-Apolipoprotein a-1 Igg Predict All-Cause Mortality and Are Associated with Fc Receptor-Like 3 Polymorphisms. Front. Immunol. 2017, 8, 437. [Google Scholar] [CrossRef]
- Carbone, F.; Satta, N.; Montecucco, F.; Virzi, J.; Burger, F.; Roth, A.; Roversi, G.; Tamborino, C.; Casetta, I.; Seraceni, S.; et al. Anti-Apoa-1 Igg Serum Levels Predict Worse Poststroke Outcomes. Eur. J. Clin. Investig. 2016, 46, 805–817. [Google Scholar] [CrossRef]
- Vuilleumier, N.; Bas, S.; Pagano, S.; Montecucco, F.; Guerne, P.A.; Finckh, A.; Lovis, C.; Mach, F.; Hochstrasser, D.; Roux-Lombard, P.; et al. Anti-Apolipoprotein a-1 Igg Predicts Major Cardiovascular Events in Patients with Rheumatoid Arthritis. Arthritis Rheum. 2010, 62, 2640–2650. [Google Scholar] [CrossRef] [PubMed]
- Vuilleumier, N.; Montecucco, F.; Spinella, G.; Pagano, S.; Bertolotto, M.; Pane, B.; Pende, A.; Galan, K.; Roux-Lombard, P.; Combescure, C.; et al. Serum Levels of Anti-Apolipoprotein a-1 Auto-Antibodies and Myeloperoxidase as Predictors of Major Adverse Cardiovascular Events after Carotid Endarterectomy. Thromb. Haemost. 2013, 109, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Vuilleumier, N.; Rossier, M.F.; Pagano, S.; Python, M.; Charbonney, E.; Nkoulou, R.; James, R.; Reber, G.; Mach, F.; Roux-Lombard, P. Anti-Apolipoprotein a-1 Igg as an Independent Cardiovascular Prognostic Marker Affecting Basal Heart Rate in Myocardial Infarction. Eur. Heart J. 2010, 31, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.C.; Pagano, S.; Virzi, J.; Dullaart, R.P.F.; Annema, W.; Kuipers, F.; Bakker, S.J.L.; Vuilleumier, N.; Tietge, U.J.F. Autoantibodies to Apolipoprotein a-1 as Independent Predictors of Cardiovascular Mortality in Renal Transplant Recipients. J. Clin. Med. 2019, 8, 948. [Google Scholar] [CrossRef]
- Vuilleumier, N.; Pagano, S.; Combescure, C.; Gencer, B.; Virzi, J.; Raber, L.; Carballo, D.; Carballo, S.; Nanchen, D.; Rodondi, N.; et al. Non-Linear Relationship between Anti-Apolipoprotein a-1 Iggs and Cardiovascular Outcomes in Patients with Acute Coronary Syndromes. J. Clin. Med. 2019, 8, 1002. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Braunersreuther, V.; Burger, F.; Lenglet, S.; Pelli, G.; Carbone, F.; Fraga-Silva, R.; Stergiopulos, N.; Monaco, C.; Mueller, C.; et al. Anti-Apoa-1 Auto-Antibodies Increase Mouse Atherosclerotic Plaque Vulnerability, Myocardial Necrosis and Mortality Triggering Tlr2 and Tlr4. Thromb. Haemost. 2015, 114, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Vuilleumier, N.; Pagano, S.; Lenglet, S.; Bertolotto, M.; Braunersreuther, V.; Pelli, G.; Kovari, E.; Pane, B.; Spinella, G.; et al. Anti-Apolipoprotein a-1 Auto-Antibodies Are Active Mediators of Atherosclerotic Plaque Vulnerability. Eur. Heart J. 2011, 32, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Pagano, S.; Carbone, F.; Burger, F.; Roth, A.; Bertolotto, M.; Pane, B.; Spinella, G.; Palombo, D.; Pende, A.; Dallegri, F.; et al. Anti-Apolipoprotein a-1 Auto-Antibodies as Active Modulators of Atherothrombosis. Thromb. Haemost. 2016, 116, 554–564. [Google Scholar] [CrossRef]
