Human Ovarian Granulosa Cells Isolated during an IVF Procedure Exhibit Differential Expression of Genes Regulating Cell Division and Mitotic Spindle Formation

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Selection and Granulosa Cell Separation
2.2. Cell Culture
2.3. Transmission Electron Microscopy Analysis
2.4. RNA Isolation
2.5. Microarray Analysis
2.6. RT-qPCR Analysis
2.7. Statistics
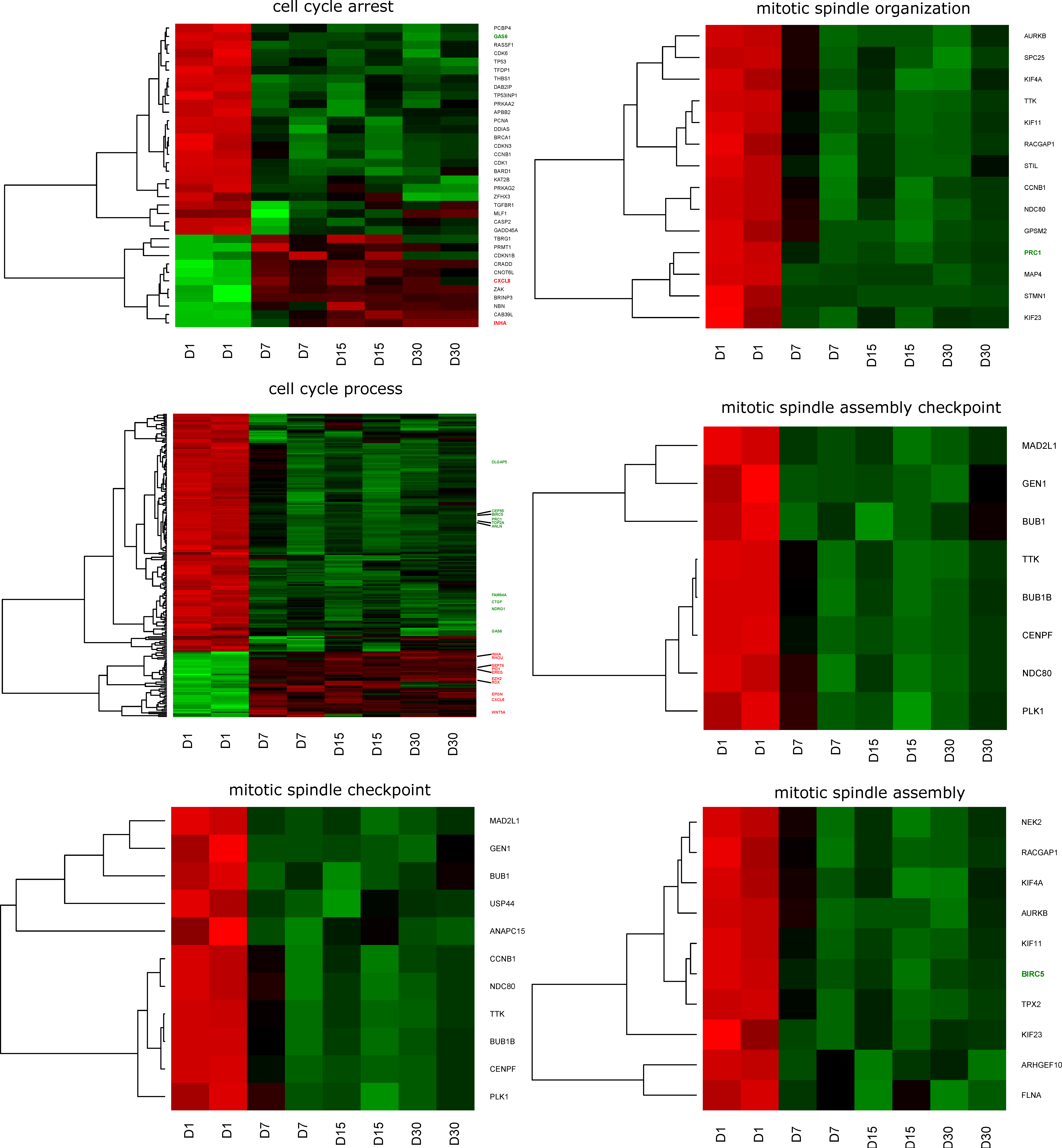
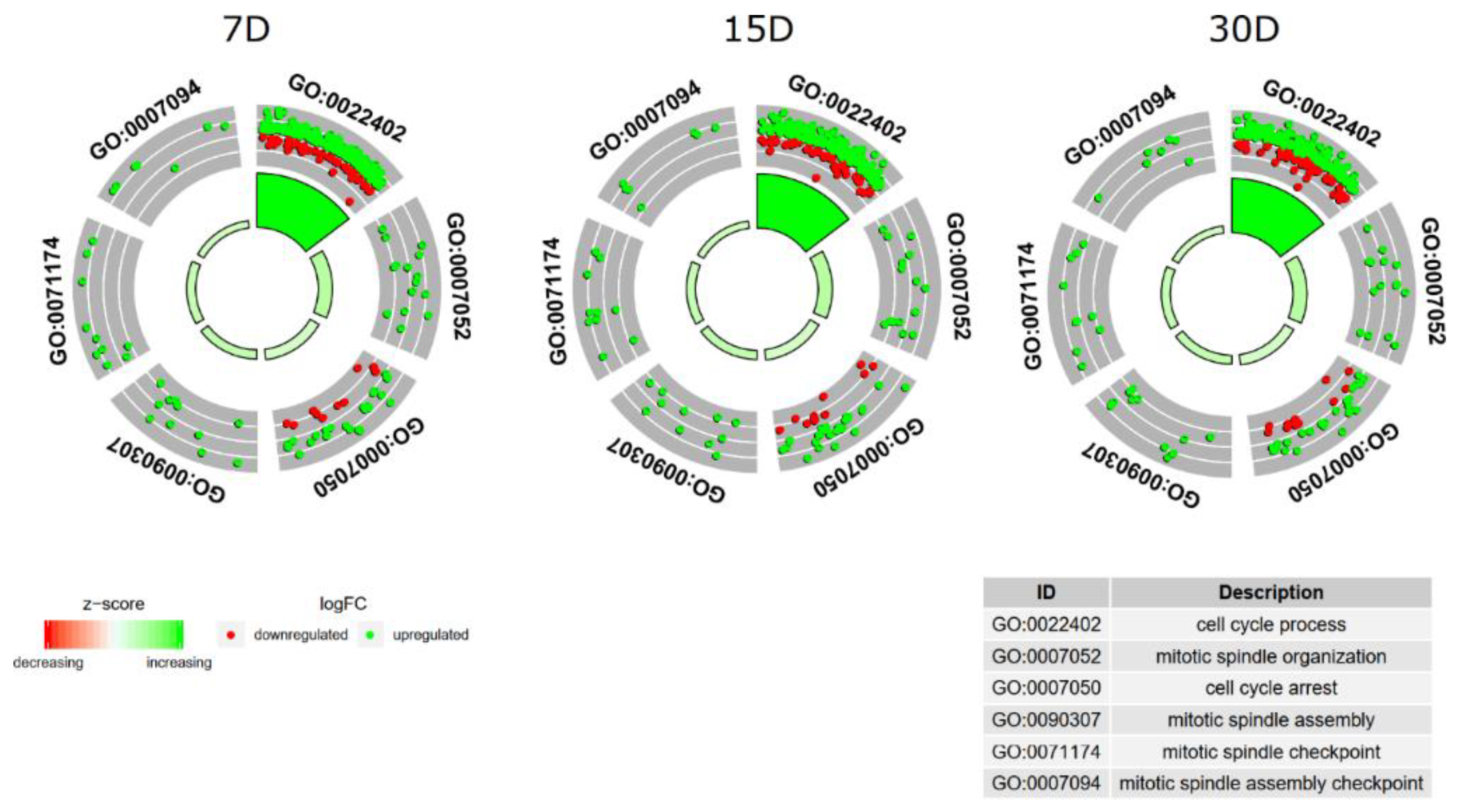
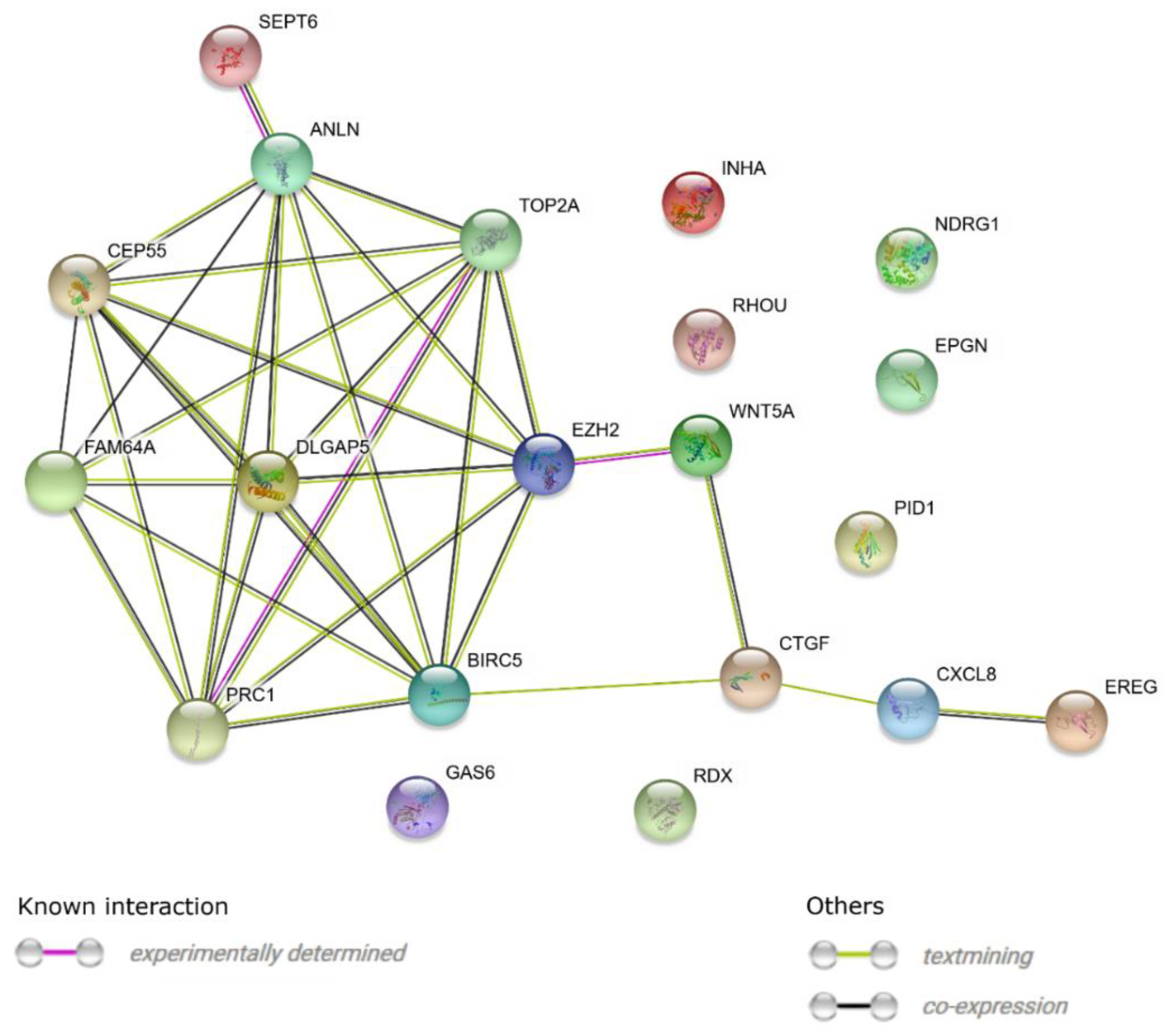
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chermuła, B.; Brązert, M.; Iżycki, D.; Ciesiółka, S.; Kranc, W.; Celichowski, P.; Ożegowska, K.; Nawrocki, M.J.; Jankowski, M.; Jeseta, M.; et al. New Gene Markers of Angiogenesis and Blood Vessels Development in Porcine Ovarian Granulosa Cells during Short-Term Primary Culture In Vitro. Biomed Res. Int. 2019, 2019, 6545210. [Google Scholar] [CrossRef]
- Rybska, M.; Knap, S.; Jankowski, M.; Jeseta, M.; Bukowska, D.; Antosik, P.; Nowicki, M.; Zabel, M.; Kempisty, B.; Jaśkowski, J.M. Characteristic of factors influencing the proper course of folliculogenesis in mammals. Med. J. Cell Biol. 2018, 6, 33–38. [Google Scholar] [CrossRef]
- Rybska, M.; Knap, S.; Jankowski, M.; Jeseta, M.; Bukowska, D. Cytoplasmic and nuclear maturation of oocytes in mammals–living in the shadow of cells developmental capability. Med. J. Cell Biol. 2018, 1. [Google Scholar] [CrossRef]
- Kossowska-Tomaszczuk, K.; De Geyter, C. Cells with stem cell characteristics in somatic compartments of the ovary. Biomed Res. Int. 2013, 2013, 310859. [Google Scholar] [CrossRef] [PubMed]
- Kossowska-Tomaszczuk, K.; Pelczar, P.; Güven, S.; Kowalski, J.; Volpi, E.; De Geyter, C.; Scherberich, A. A novel three-dimensional culture system allows prolonged culture of functional human granulosa cells and mimics the ovarian environment. Tissue Eng. Part A 2010, 16, 2063–2073. [Google Scholar] [CrossRef]
- Kossowska-Tomaszczuk, K.; De Geyter, C.; De Geyter, M.; Martin, I.; Holzgreve, W.; Scherberich, A.; Zhang, H. The Multipotency of Luteinizing Granulosa Cells Collected from Mature Ovarian Follicles. Stem Cells 2009, 27, 210–219. [Google Scholar] [CrossRef]
- Moncrieff, L.; Mozdziak, P.; Jeseta, M.; Machatkova, M.; Kranc, W.; Kempisty, B. Ovarian follicular cells–living in the shadow of stemness cellular competence. Med. J. Cell Biol. 2019, 7, 134–140. [Google Scholar] [CrossRef]
