The Role of Protein SUMOylation in the Pathogenesis of Atherosclerosis


 ,
,  ,
, 
Abstract
1. Introduction
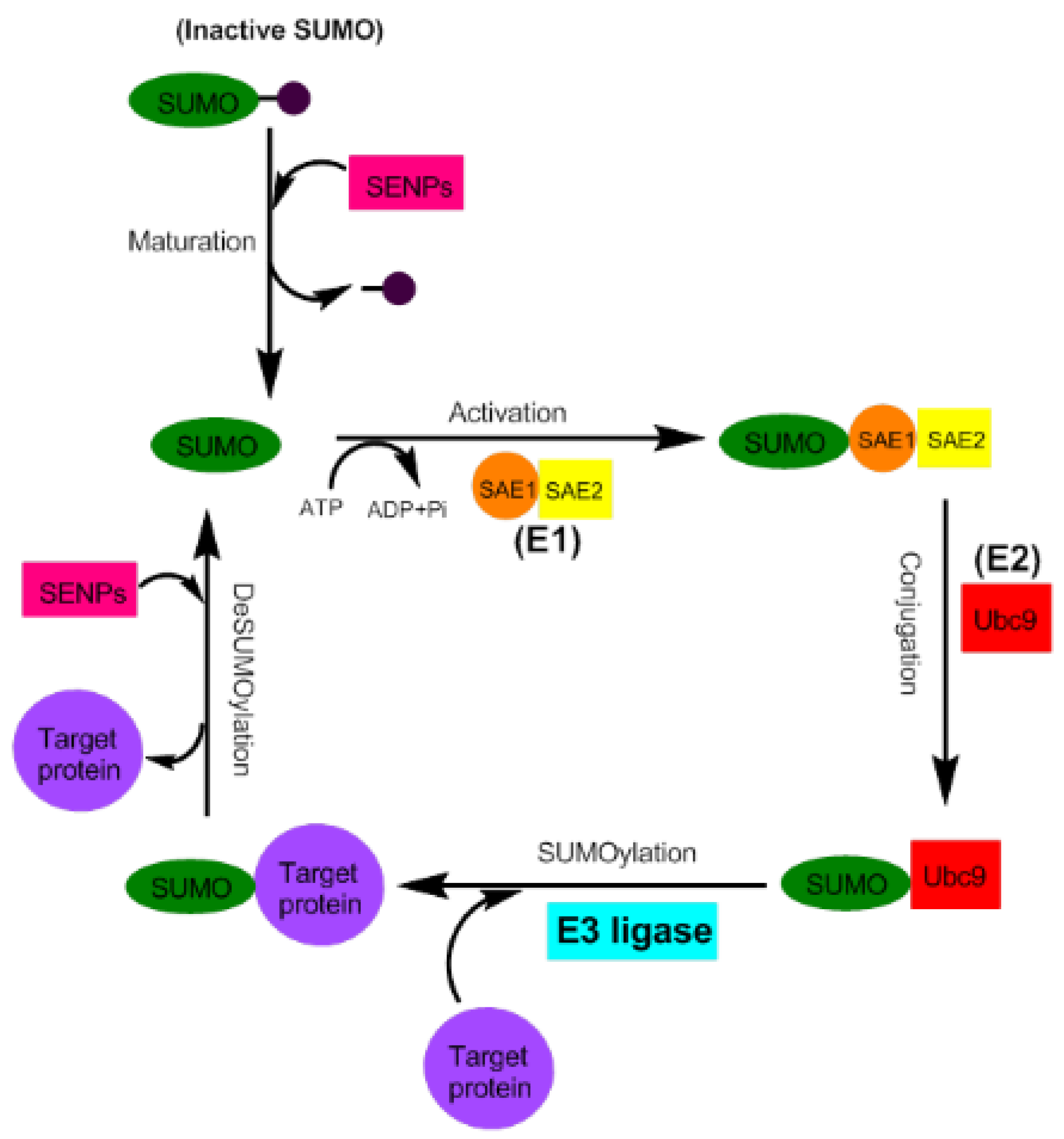
2. Small Ubiquitin-Like Modifier (SUMO) Proteins and the Process of SUMOylation
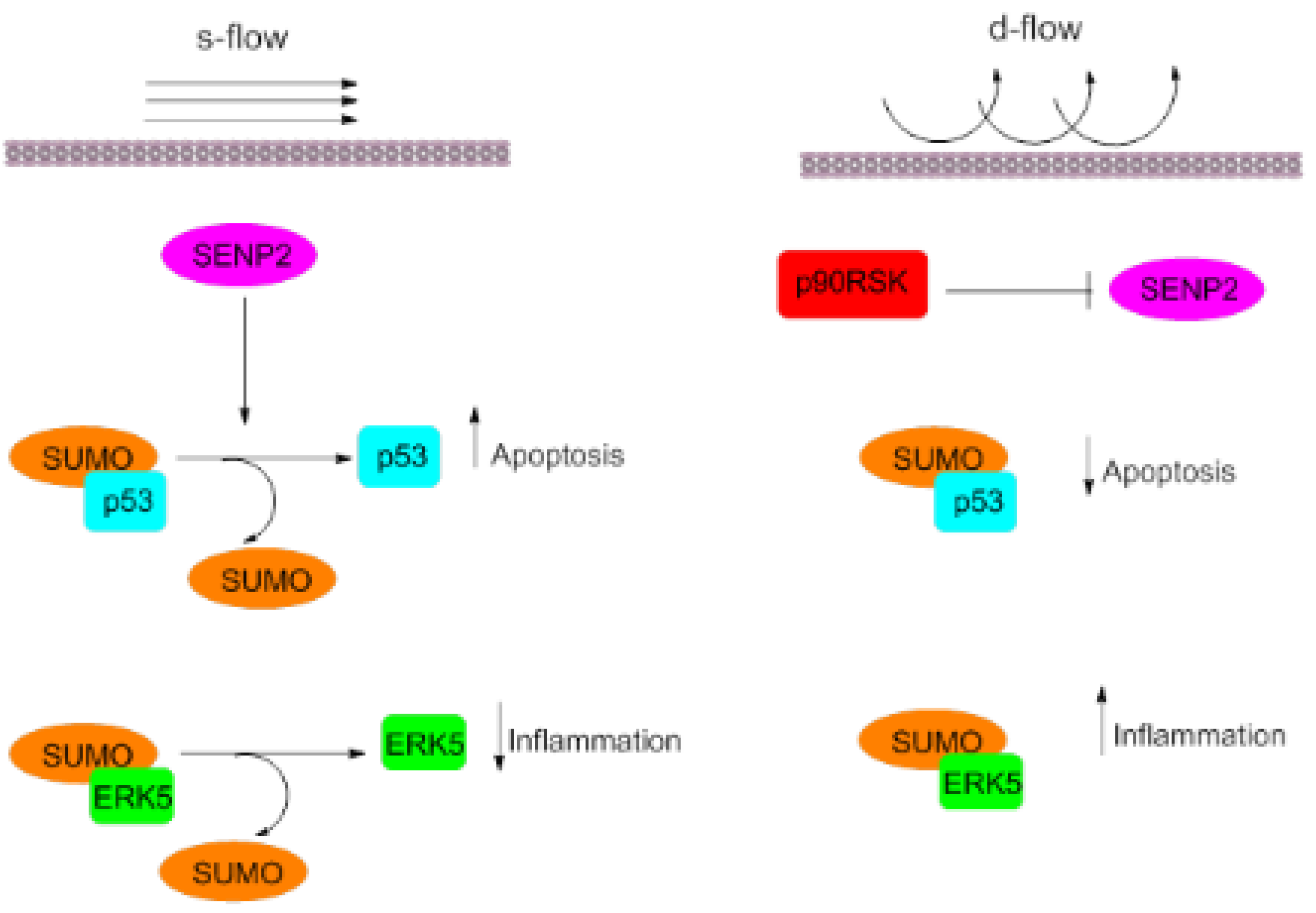
3. Blood Flows and Atherosclerosis
4. NFκB Is a Regulator of Inflammation in ECs and a SUMOylation Target
5. Other Targets of SUMOylation Involved in the Pathogenesis of Atherosclerosis
5.1. MAPK-Activated Protein Kinase-2 (MK2)
5.2. ERK5
5.3. P90 Ribosomal s6 Kinase (p90RSK)
6. KLF
7. Membrane-Associated Guanylate Kinase with Inverted Domain Structure-1 (MAGI-1)
8. p53
9. Adenosine Monophosphate-Activated Protein Kinase (AMPK)
10. Liver Receptor Homolog-1 (LRH-1)
11. Conclusions
Funding
Conflicts of Interest
References
- Björkbacka, H.; Nilsson, J. Innate immunity in atherosclerosis. J. Innate Immun. 2010, 2, 305. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Nicorescu, I.; Dallinga, G.M.; de Winther, M.P.; Stroes, E.S.; Bahjat, M. Potential epigenetic therapeutics for atherosclerosis treatment. Atherosclerosis 2018, 281, 189–197. [Google Scholar] [CrossRef]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204. [Google Scholar] [CrossRef]
- Gisterå, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317. [Google Scholar] [CrossRef]
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef]
- Skålén, K.; Gustafsson, M.; Rydberg, E.K.; Hultén, L.M.; Wiklund, O.; Innerarity, T.L.; Borén, J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 2002, 417, 750. [Google Scholar] [CrossRef]
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–561. [Google Scholar] [CrossRef]
- Libby, P.; Lichtman, A.H.; Hansson, G.K. Immune effector mechanisms implicated in atherosclerosis: From mice to humans. Immunity 2013, 38, 1092–1104. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Raines, E.W.; Plump, A.S.; Breslow, J.L.; Ross, R. Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Cybulsky, M.I.; Gimbrone, M.A. Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science 1991, 251, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef] [PubMed]
- Pentikäinen, M.; Öörni, K.; Ala-Korpela, M.; Kovanen, P. Modified LDL—Trigger of atherosclerosis and inflammation in the arterial intima. J. Intern. Med. 2000, 247, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M.; Febbraio, M.; Silverstein, R.L. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J. Clin. Investig. 2009, 119, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef]
- Joehanes, R.; Just, A.C.; Marioni, R.E.; Pilling, L.C.; Reynolds, L.M.; Mandaviya, P.R.; Guan, W.; Xu, T.; Elks, C.E.; Aslibekyan, S. Epigenetic signatures of cigarette smoking. Circ. Cardiovasc. Genet. 2016, 9, 436–447. [Google Scholar] [CrossRef]
- Xu, S.; Pelisek, J.; Jin, Z.G. Atherosclerosis is an epigenetic disease. Trends Endocrinol. Metab. 2018, 29, 739–742. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef]