- Bridge, S.H.; Pagano, S.; Jones, M.; Foster, G.R.; Neely, D.; Vuilleumier, N.; Bassendine, M.F. Autoantibody to Apolipoprotein a-1 in Hepatitis C Virus Infection: A Role in Atherosclerosis? Hepatol. Int. 2018, 12, 17–25. [Google Scholar] [CrossRef]
- Quercioli, A.; Montecucco, F.; Galan, K.; Ratib, O.; Roux-Lombard, P.; Pagano, S.; Mach, F.; Schindler, T.H.; Vuilleumier, N. Anti-Apolipoprotein a-1 Igg Levels Predict Coronary Artery Calcification in Obese but Otherwise Healthy Individuals. Mediat. Inflamm. 2012, 2012, 243158. [Google Scholar] [CrossRef]
- Pagano, S.; Satta, N.; Werling, D.; Offord, V.; de Moerloose, P.; Charbonney, E.; Hochstrasser, D.; Roux-Lombard, P.; Vuilleumier, N. Anti-Apolipoprotein a-1 Igg in Patients with Myocardial Infarction Promotes Inflammation through Tlr2/Cd14 Complex. J. Intern. Med. 2012, 272, 344–357. [Google Scholar] [CrossRef]
- Dullaart, R.P.F.; Pagano, S.; Perton, F.G.; Vuilleumier, N. Antibodies against the C-Terminus of Apoa-1 Are Inversely Associated with Cholesterol Efflux Capacity and Hdl Metabolism in Subjects with and without Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2019, 20, 732. [Google Scholar] [CrossRef]
- Vuilleumier, N.; Pagano, S.; Montecucco, F.; Quercioli, A.; Schindler, T.H.; Mach, F.; Cipollari, E.; Ronda, N.; Favari, E. Relationship between Hdl Cholesterol Efflux Capacity, Calcium Coronary Artery Content, and Antibodies against Apolipoproteina-1 in Obese and Healthy Subjects. J. Clin. Med. 2019, 8, E1225. [Google Scholar] [CrossRef]
- Hansson, G.K.; Hermansson, A. The Immune System in Atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. A Century of Cholesterol and Coronaries: From Plaques to Genes to Statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Marquart, T.J.; Allen, R.M.; Ory, D.S.; Baldan, A. Mir-33 Links Srebp-2 Induction to Repression of Sterol Transporters. Proc. Natl. Acad. Sci. USA 2010, 107, 12228–12232. [Google Scholar] [CrossRef] [PubMed]
- Najafi-Shoushtari, S.H.; Kristo, F.; Li, Y.; Shioda, T.; Cohen, D.E.; Gerszten, R.E.; Naar, A.M. Microrna-33 and the Srebp Host Genes Cooperate to Control Cholesterol Homeostasis. Science 2010, 328, 1566–1569. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J.; Sheedy, F.J.; Esau, C.C.; Hussain, F.N.; Temel, R.E.; Parathath, S.; van Gils, J.M.; Rayner, A.J.; Chang, A.N.; Suarez, Y.; et al. Antagonism of Mir-33 in Mice Promotes Reverse Cholesterol Transport and Regression of Atherosclerosis. J. Clin. Investig. 2011, 121, 2921–2931. [Google Scholar] [CrossRef] [PubMed]
- Rotllan, N.; Ramirez, C.M.; Aryal, B.; Esau, C.C.; Fernandez-Hernando, C. Therapeutic Silencing of Microrna-33 Inhibits the Progression of Atherosclerosis in Ldlr-/-Mice—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1973–1977. [Google Scholar] [CrossRef]
- Price, N.L.; Singh, A.K.; Rotllan, N.; Goedeke, L.; Wing, A.; Canfran-Duque, A.; Diaz-Ruiz, A.; Araldi, E.; Baldan, A.; Camporez, J.P.; et al. Genetic Ablation of Mir-33 Increases Food Intake, Enhances Adipose Tissue Expansion, and Promotes Obesity and Insulin Resistance. Cell Rep. 2018, 22, 2133–2145. [Google Scholar] [CrossRef]