- Kranc, W.; Brązert, M.; Budna, J.; Celichowski, P.; Bryja, A.; Nawrocki, M.J.; Ożegowska, K.; Jankowski, M.; Chermuła, B.; Dyszkiewicz-Konwińska, M.; et al. Genes responsible for proliferation, differentiation, and junction adhesion are significantly up-regulated in human ovarian granulosa cells during a long-term primary in vitro culture. Histochem. Cell Biol. 2019, 151, 125–143. [Google Scholar] [CrossRef]
- Bryja, A.; Dyszkiewicz-Konwińska, M.; Jankowski, M.; Celichowski, P.; Stefańska, K.; Chamier-Gliszczyńska, A.; Popis, M.; Mehr, K.; Bukowska, D.; Antosik, P.; et al. Ion homeostasis and transport are regulated by genes differentially expressed in porcine buccal pouch mucosal cells during long-term culture in vitro-a microarray approach. Bryja al. Med. J. Cell Biol. 2018. [Google Scholar] [CrossRef]
- Nawrocki, M.J.; Celichowski, P.; Jankowski, M.; Kranc, W.; Bryja, A.; Borys-Wójcik, S.; Jeseta, M.; Antosik, P.; Bukowska, D.; Bruska, M.; et al. Ontology groups representing angiogenesis and blood vessels development are highly up-regulated during porcine oviductal epithelial cells long-term real-time proliferation—A a primary cell culture approach. Med. J. Cell Biol. 2018, 6, 186–194. [Google Scholar] [CrossRef]
- Chermuła, B.; Brązert, M.; Jeseta, M.; Ożegowska, K.; Kocherova, I.; Jankowski, M.; Celichowski, P.; Sujka-Kordowska, P.; Konwerska, A.; Piotrowska-Kempisty, H.; et al. Transcriptomic Pattern of Genes Regulating Protein Response and Status of Mitochondrial Activity Are Related to Oocyte Maturational Competence—A Transcriptomic Study. Int. J. Mol. Sci. 2019, 20, 2238. [Google Scholar] [CrossRef] [PubMed]
- Budna, J.; Bryja, A.; Celichowski, P.; Kranc, W.; Ciesiółka, S.; Borys, S.; Rybska, M.; Kolecka-Bednarczyk, A.; Jeseta, M.; Bukowska, D.; et al. “Bone Development” Is an Ontology Group Upregulated in Porcine Oocytes before In Vitro Maturation: A Microarray Approach. DNA Cell Biol. 2017, 36, 638–646. [Google Scholar] [CrossRef] [PubMed]
- D’Aurora, M.; Sperduti, S.; Di Emidio, G.; Stuppia, L.; Artini, P.G.; Gatta, V. Inside the granulosa transcriptome. Gynecol. Endocrinol. 2016, 32, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Ferraretti, A.P.; La Marca, A.; Fauser, B.C.J.M.; Tarlatzis, B.; Nargund, G.; Gianaroli, L. ESHRE working group on Poor Ovarian Response Definition ESHRE consensus on the definition of “poor response” to ovarian stimulation for in vitro fertilization: The Bologna criteria. Hum. Reprod. 2011, 26, 1616–1624. [Google Scholar] [CrossRef] [PubMed]