- Becares, N.; Gage, M.C.; Pineda-Torra, I. Posttranslational modifications of lipid-activated nuclear receptors: Focus on metabolism. Endocrinology 2016, 158, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-T.; Huang, K.-Y.; Su, M.-G.; Lee, T.-Y.; Bretana, N.A.; Chang, W.-C.; Chen, Y.-J.; Chen, Y.-J.; Huang, H.-D. DbPTM 3.0: An informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic Acids Res. 2012, 41, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-wide post-translational modification statistics: Frequency analysis and curation of the swiss-prot database. Sci. Rep. 2011, 1, 90. [Google Scholar] [CrossRef] [PubMed]
- Beltrao, P.; Bork, P.; Krogan, N.J.; van Noort, V. Evolution and functional cross-talk of protein post-translational modifications. Mol. Syst. Biol. 2013, 9, 714. [Google Scholar] [CrossRef]
- Apweiler, R.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alam-Faruque, Y.; Antunes, R.; Barrell, D.; Bely, B.; Bingley, M.; Binns, D. The universal protein resource (UniProt) in 2010. Nucleic Acids Res. 2010, 38, 142–148. [Google Scholar]
- Adorisio, S.; Fierabracci, A.; Muscari, I.; Liberati, A.M.; Ayroldi, E.; Migliorati, G.; Thuy, T.T.; Riccardi, C.; Delfino, D.V. SUMO proteins: Guardians of immune system. J. Autoimmun. 2017, 84, 21–28. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Q.; Chen, W.; Wang, C. Dynamic regulation of innate immunity by ubiquitin and ubiquitin-like proteins. Cytokine Growth Factor Rev. 2013, 24, 559–570. [Google Scholar] [CrossRef]
- Chang, E.; Abe, J. Kinase-SUMO networks in diabetes-mediated cardiovascular disease. Metab. Clin. Exp. 2016, 65, 623–633. [Google Scholar] [CrossRef]
- Guo, B.; Yang, S.-H.; Witty, J.; Sharrocks, A. Signalling Pathways and the Regulation of SUMO Modification; Portland Press Limited: London, UK, 2007. [Google Scholar]
- Le, N.T.; Corsetti, J.P.; Dehoff-Sparks, J.L.; Sparks, C.E.; Fujiwara, K.; Abe, J. Reactive Oxygen Species, SUMOylation, and Endothelial Inflammation. Int. J. Inflamm. 2012, 2012, 678190. [Google Scholar] [CrossRef][Green Version]
- Gill, G. SUMO and ubiquitin in the nucleus: Different functions, similar mechanisms? Genes Dev. 2004, 18, 2046–2059. [Google Scholar] [CrossRef]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947. [Google Scholar] [CrossRef] [PubMed]
- Le, N.-T.; Sandhu, U.G.; Quintana-Quezada, R.A.; Hoang, N.M.; Fujiwara, K.; Abe, J.-I. Flow signaling and atherosclerosis. Cell. Mol. Life Sci. 2017, 74, 1835–1858. [Google Scholar] [CrossRef]
- Dehnavi, S.; Sadeghi, M.; Johnston, T.P.; Barreto, G.; Shohan, M.; Sahebkar, A. The role of protein SUMOylation in rheumatoid arthritis. J. Autoimmun. 2019, 102, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hickey, C.M.; Wilson, N.R.; Hochstrasser, M. Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell Biol. 2012, 13, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Schwertman, P.; Bekker-Jensen, S.; Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016, 17, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Huang, W.; Kanasaki, K.; Xu, Y. The role of ubiquitination and sumoylation in diabetic nephropathy. Biomed. Res. Int. 2014, 2014, 160692. [Google Scholar] [CrossRef]