- Campagna, F.; Martino, F.; Bifolco, M.; Montali, A.; Martino, E.; Morrone, F.; Antonini, R.; Cantafora, A.; Verna, R.; Arca, M. Detection of Familial Hypercholesterolemia in a Cohort of Children with Hypercholesterolemia: Results of a Family and DNA-Based Screening. Atherosclerosis 2008, 196, 356–364. [Google Scholar] [CrossRef]
- Moore, K.J.; Kunjathoor, V.V.; Koehn, S.L.; Manning, J.J.; Tseng, A.A.; Silver, J.M.; McKee, M.; Freeman, M.W. Loss of Receptor-Mediated Lipid Uptake Via Scavenger Receptor a or Cd36 Pathways Does Not Ameliorate Atherosclerosis in Hyperlipidemic Mice. J. Clin. Investig. 2005, 115, 2192–2201. [Google Scholar] [CrossRef]
- Ye, Q.; Chen, Y.; Lei, H.; Liu, Q.; Moorhead, J.F.; Varghese, Z.; Ruan, X.Z. Inflammatory Stress Increases Unmodified Ldl Uptake Via Ldl Receptor: An Alternative Pathway for Macrophage Foam-Cell Formation. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. [et al.] 2009, 58, 809–818. [Google Scholar] [CrossRef]
- Haase, A.; Goldberg, A.C. Identification of People with Heterozygous Familial Hypercholesterolemia. Curr. Opin. Lipidol. 2012, 23, 282–289. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate Normalization of Real-Time Quantitative Rt-Pcr Data by Geometric Averaging of Multiple Internal Control Genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative Pcr and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kellner-Weibel, G.; de La Llera-Moya, M.; Connelly, M.A.; Stoudt, G.; Christian, A.E.; Haynes, M.P.; Williams, D.L.; Rothblat, G.H. Expression of Scavenger Receptor Bi in Cos-7 Cells Alters Cholesterol Content and Distribution. Biochemistry 2000, 39, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Ho, Y.K.; Goldstein, J.L. The Cholesteryl Ester Cycle in Macrophage Foam Cells. Continual Hydrolysis and Re-Esterification of Cytoplasmic Cholesteryl Esters. J. Biol. Chem. 1980, 255, 9344–9352. [Google Scholar] [PubMed]
- Barberio, M.D.; Kasselman, L.J.; Playford, M.P.; Epstein, S.B.; Renna, H.A.; Goldberg, M.; DeLeon, J.; Voloshyna, I.; Barlev, A.; Salama, M.; et al. Cholesterol Efflux Alterations in Adolescent Obesity: Role of Adipose-Derived Extracellular Vesical Micrornas. J. Transl. Med. 2019, 17, 232. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C. Molecular Mechanisms of Cellular Cholesterol Efflux. J. Biol. Chem. 2014, 289, 24020–24029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayner, K.J.; Suarez, Y.; Davalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernandez-Hernando, C. Mir-33 Contributes to the Regulation of Cholesterol Homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, D.; Thrush, A.B.; Nguyen, M.A.; Richards, L.; Geoffrion, M.; Singaravelu, R.; Ramphos, E.; Shangari, P.; Ouimet, M.; Pezacki, J.P.; et al. Macrophage Mitochondrial Energy Status Regulates Cholesterol Efflux and Is Enhanced by Anti-Mir33 in Atherosclerosis. Circ. Res. 2015, 117, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Niesor, E.J.; Schwartz, G.G.; Perez, A.; Stauffer, A.; Durrwell, A.; Bucklar-Suchankova, G.; Benghozi, R.; Abt, M.; Kallend, D. Statin-Induced Decrease in Atp-Binding Cassette Transporter A1 Expression Via Microrna33 Induction May Counteract Cholesterol Efflux to High-Density Lipoprotein. Cardiovasc. Drugs Ther. 2015, 29, 7–14. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A Proteolytic Pathway That Controls the Cholesterol Content of Membranes, Cells, and Blood. Proc. Natl. Acad. Sci. USA 1999, 96, 11041–11048. [Google Scholar] [CrossRef] [Green Version]
- Buhman, K.F.; Accad, M.; Farese, R.V. Mammalian Acyl-Coa:Cholesterol Acyltransferases. Biochim. Biophys. Acta 2000, 1529, 142–154. [Google Scholar] [CrossRef]
- Adorni, M.P.; Zimetti, F.; Billheimer, J.T.; Wang, N.; Rader, D.J.; Phillips, M.C.; Rothblat, G.H. The Roles of Different Pathways in the Release of Cholesterol from Macrophages. J. Lipid. Res. 2007, 48, 2453–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruijm, M.; Schmidtko, J.; Aho, A.; Pagano, S.; Roux-Lombard, P.; Teta, D.; Burnier, M.; Vuilleumier, N. High Prevalence of Anti-Apolipoprotein/a-1 Autoantibodies in Maintenance Hemodialysis and Association with Dialysis Vintage. Ther. Apher. Dial. Off. Peer-Rev. J. Int. Soc. Apher. Jpn. Soc. Apher. Jpn. Soc. Dial. Ther. 2012, 16, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.Y.; Chang, C.C.; Cheng, D. Acyl-Coenzyme A:Cholesterol Acyltransferase. Annu. Rev. Biochem. 1997, 66, 613–638. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Varghese, Z.; Moorhead, J.F.; Lee, C.T.; Chen, J.B.; Ruan, X.Z. Cross-Talk between Tlr4-Myd88-Nf-Kappab and Scap-Srebp2 Pathways Mediates Macrophage Foam Cell Formation. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H874–H884. [Google Scholar] [CrossRef]
- Li, Y.J.; Zhu, P.; Liang, Y.; Yin, W.G.; Xiao, J.H. Hepatitis B Virus Induces Expression of Cholesterol Metabolism-Related Genes Via Tlr2 in Hepg2 Cells. World J. Gastroenterol. 2013, 19, 2262–2269. [Google Scholar] [CrossRef]
- Lai, L.; Azzam, K.M.; Lin, W.C.; Rai, P.; Lowe, J.M.; Gabor, K.A.; Madenspacher, J.H.; Aloor, J.J.; Parks, J.S.; Naar, A.M.; et al. Microrna-33 Regulates the Innate Immune Response Via Atp Binding Cassette Transporter-Mediated Remodeling of Membrane Microdomains. J. Biol. Chem. 2016, 291, 19651–19660. [Google Scholar] [CrossRef] [Green Version]
- Horie, T.; Ono, K.; Horiguchi, M.; Nishi, H.; Nakamura, T.; Nagao, K.; Kinoshita, M.; Kuwabara, Y.; Marusawa, H.; Iwanaga, Y.; et al. Microrna-33 Encoded by an Intron of Sterol Regulatory Element-Binding Protein 2 (Srebp2) Regulates Hdl in Vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 17321–17326. [Google Scholar] [CrossRef] [Green Version]
- Afonso, M.S.; Machado, R.M.; Lavrador, M.S.; Quintao, E.C.R.; Moore, K.J.; Lottenberg, A.M. Molecular Pathways Underlying Cholesterol Homeostasis. Nutrients 2018, 10, 760. [Google Scholar] [CrossRef] [Green Version]