- Kranc, W.; Budna, J.; Kahan, R.; Chachuła, A.; Bryja, A.; Ciesiółka, S.; Borys, S.; Antosik, M.P.; Bukowska, D.; Brussow, K.P.; et al. Molecular basis of growth, proliferation, and differentiation of mammalian follicular granulosa cells. J. Biol. Regul. Homeost. Agents 2017, 31, 1–8. [Google Scholar] [PubMed]
- Kranc, W.; Brązert, M.; Celichowski, P.; Bryja, A.; Nawrocki, M.; Ożegowska, K.; Jankowski, M.; Jeseta, M.; Pawelczyk, L.; Bręborowicz, A.; et al. ‘Heart development and morphogenesis’ is a novel pathway for human ovarian granulosa cell differentiation during long‑term in vitro cultivation—A microarray approach. Mol. Med. Rep. 2019, 19, 1705–1715. [Google Scholar] [CrossRef]
- Brązert, M.; Iżycki, D.; Kranc, W.; Borowiec, B.; Popis, M.; Ożegowska, K.; Bręborowicz, A.; Rachoń, D.; Nowicki, M.; Kempisty, B. Genes involved in hormone metabolism and cellular response in human ovarian granulosa cells. J. Biol. Regul. Homeost. Agents 2019, 33, 461–468. [Google Scholar]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Kranc, W.; Brązert, M.; Ożegowska, K.; Nawrocki, M.M.J.; Budna, J.; Celichowski, P.; Dyszkiewicz-Konwińska, M.; Jankowski, M.; Jeseta, M.; Pawelczyk, L.; et al. Expression Profile of Genes Regulating Steroid Biosynthesis and Metabolism in Human Ovarian Granulosa Cells—A Primary Culture Approach. Int. J. Mol. Sci. 2017, 18, 2673. [Google Scholar] [CrossRef]
- Budna, J.; Celichowski, P.; Knap, S.; Jankowski, M.; Magas, M.; Nawrocki, M.J.; Ramlau, P.; Nowicki, A.; Rojewska, M.; Chermuła, B.; et al. Fatty Acids Related Genes Expression Undergo Substantial Changes in Porcine Oviductal Epithelial Cells During Long-Term Primary Culture. Med. J. Cell Biol. 2018, 6, 39–47. [Google Scholar] [CrossRef]
- Kranc, W.; Brązert, M.; Ożegowska, K.; Budna-Tukan, J.; Celichowski, P.; Jankowski, M.; Bryja, A.; Nawrocki, M.J.; Popis, M.; Jeseta, M.; et al. Response to abiotic and organic substances stimulation belongs to ontologic groups significantly up-regulated in porcine immature oocytes. Med. J. Cell Biol. 2018, 6, 91–100. [Google Scholar] [CrossRef]