- Yeh, E.T. SUMOylation and De-SUMOylation: Wrestling with life’s processes. J. Biol. Chem. 2009, 284, 8223–8237. [Google Scholar] [CrossRef]
- Yang, L.; Lin, C.; Liu, W.; Zhang, J.; Ohgi, K.A.; Grinstein, J.D.; Dorrestein, P.C.; Rosenfeld, M.G. ncRNA-and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell 2011, 147, 773–788. [Google Scholar] [CrossRef]
- Dutchak, P.A.; Katafuchi, T.; Bookout, A.L.; Choi, J.H.; Ruth, T.Y.; Mangelsdorf, D.J.; Kliewer, S.A. Fibroblast growth factor-21 regulates PPARγ activity and the antidiabetic actions of thiazolidinediones. Cell 2012, 148, 556–567. [Google Scholar] [CrossRef]
- Rosonina, E.; Akhter, A.; Dou, Y.; Babu, J.; Sri Theivakadadcham, V.S. Regulation of transcription factors by sumoylation. Transcription. 2017, 8, 220–231. [Google Scholar] [CrossRef]
- Hilgarth, R.S.; Murphy, L.A.; Skaggs, H.S.; Wilkerson, D.C.; Xing, H.; Sarge, K.D. Regulation and function of SUMO modification. J. Biol. Chem. 2004, 279, 53899–53902. [Google Scholar] [CrossRef]
- Abe, J.-I.; Le, N.-T.; Heo, K.-S. Role for SUMOylation in disturbed flow-induced atherosclerotic plaque formation. Biomed. Eng. Lett. 2015, 5, 162–171. [Google Scholar] [CrossRef]
- Davies, P.F.; Civelek, M.; Fang, Y.; Guerraty, M.A.; Passerini, A.G. Seminars in Thrombosis and Hemostasis. In Endothelial Heterogeneity Associated with Regional Athero-Susceptibility and Adaptation to Disturbed Blood Flow In Vivo; Thieme Medical Publishers: New York, NY, USA, 2010; pp. 265–275. [Google Scholar]
- Heo, K.-S.; Chang, E.; Le, N.-T.; Cushman, H.; Yeh, E.T.; Fujiwara, K.; Abe, J.-I. De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow—Induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ. Res. 2013, 112, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Heo, K.-S.; Fujiwara, K.; Abe, J.-I. Disturbed-flow-mediated vascular reactive oxygen species induce endothelial dysfunction. Circ. J. 2011, 75, 2722–2730. [Google Scholar] [CrossRef]
- Heo, K.-S.; Le, N.-T.; Cushman, H.J.; Giancursio, C.J.; Chang, E.; Woo, C.-H.; Sullivan, M.A.; Taunton, J.; Yeh, E.T.; Fujiwara, K. Disturbed flow-activated p90RSK kinase accelerates atherosclerosis by inhibiting SENP2 function. J. Clin. Investig. 2015, 125, 1299–1310. [Google Scholar] [CrossRef]
- Urbich, C.; Stein, M.; Reisinger, K.; Kaufmann, R.; Dimmeler, S.; Gille, J. Fluid shear stress-induced transcriptional activation of the vascular endothelial growth factor receptor-2 gene requires Sp1-dependent DNA binding. FEBS Lett. 2003, 535, 87–93. [Google Scholar] [CrossRef]
- Huddleson, J.P.; Srinivasan, S.; Ahmad, N.; Lingrel, J.B. Fluid shear stress induces endothelial KLF2 gene expression through a defined promoter region. Biol. Chem. 2004, 385, 723–729. [Google Scholar] [CrossRef]
- Reinhart-King, C.A.; Fujiwara, K.; Berk, B.C. Physiologic stress-mediated signaling in the endothelium. Methods Enzymol. 2008, 443, 25–44. [Google Scholar]
- Abe, J.-I.; Berk, B.C. Novel mechanisms of endothelial mechanotransduction. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2378–2386. [Google Scholar] [CrossRef]