- Kam, N.T.; Albright, E.; Mathur, S.; Field, F.J. Effect of Lovastatin on Acyl-Coa: Cholesterol O-Acyltransferase (Acat) Activity and the Basolateral-Membrane Secretion of Newly Synthesized Lipids by Caco-2 Cells. Biochem. J. 1990, 272, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Akioyamen, L.E.; Genest, J.; Shan, S.D.; Reel, R.L.; Albaum, J.M.; Chu, A.; Tu, J.V. Estimating the Prevalence of Heterozygous Familial Hypercholesterolaemia: A Systematic Review and Meta-Analysis. BMJ Open 2017, 7, e016461. [Google Scholar] [CrossRef] [PubMed]
- Ameres, S.L.; Horwich, M.D.; Hung, J.H.; Xu, J.; Ghildiyal, M.; Weng, Z.; Zamore, P.D. Target Rna-Directed Trimming and Tailing of Small Silencing Rnas. Science 2010, 328, 1534–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruegger, S.; Grosshans, H. Microrna Turnover: When, How, and Why. Trends Biochem. Sci. 2012, 37, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. Scientific Document. 2019 Esc/Eas Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk. Eur. Heart J. 2019. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Healthy Children (n = 25) | FH Children (n = 29) | * p-Value |
|---|---|---|---|
| Age, year (IQR) | 8.5 (7–11) | 9 (6–11) | 0.89 |
| Sex (male %) | 45 | 48.2 | 0.88 |
| BMI (kg/m2) | 19 (16.6–20.2) | 17.8 (15.6–20) | 0.55 |
| Systolic BP *, mmHg (IQR) | 105 (95–110) | 110 (100–120) | 0.14 |
| Diastolic BP, mmHg (IQR) | 60 (60–70) | 60 (60–70) | 0.75 |
| Waist circumference, cm (IQR) | 61.5 (53–69) | 60 (53–68) | 0.94 |
| Hip circumference, cm (IQR) | 66 (61–76) | 64 (57–73) | 0.85 |
| Arm circumference, cm (IQR) | 20 (18–22) | 20 (17–23) | 0.83 |
| Total cholesterol (mg/dL) | 152.5 (138–167.5) | 231 (213–280) | <0.0001 |
| LDL cholesterol (mg/dL) | 91.4 (79.5–103) | 156 (137.6–211.6) | <0.0001 |
| HDL cholesterol (mg/dL) | 52 (43.5–58.5) | 57 (51–66.5) | 0.02 |
| Triglycerides (mg/dL) | 56.5 (45.5–63) | 64 (47–75) | 0.17 |
| Apolipoprotein B (mg/dL) | 65 (49–81.5) | 93 (86–134) | <0.0001 |
| Anti-ApoA-1 IgG (OD value) | 0.53 (0.4–0.77) | 0.55 (0.5–0.68) | 1 |
| Anti-ApoA-1 IgG positivity (%) | 38.8 | 37.9 | 0.9 |
| miR-33a (2^-ΔCt) | 0.05 (0.03–0.2) | 0.4 (0.19–1) | <0.0001 |
| Characteristic | Healthy Children (n = 25) | Anti-ApoA-1 Negative (n = 15) | Anti-ApoA-1 Positive (n = 10) | * p-Value |
|---|---|---|---|---|
| Age, year (IQR) | 8.5 (7–11) | 10 (7–11) | 7 (7–9) | 0.08 |
| Sex (male %) | 45 | 42.8 | 50 | 0.28 |
| BMI (kg/m2) | 19 (16.6–20.2) | 19.2 (17–20.2) | 17.2 (15.4–19.5) | 0.02 |
| Systolic BP *, mmHg (IQR) | 105 (95–110) | 100 (90–105) | 110 (100–120) | 0.7 |
| Diastolic BP, mmHg (IQR) | 60 (60–70) | 60 (60–75) | 62.5 (60–70) | 0.53 |
| Waist circumference, cm (IQR) | 61.5 (53–69) | 66 (51–69) | 61 (54–64) | 0.03 |
| Hip circumference, cm (IQR) | 66 (61–76) | 70 (61–76) | 66 (61–67) | 0.09 |
| Arm circumference, cm (IQR) | 20 (18–22) | 20 (17–22) | 19.5 (18–21) | 0.01 |
| Total cholesterol (mg/dL) | 151 (138.5–167.5) | 147 (135–167) | 163 (148–168) | 0.27 |
| LDL cholesterol (mg/dL) | 91.4 (79.5–103) | 89.4 (72–95) | 101.8 (87–108) | 0.11 |