- Stefańska, K.; Chamier-Gliszczyńska, A.; Jankowski, M.; Celichowski, P.; Kulus, M.; Rojewska, M.; Antosik, P.; Bukowska, D.; Bruska, M.; Nowicki, M.; et al. Epithelium morphogenesis and oviduct development are regulated by significant increase of expression of genes after long-term in vitro primary culture—A microarray assays. Med. J. Cell Biol. 2018, 6, 195–204. [Google Scholar] [CrossRef]
- Stefańska, K.; Kocherova, I.; Knap, S.; Kulus, M.; Celichowski, P.; Jeseta, M. The genes regulating maintenance of cellular protein location are differentially expressed in porcine epithelial oviductal cells during longterm in vitro cultivation. Med. J. Cell Biol. 2019, 7, 77–85. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef]
- Von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2004, 33, D433–D437. [Google Scholar] [CrossRef]
- Walter, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- Chamier-Gliszczyńska, A.; Brązert, M.; Sujka-Kordowska, P.; Popis, M.; Ożegowska, K.; Stefańska, K.; Kocherova, I.; Celichowski, P.; Kulus, M.; Bukowska, D.; et al. Genes involved in angiogenesis and circulatory system development are differentially expressed in porcine epithelial oviductal cells during long-term primary in vitro culture—A transcriptomic study. Med. J. Cell Biol. 2018, 6, 163–173. [Google Scholar] [CrossRef]
- Kocherova, I.; Kulus, M.; Dompe, C.; Antosik, P.; Bukowska, D. Biochemical properties of cofactor and coenzyme metabolism in porcine oviductal epithelial cells —A microarray study. 2019. [Google Scholar] [CrossRef]
- Morey, J.S.; Ryan, J.C.; Van Dolah, F.M. Microarray validation: Factors influencing correlation between oligonucleotide microarrays and real-time PCR. Biol. Proced. Online 2006, 8, 175–193. [Google Scholar] [CrossRef]
- Thompson, W.E.; Branch, A.; Whittaker, J.A.; Lyn, D.; Zilberstein, M.; Mayo, K.E.; Thomas, K. Characterization of Prohibitin in a Newly Established Rat Ovarian Granulosa Cell Line. Endocrinology 2001, 142, 4076–4085. [Google Scholar] [CrossRef]
- Rotmensch, S.; Dor, J.; Furman, A.; Rudak, M.E.; Mashiach, S.; Amsterdam, A. Ultrastructural characterization of human granulosa cells in stimulated cycles: Correlation with oocyte fertilizability. Fertil. Steril. 1986, 45, 671–679. [Google Scholar] [CrossRef]