- Abe, J.-I.; Ko, K.A.; Kotla, S.; Wang, Y.; Paez-Mayorga, J.; Shin, I.J.; Imanishi, M.; Vu, H.T.; Tao, Y.; Leiva-Juarez, M.M. MAGI1 as a link between endothelial activation and ER stress drives atherosclerosis. JCI Insight 2019, 4, e125570. [Google Scholar] [CrossRef]
- Nigro, P.; Abe, J.; Berk, B.C. Flow shear stress and atherosclerosis: A matter of site specificity. Antioxid. Redox. Signal. 2011, 15, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.-W.; Qiu, H.; Rezvan, A.; Kwon, K.; Nam, D.; Son, D.J.; Visvader, J.E.; Jo, H. Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood 2010, 116, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Gerstberger, S.; Carlson, L.; Franzoso, G.; Siebenlist, U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science 1995, 267, 1485–1488. [Google Scholar] [CrossRef]
- Xiao, L.; Liu, Y.; Wang, N. New paradigms in inflammatory signaling in vascular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, 317–325. [Google Scholar] [CrossRef]
- Vatsyayan, J.; Qing, G.; Xiao, G.; Hu, J. SUMO1 modification of NF-kappaB2/p100 is essential for stimuli-induced p100 phosphorylation and processing. EMBO Rep. 2008, 9, 885–890. [Google Scholar] [CrossRef]
- Schmidt-Supprian, M.; Bloch, W.; Courtois, G.; Addicks, K.; Israël, A.; Rajewsky, K.; Pasparakis, M. NEMO/IKKγ-deficient mice model incontinentia pigmenti. Mol. Cell 2000, 5, 981–992. [Google Scholar] [CrossRef]
- Gareus, R.; Kotsaki, E.; Xanthoulea, S.; van der Made, I.; Gijbels, M.J.; Kardakaris, R.; Polykratis, A.; Kollias, G.; de Winther, M.P.; Pasparakis, M. Endothelial cell-specific NF-κB inhibition protects mice from atherosclerosis. Cell Metab. 2008, 8, 372–383. [Google Scholar] [CrossRef]
- Desterro, J.M.; Rodriguez, M.S.; Hay, R.T. SUMO-1 modification of IκBα inhibits NF-κB activation. Mol. Cell 1998, 2, 233–239. [Google Scholar] [CrossRef]
- Lee, M.H.; Mabb, A.M.; Gill, G.B.; Yeh, E.T.; Miyamoto, S. NF-κB induction of the SUMO protease SENP2: A negative feedback loop to attenuate cell survival response to genotoxic stress. Mol. Cell 2011, 43, 180–191. [Google Scholar] [CrossRef]
- Yang, Y.; Xia, F.; Hermance, N.; Mabb, A.; Simonson, S.; Morrissey, S.; Gandhi, P.; Munson, M.; Miyamoto, S.; Kelliher, M.A. A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-κB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol. Cell. Biol. 2011, 31, 2774–2786. [Google Scholar] [CrossRef]
- Miyamoto, S. Nuclear initiated NF-κB signaling: NEMO and ATM take center stage. Cell Res. 2011, 21, 116. [Google Scholar] [CrossRef] [PubMed]
- Wuerzberger-Davis, S.; Nakamura, Y.; Seufzer, B.; Miyamoto, S. NF-κB activation by combinations of NEMO SUMOylation and ATM activation stresses in the absence of DNA damage. Oncogene 2007, 26, 641. [Google Scholar] [CrossRef] [PubMed]
- Jagavelu, K.; Tietge, U.J.; Gaestel, M.; Drexler, H.; Schieffer, B.; Bavendiek, U. Systemic Deficiency of the MAP Kinase—Activated Protein Kinase 2 Reduces Atherosclerosis in Hypercholesterolemic Mice. Circ. Res. 2007, 101, 1104–1112. [Google Scholar] [CrossRef]