| HDL cholesterol (mg/dL) | 52 (43.5–58.5) | 50.5 (39–55) | 56 (47–62) | 0.12 |
| Triglycerides (mg/dL) | 56.5 (45.5–63) | 58.5 (47–70) | 55 (41–58) | 0.97 |
| Apolipoprotein B (mg/dL) | 65 (49–81.5) | 53.5 (40–66) | 79.5 (69–85) | 0.19 |
| miR-33a (2^-ΔCt) | 0.05 (0.03–0.25) | 0.1 (0.03–0.26) | 0.05 (0.04–0.1) | 0.4 |
| Characteristic | FH Children (n = 29) | Anti-ApoA-1 Negative (n = 18) | Anti-ApoA-1 Positive (n = 11) | * p-Value |
|---|---|---|---|---|
| Age, year (IQR) | 9 (6–11) | 8 (7–10) | 10 (6–13) | 0.45 |
| Sex (male %) | 48 | 54.5 | 72.7 | 0.08 |
| BMI (kg/m2) | 17.8 (15.6–20) | 16.8 (15.3–18.2) | 19.3 (17.4–21.9) | 0.05 |
| Systolic BP*, mmHg (IQR) | 110 (100–120) | 110 (100–115) | 110 (100–125) | 0.39 |
| Diastolic BP, mmHg (IQR) | 60 (60–70) | 62.5 (60–70) | 60 (60–70) | 0.58 |
| Waist circumference, cm (IQR) | 60 (53–68) | 56.5 (53–63) | 66 (61–70) | 0.05 |
| Hip circumference, cm (IQR) | 64 (57–73) | 72 (64–80) | 62.5 (57–71) | 0.10 |
| Arm circumference, cm (IQR) | 20 (17–23) | 22 (22–23) | 19 (17–22) | 0.14 |
| Total cholesterol (mg/dL) | 213 (213–280) | 263 (219–305) | 209 (188–240) | 0.04 |
| LDL cholesterol (mg/dL) | 156.6 (137.6–211.6) | 191.2 (144.6–228) | 124.4 (113.8–168.8) | 0.04 |
| HDL cholesterol (mg/dL) | 57 (51–66.5) | 61 (55–67) | 55 (50–58) | 0.24 |
| Triglycerides mg/dL) | 64 (47–75) | 60 (47–66) | 66 (35–78) | 0.35 |
| Apolipoprotein B (mg/dL) | 93 (86–134) | 109 (91–143) | 86 (76–103) | 0.05 |
| miR-33a (2^-Ct) | 0.41(0.19–1.03) | 0.85 (0.36–1.2) | 0.28 (0.14–0.5) | 0.04 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagano, S.; Magenta, A.; D’Agostino, M.; Martino, F.; Barillà, F.; Satta, N.; Frias, M.A.; Ronca, A.; Mach, F.; Gencer, B.; et al. Anti-ApoA-1 IgGs in Familial Hypercholesterolemia Display Paradoxical Associations with Lipid Profile and Promote Foam Cell Formation. J. Clin. Med. 2019, 8, 2035. https://doi.org/10.3390/jcm8122035
Pagano S, Magenta A, D’Agostino M, Martino F, Barillà F, Satta N, Frias MA, Ronca A, Mach F, Gencer B, et al. Anti-ApoA-1 IgGs in Familial Hypercholesterolemia Display Paradoxical Associations with Lipid Profile and Promote Foam Cell Formation. Journal of Clinical Medicine. 2019; 8(12):2035. https://doi.org/10.3390/jcm8122035
Chicago/Turabian StylePagano, Sabrina, Alessandra Magenta, Marco D’Agostino, Francesco Martino, Francesco Barillà, Nathalie Satta, Miguel A. Frias, Annalisa Ronca, François Mach, Baris Gencer, and et al. 2019. "Anti-ApoA-1 IgGs in Familial Hypercholesterolemia Display Paradoxical Associations with Lipid Profile and Promote Foam Cell Formation" Journal of Clinical Medicine 8, no. 12: 2035. https://doi.org/10.3390/jcm8122035
APA StylePagano, S., Magenta, A., D’Agostino, M., Martino, F., Barillà, F., Satta, N., Frias, M. A., Ronca, A., Mach, F., Gencer, B., Favari, E., & Vuilleumier, N. (2019). Anti-ApoA-1 IgGs in Familial Hypercholesterolemia Display Paradoxical Associations with Lipid Profile and Promote Foam Cell Formation. Journal of Clinical Medicine, 8(12), 2035. https://doi.org/10.3390/jcm8122035