- Nottola, S.A.; Heyn, R.; Camboni, A.; Correr, S.; Macchiarelli, G. Ultrastructural characteristics of human granulosa cells in a coculture system for in vitro fertilization. Microsc. Res. Tech. 2006, 69, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Archangelo, L.F.; Greif, P.A.; Maucuer, A.; Manceau, V.; Koneru, N.; Bigarella, C.L.; Niemann, F.; dos Santos, M.T.; Kobarg, J.; Bohlander, S.K.; et al. The CATS (FAM64A) protein is a substrate of the Kinase Interacting Stathmin (KIS). Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Han, Y.; Huang, H.; Min, L.; Qu, L.; Shou, C. Integrated analysis of expression profiling data identifies three genes in correlation with poor prognosis of triple-negative breast cancer. Int. J. Oncol. 2014, 44, 2025–2033. [Google Scholar] [CrossRef]
- Piekny, A.J.; Maddox, A.S. The myriad roles of Anillin during cytokinesis. Semin. Cell Dev. Biol. 2010, 21, 881–891. [Google Scholar] [CrossRef]
- Wang, D.; Chadha, G.K.; Feygin, A.; Ivanov, A.I. F-actin binding protein, anillin, regulates integrity of intercellular junctions in human epithelial cells. Cell. Mol. Life Sci. 2015, 72, 3185–3200. [Google Scholar] [CrossRef][Green Version]
- Hall, P.A.; Todd, C.B.; Hyland, P.L.; McDade, S.S.; Grabsch, H.; Dattani, M.; Hillan, K.J.; Russell, S.E.H. The septin-binding protein anillin is overexpressed in diverse human tumors. Clin. Cancer Res. 2005, 11, 6780–6786. [Google Scholar] [CrossRef]
- Ghisoni, E.; Maggiorotto, F.; Borella, F.; Mittica, G.; Genta, S.; Giannone, G.; Katsaros, D.; Sciarrillo, A.; Ferrero, A.; Sarotto, I.; et al. TOP2A as marker of response to pegylated lyposomal doxorubicin (PLD) in epithelial ovarian cancers. J. Ovarian Res. 2019, 12, 17. [Google Scholar] [CrossRef]
- Miles, G.D.; Seiler, M.; Rodriguez, L.; Rajagopal, G.; Bhanot, G. Identifying microRNA/mRNA dysregulations in ovarian cancer. BMC Res. Notes 2012, 5, 164. [Google Scholar] [CrossRef]
- Wandji, S.A.; Gadsby, J.E.; Barber, J.A.; Hammond, J.M. Messenger ribonucleic acids for MAC25 and connective tissue growth factor (CTGF) are inversely regulated during folliculogenesis and early luteogenesis. Endocrinology 2000, 141, 2648–2657. [Google Scholar] [CrossRef]
- Cheng, J.-C.; Chang, H.-M.; Fang, L.; Sun, Y.-P.; Leung, P.C.K. TGF-β1 Up-Regulates Connective Tissue Growth Factor Expression in Human Granulosa Cells through Smad and ERK1/2 Signaling Pathways. PLoS ONE 2015, 10, e0126532. [Google Scholar] [CrossRef] [PubMed]
- Kalimutho, M.; Sinha, D.; Jeffery, J.; Nones, K.; Srihari, S.; Fernando, W.C.; Duijf, P.H.; Vennin, C.; Raninga, P.; Nanayakkara, D.; et al. CEP55 is a determinant of cell fate during perturbed mitosis in breast cancer. EMBO Mol. Med. 2018, 10, e8566. [Google Scholar] [CrossRef] [PubMed]