- Chang, E.; Heo, K.-S.; Woo, C.-H.; Lee, H.; Le, N.-T.; Thomas, T.N.; Fujiwara, K.; Abe, J.-I. MK2 SUMOylation regulates actin filament remodeling and subsequent migration in endothelial cells by inhibiting MK2 kinase and HSP27 phosphorylation. Blood 2011, 117, 2527–2537. [Google Scholar] [CrossRef]
- Akaike, M.; Che, W.; Marmarosh, N.-L.; Ohta, S.; Osawa, M.; Ding, B.; Berk, B.C.; Yan, C.; Abe, J.-I. The hinge-helix 1 region of peroxisome proliferator-activated receptor γ1 (PPARγ1) mediates interaction with extracellular signal-regulated kinase 5 and PPARγ1 transcriptional activation: Involvement in flow-induced PPARγ activation in endothelial cells. Mol. Cell. Biol. 2004, 24, 8691–8704. [Google Scholar] [CrossRef]
- Kasler, H.G.; Victoria, J.; Duramad, O.; Winoto, A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell. Biol. 2000, 20, 8382–8389. [Google Scholar] [CrossRef]
- Le, N.-T.; Heo, K.-S.; Takei, Y.; Lee, H.; Woo, C.-H.; Chang, E.; McClain, C.; Hurley, C.; Wang, X.; Li, F. A crucial role for p90RSK-mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation 2013, 127, 486–499. [Google Scholar] [CrossRef]
- Woo, C.-H.; Shishido, T.; McClain, C.; Lim, J.H.; Li, J.-D.; Yang, J.; Yan, C.; Abe, J.-I. Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress—Induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ. Res. 2008, 102, 538–545. [Google Scholar] [CrossRef]
- Lee, C.-H.; Chawla, A.; Urbiztondo, N.; Liao, D.; Boisvert, W.A.; Evans, R.M. Transcriptional repression of atherogenic inflammation: Modulation by PPARδ. Science 2003, 302, 453–457. [Google Scholar] [CrossRef]
- Barish, G.D.; Ruth, T.Y.; Karunasiri, M.S.; Becerra, D.; Kim, J.; Tseng, T.W.; Tai, L.-J.; LeBlanc, M.; Diehl, C.; Cerchietti, L. The Bcl6-SMRT/NCoR cistrome represses inflammation to attenuate atherosclerosis. Cell Metab. 2012, 15, 554–562. [Google Scholar] [CrossRef]
- Ohnesorge, N.; Viemann, D.; Schmidt, N.; Czymai, T.; Spiering, D.; Schmolke, M.; Ludwig, S.; Roth, J.; Goebeler, M.; Schmidt, M. Erk5 activation elicits a vasoprotective endothelial phenotype via induction of Krüppel-like factor 4 (KLF4). J. Biol. Chem. 2010, 285, 26199–26210. [Google Scholar] [CrossRef] [PubMed]
- Parmar, K.M.; Larman, H.B.; Dai, G.; Zhang, Y.; Wang, E.T.; Moorthy, S.N.; Kratz, J.R.; Lin, Z.; Jain, M.K.; Gimbrone, M.A. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J. Clin. Investig. 2006, 116, 49–58. [Google Scholar] [CrossRef]
- Zhou, A.-X.; Wang, X.; Lin, C.S.; Han, J.; Yong, J.; Nadolski, M.J.; Borén, J.; Kaufman, R.J.; Tabas, I. C/EBP-homologous protein (CHOP) in vascular smooth muscle cells regulates their proliferation in aortic explants and atherosclerotic lesions. Circ. Res. 2015, 116, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.-J.; Hui, L.Y.; Zhang, X.-H.; Wang, Z.-P.; Jiang, W.; Zhang, Y.; Yin, W.-N.; Zhang, Y.; Shi, H.-J.; Liu, Y. SUMOylation of KLF4 acts as a switch in transcriptional programs that control VSMC proliferation. Exp. Cell Res. 2016, 342, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, I.; Giudice, V.; Delahaye, E.; Wianny, F.; Aubry, M.; Mure, M.; Chen, J.; Jauch, R.; Bogu, G.K.; Nolden, T. Klf4 and Klf5 differentially inhibit mesoderm and endoderm differentiation in embryonic stem cells. Nat. Commun. 2014, 5, 3719. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628. [Google Scholar] [CrossRef]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of induced pluripotent stem cell generation by the p53–p21 pathway. Nature 2009, 460, 1132. [Google Scholar] [CrossRef]
- Li, H.; Niu, H.; Peng, Y.; Wang, J.; He, P. Ubc9 promotes invasion and metastasis of lung cancer cells. Oncol. Rep. 2013, 29, 1588–1594. [Google Scholar] [CrossRef]
- SenBanerjee, S.; Lin, Z.; Atkins, G.B.; Greif, D.M.; Rao, R.M.; Kumar, A.; Feinberg, M.W.; Chen, Z.; Simon, D.I.; Luscinskas, F.W. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J. Exp. Med. 2004, 199, 1305–1315. [Google Scholar] [CrossRef]
- Sen-Banerjee, S.; Mir, S.; Lin, Z.; Hamik, A.; Atkins, G.B.; Das, H.; Banerjee, P.; Kumar, A.; Jain, M.K. Kruppel-like factor 2 as a novel mediator of statin effects in endothelial cells. Circ. Hagertown 2005, 112, 720–726. [Google Scholar] [CrossRef]
- Lin, Z.; Natesan, V.; Shi, H.; Dong, F.; Kawanami, D.; Mahabeleshwar, G.H.; Atkins, G.B.; Nayak, L.; Cui, Y.; Finigan, J.H. Kruppel-like factor 2 regulates endothelial barrier function. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1952–1959. [Google Scholar] [CrossRef] [PubMed]
- Arnau-Soler, A.; Adams, M.J.; Hayward, C.; Thomson, P.A. Genome-wide interaction study of a proxy for stress-sensitivity and its prediction of major depressive disorder. PLoS ONE 2018, 13, e0209160. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Domenech, E.; Julia, A.; Panes, J.; Garcia-Sanchez, V.; Mateu, P.N.; Gutierrez, A.; Gomollon, F.; Mendoza, J.L.; Garcia-Planella, E.; et al. Identification of risk loci for Crohn’s disease phenotypes using a genome-wide association study. Gastroenterology 2015, 148, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Julia, A.; Pinto, J.A.; Gratacos, J.; Queiro, R.; Ferrandiz, C.; Fonseca, E.; Montilla, C.; Torre-Alonso, J.C.; Puig, L.; Perez Venegas, J.J.; et al. A deletion at ADAMTS9-MAGI1 locus is associated with psoriatic arthritis risk. Ann. Rheum. Dis. 2015, 74, 1875–1881. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M.; Carlson, P.; Valentin, N.; Acosta, A.; O’Neill, J.; Eckert, D.; Dyer, R.; Na, J.; Klee, E.W.; Murray, J.A. Pilot study of small bowel mucosal gene expression in patients with irritable bowel syndrome with diarrhea. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Jia, S.; Martin, T.A.; Jiang, W.G. Regulation and involvement in cancer and pathological conditions of MAGI1, a tight junction protein. Anticancer Res. 2014, 34, 3251–3256. [Google Scholar]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Teodoro, J.G.; Parker, A.E.; Zhu, X.; Green, M.R. p53-mediated inhibition of angiogenesis through up-regulation of a collagen prolyl hydroxylase. Science 2006, 313, 968–971. [Google Scholar] [CrossRef]
- Takabe, W.; Alberts-Grill, N.; Jo, H. Disturbed flow: p53 SUMOylation in the turnover of endothelial cells. J. Cell Biol. 2011, 193, 805–807. [Google Scholar] [CrossRef]