- Brodská, B.; Otevřelová, P.; Holoubek, A. Decitabine and SAHA-induced apoptosis is accompanied by survivin downregulation and potentiated by ATRA in p53-deficient cells. Oxid. Med. Cell. Longev. 2014, 2014, 165303. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guo, J.; Zhang, T.; Guo, Y.; Sun, T.; Li, H.; Zhang, X.; Yin, H.; Cao, G.; Yin, Y.; Wang, H.; et al. Oocyte stage-specific effects of MTOR determine granulosa cell fate and oocyte quality in mice. Proc. Natl. Acad. Sci. USA. 2018, 115, E5326–E5333. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Zhou, F.; Cui, X.; Liu, M.; Li, Y.; Liu, S.; Tan, J.; Yan, Q. Novel Serum Biomarkers Detected by Protein Array in Polycystic Ovary Syndrome with Low Progesterone Level. Cell. Physiol. Biochem. 2018, 46, 2297–2310. [Google Scholar] [CrossRef]
- Yin, C.; Liu, W.; hua; Liu, Y.; Wang, L.; Xiao, Y. PID1 alters the antilipolytic action of insulin and increases lipolysis via inhibition of AKT/PKA pathway activation. PLoS ONE 2019, 14, e0214606. [Google Scholar] [CrossRef]
- Kong, D.; Guan, Q.; Li, G.; Xin, W.; Qi, X.; Guo, Y.; Zhao, J.; Xu, J.; Sun, S.; Gao, L. Expression of FSHR in chondrocytes and the effect of FSH on chondrocytes. Biochem. Biophys. Res. Commun. 2018, 495, 587–593. [Google Scholar] [CrossRef]
- Gatla, H.R.; Zou, Y.; Uddin, M.M.; Singha, B.; Bu, P.; Vancura, A.; Vancurova, I. Histone Deacetylase (HDAC) Inhibition Induces IκB Kinase (IKK)-dependent Interleukin-8/CXCL8 Expression in Ovarian Cancer Cells. J. Biol. Chem. 2017, 292, 5043–5054. [Google Scholar] [CrossRef]
- O’Neill, R.S.; Clark, D.V. Partial Functional Diversification of Drosophila melanogaster Septin Genes Sep2 and Sep5. G3 (Bethesda) 2016, 6, 1947–1957. [Google Scholar] [CrossRef]
- Carletti, M.Z.; Christenson, L.K. Rapid effects of LH on gene expression in the mural granulosa cells of mouse periovulatory follicles. Reproduction 2009, 137, 843–855. [Google Scholar] [CrossRef]
- Jin, P.; Song, Y.; Yu, G. The Role of Abnormal Methylation of Wnt5a Gene Promoter Regions in Human Epithelial Ovarian Cancer: A Clinical and Experimental Study. Anal. Cell. Pathol. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Shi, H. An In-Depth Look at Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT): Clinical Implications from Recent Molecular Findings. J. Cancer 2019, 10, 223–237. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequence (5′–3′) | Product Size (bp) |
|---|---|---|
| NDRG1 | F: ACAACCCTGAGATGGTGGAG R: TGTGGACCACTTCCACGTTA | 174 |
| GAS6 | F: ATCAAGGTCAACAGGGATGC R: CTTCTCCGTTCAGCCAGTTC | 187 |
| DLGAP5 | F: CCAGTCGACACAGGAAGGAT R: CATTGCCCTTGGCTTAACAT | 227 |
| PRC1 | F: TAGACCACACCCCAGACACA R: GTGGCCACAGCTTCTCTTTC | 223 |
| BIRC5 | F: GCCTTTCCTTAAAGGCCATC R: AACCCTTCCCAGACTCCACT | 187 |
| CEP55 | F: CAGTATCCAGCCACTGAGCA R: GGGAGGTATCACTGCCAAGA | 245 |
| CTGF | F: GGAAAAGATTCCCACCCAAT R: TGCTCCTAAAGCCACACCTT | 153 |
| TOP2A | F: AATCTCAGAGCTTCCCGTCA R: TGCCTCTGCCAGTTTTTCTT | 175 |
| ANLN | F: ATGCAGTGTGGTGCACATTT R: AACCCAAACACTTTGGCAAG | 195 |
| FAM64A | F: GGAGATCTCTCCAGCACCAG R: GCACCCAAAGCACTCTTAGC | 244 |
| EZH2 | F: AGGACGGCTCCTCTAACCAT R: CTTGGTGTTGCACTGTGCTT | 179 |
| WNT5A | F: TGGCTTTGGCCATATTTTTC R: CCGATGTACTGCATGTGGTC | 199 |
| RDX | F: CCCAGAGACTCTTCTTCTTGC R: TACACGCTGGGGTAGGAGTC | 179 |
| EPGN | F: TGACAGCACTGACCGAAGAG R: CTCATGGTGGAATGCACAAG | 188 |
| SEPT6 | F: TCTGCTTCAACATCCTGTGC R: GCTTTAGCCTCACGTTGCTC | 168 |
| CXCL8 | F: AAGAAACCACCGGAAGGAAC R: AAATTTGGGGTGGAAAGGTT | 183 |
| RHOU | F: AGGCCTCTCTGCTACACCAA R: TCAGGCACTGGCTTTTCTTT | 215 |
| INHA | F: CCAGCTGTGAGGACAAGTCA R: CTAGCAGGGGCTCAGAGCTA | 186 |
| PID1 | F: TACCTGGGCAAAGTCTCCAC R: TTTGTGGTCGAGATGATGGA | 171 |
| EREG | F: CCAAGGACGGAAAATGCTTA R: AAAATTAGCTGGGCATGGTG | 237 |
| GAPDH | F: TCAGCCGCATCTTCTTTTGC R: ACGACCAAATCCGTTGACTC | 90 |
| ACTB | F: AAAGACCTGTACGCCAACAC R: CTCAGGAGGAGCAATGATCTTG | 132 |
| HPRT1 | F: TGGCGTCGTGATTAGTGATG R: ACATCTCGAGCAAGACGTTC | 141 |
| Gene Symbol | Fold Change D7/D1 | Fold Change D15/D1 | Fold Change D30/D1 | Adjusted p-Value D7/D1 | Adjusted p-Value D15/D1 | Adjusted p-Value D30/D1 | Entrez Gene ID |
|---|---|---|---|---|---|---|---|
| EREG | 0.010 | 0.008 | 0.008 | 0.0002 | 0.0002 | 0.0002 | 2069 |
| PID1 | 0.076 | 0.068 | 0.071 | 0.0024 | 0.0020 | 0.0017 | 55022 |
| INHA | 0.204 | 0.090 | 0.063 | 0.0266 | 0.0066 | 0.0036 | 3623 |
| RHOU | 0.236 | 0.167 | 0.073 | 0.0481 | 0.0226 | 0.0061 | 58480 |
| CXCL8 | 0.116 | 0.122 | 0.191 | 0.0189 | 0.0180 | 0.0337 | 3576 |
| SEPT6 | 0.172 | 0.156 | 0.179 | 0.0006 | 0.0005 | 0.0005 | 23157 |
| EPGN | 0.169 | 0.220 | 0.149 | 0.0437 | 0.0621 | 0.0289 | 255324 |
| RDX | 0.306 | 0.273 | 0.129 | 0.0227 | 0.0153 | 0.0033 | 5962 |
| WNT5A | 0.158 | 0.347 | 0.268 | 0.0184 | 0.0825 | 0.0407 | 7474 |
| EZH2 | 0.259 | 0.369 | 0.185 | 0.0016 | 0.0031 | 0.0006 | 2146 |