- Sanz-González, S.M.; Barquín, L.; García-Cao, I.; Roque, M.; González, J.M.; Fuster, J.J.; Castells, M.T.; Flores, J.M.; Serrano, M.; Andrés, V. Increased p53 gene dosage reduces neointimal thickening induced by mechanical injury but has no effect on native atherosclerosis. Cardiovasc. Res. 2007, 75, 803–812. [Google Scholar] [CrossRef]
- Kumar, A.; Kim, C.-S.; Hoffman, T.A.; Naqvi, A.; DeRicco, J.; Jung, S.-B.; Lin, Z.; Jain, M.K.; Irani, K. p53 impairs endothelial function by transcriptionally repressing Kruppel-Like Factor 2. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Guevara, N.V.; Kim, H.-S.; Antonova, E.I.; Chan, L. The absence of p53 accelerates atherosclerosis by increasing cell proliferation in vivo. Nat. Med. 1999, 5, 335. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Mahmoudi, M.; Bennett, M. DNA damage, p53, apoptosis and vascular disease. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2007, 621, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Bennett, M. The role of p53 in atherosclerosis. Cell Cycle Georget. Tex. 2006, 5, 1907–1909. [Google Scholar] [CrossRef]
- Khanna, A.K. Enhanced susceptibility of cyclin kinase inhibitor p21 knockout mice to high fat diet induced atherosclerosis. J. Biomed. Sci. 2009, 16, 66. [Google Scholar] [CrossRef]
- Santiago, A.; Li, D.; Zhao, L.Y.; Godsey, A.; Liao, D. p53 SUMOylation promotes its nuclear export by facilitating its release from the nuclear export receptor CRM1. Mol. Biol. Cell 2013, 24, 2739–2752. [Google Scholar] [CrossRef]
- Heo, K.-S.; Lee, H.; Nigro, P.; Thomas, T.; Le, N.-T.; Chang, E.; McClain, C.; Reinhart-King, C.A.; King, M.R.; Berk, B.C. PKCζ mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J. Cell Biol. 2011, 193, 867–884. [Google Scholar] [CrossRef]
- Johnson, E.S. Protein modification by SUMO. Annu. Rev. Biochem. 2004, 73, 355–382. [Google Scholar] [CrossRef]
- Nigro, P.; Abe, J.-I.; Woo, C.-H.; Satoh, K.; McClain, C.; O’Dell, M.R.; Lee, H.; Lim, J.-H.; Li, J.-D.; Heo, K.-S. PKCζ decreases eNOS protein stability via inhibitory phosphorylation of ERK5. Blood 2010, 116, 1971–1979. [Google Scholar] [CrossRef]
- Zaha, V.G.; Young, L.H. AMP-activated protein kinase regulation and biological actions in the heart. Circ. Res. 2012, 111, 800–814. [Google Scholar] [CrossRef]
- Zhang, Y.; Lee, T.-S.; Kolb, E.M.; Sun, K.; Lu, X.; Sladek, F.M.; Kassab, G.S.; Garland, T.; Shyy, J.Y.-J. AMP-activated protein kinase is involved in endothelial NO synthase activation in response to shear stress. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1281–1287. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhang, M.; Liang, B.; Xie, Z.; Zhao, Z.; Asfa, S.; Choi, H.C.; Zou, M.-H. Reduction of AMP-activated protein kinase alpha 2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 2010, 121, 792. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, M.; Liang, B.; Shirwany, N.; Zhu, Y.; Zou, M.-H. Activation of AMP-activated protein kinase is required for berberine-induced reduction of atherosclerosis in mice: The role of uncoupling protein 2. PLoS ONE 2011, 6, e25436. [Google Scholar] [CrossRef] [PubMed]