| NDRG1 | 11.930 | 22.621 | 20.319 | 0.0022 | 0.0011 | 0.0009 | 10397 |
| GAS6 | 13.923 | 14.854 | 26.709 | 0.0059 | 0.0051 | 0.0024 | 2621 |
| DLGAP5 | 14.126 | 22.518 | 20.818 | 0.0230 | 0.0124 | 0.0120 | 9787 |
| PRC1 | 18.098 | 23.764 | 18.552 | 0.0016 | 0.0012 | 0.0012 | 9055 |
| BIRC5 | 18.745 | 25.651 | 18.849 | 0.0029 | 0.0022 | 0.0023 | 332 |
| CEP55 | 26.885 | 33.896 | 27.873 | 0.0056 | 0.0041 | 0.0042 | 55165 |
| CTGF | 36.067 | 38.456 | 56.198 | 0.0028 | 0.0025 | 0.0015 | 1490 |
| TOP2A | 45.408 | 58.461 | 50.754 | 0.0026 | 0.0020 | 0.0018 | 7153 |
| ANLN | 59.052 | 63.936 | 59.669 | 0.0016 | 0.0013 | 0.0011 | 54443 |
| FAM64A | 71.846 | 58.770 | 65.148 | 0.0009 | 0.0009 | 0.0007 | 54478 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brązert, M.; Kranc, W.; Chermuła, B.; Kowalska, K.; Jankowski, M.; Celichowski, P.; Jeseta, M.; Piotrowska-Kempisty, H.; Pawelczyk, L.; Zabel, M.; et al. Human Ovarian Granulosa Cells Isolated during an IVF Procedure Exhibit Differential Expression of Genes Regulating Cell Division and Mitotic Spindle Formation. J. Clin. Med. 2019, 8, 2026. https://doi.org/10.3390/jcm8122026
Brązert M, Kranc W, Chermuła B, Kowalska K, Jankowski M, Celichowski P, Jeseta M, Piotrowska-Kempisty H, Pawelczyk L, Zabel M, et al. Human Ovarian Granulosa Cells Isolated during an IVF Procedure Exhibit Differential Expression of Genes Regulating Cell Division and Mitotic Spindle Formation. Journal of Clinical Medicine. 2019; 8(12):2026. https://doi.org/10.3390/jcm8122026
Chicago/Turabian StyleBrązert, Maciej, Wiesława Kranc, Błażej Chermuła, Katarzyna Kowalska, Maurycy Jankowski, Piotr Celichowski, Michal Jeseta, Hanna Piotrowska-Kempisty, Leszek Pawelczyk, Maciej Zabel, and et al. 2019. "Human Ovarian Granulosa Cells Isolated during an IVF Procedure Exhibit Differential Expression of Genes Regulating Cell Division and Mitotic Spindle Formation" Journal of Clinical Medicine 8, no. 12: 2026. https://doi.org/10.3390/jcm8122026
APA StyleBrązert, M., Kranc, W., Chermuła, B., Kowalska, K., Jankowski, M., Celichowski, P., Jeseta, M., Piotrowska-Kempisty, H., Pawelczyk, L., Zabel, M., Mozdziak, P., & Kempisty, B. (2019). Human Ovarian Granulosa Cells Isolated during an IVF Procedure Exhibit Differential Expression of Genes Regulating Cell Division and Mitotic Spindle Formation. Journal of Clinical Medicine, 8(12), 2026. https://doi.org/10.3390/jcm8122026