- Rubio, T.; Vernia, S.; Sanz, P. Sumoylation of AMPKβ2 subunit enhances AMP-activated protein kinase activity. Mol. Biol. Cell 2013, 24, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Fayard, E.; Auwerx, J.; Schoonjans, K. LRH-1: An orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol. 2004, 14, 250–260. [Google Scholar] [CrossRef]
- Stein, S.; Oosterveer, M.H.; Mataki, C.; Xu, P.; Lemos, V.; Havinga, R.; Dittner, C.; Ryu, D.; Menzies, K.J.; Wang, X. SUMOylation-dependent LRH-1/PROX1 interaction promotes atherosclerosis by decreasing hepatic reverse cholesterol transport. Cell Metab. 2014, 20, 603–613. [Google Scholar] [CrossRef]
- Lee, Y.-K.; Moore, D.D. Liver receptor homolog-1, an emerging metabolic modulator. Front. Biosci. 2008, 13, 5950–5958. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Auwerx, J.; Schoonjans, K. Emerging actions of the nuclear receptor LRH-1 in the gut. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 947–955. [Google Scholar] [CrossRef]
- Chalkiadaki, A.; Talianidis, I. SUMO-dependent compartmentalization in promyelocytic leukemia protein nuclear bodies prevents the access of LRH-1 to chromatin. Mol. Cell. Biol. 2005, 25, 5095–5105. [Google Scholar] [CrossRef]
- Lee, M.B.; Lebedeva, L.A.; Suzawa, M.; Wadekar, S.A.; Desclozeaux, M.; Ingraham, H.A. The DEAD-box protein DP103 (Ddx20 or Gemin-3) represses orphan nuclear receptor activity via SUMO modification. Mol. Cell. Biol. 2005, 25, 1879–1890. [Google Scholar] [CrossRef]
- Talamillo, A.; Herboso, L.; Pirone, L.; Pérez, C.; Gonzalez, M.; Sánchez, J.; Mayor, U.; Lopitz-Otsoa, F.; Rodriguez, M.S.; Sutherland, J.D. Scavenger receptors mediate the role of SUMO and Ftz-f1 in Drosophila steroidogenesis. PLoS Genet. 2013, 9, e1003473. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.D.; Bojanala, N.; Bernal, T.; Ashrafi, K.; Asahina, M.; Yamamoto, K.R. Sumoylated NHR-25/NR5A regulates cell fate during C. elegans vulval development. PLoS Genet. 2013, 9, e1003992. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Protein | General Characteristics and Association with Pathogenesis of Atherosclerosis | Consequences of SUMO Modifications | Ref |
|---|---|---|---|
| NFκB | A family of transcriptional factors localized in the cytoplasm in the inactive form that require stimulation by an inflammatory factor to enable nuclear localization and subsequent activation of pro-inflammatory genes involved in atherogenesis | Down-regulation of NFκB pathway by SUMO1 modification of IκBα Up-regulation of NFκB pathway by SUMO2/3 modification of IκBα | [28,56,57] |
| MK2 | A pro-inflammatory kinase that increases NFκB activity | Inhibition of its kinase activity by K339 SUMOylation | [33,66,67] |
| ERK5 | A family of serine/threonine kinases that regulates various cellular functions especially in endothelial hemostasis and is protective against atherosclerosis | D-flow-induced ERK5 SUMOylation and EC dysfunction | [43,51,67,68,69] |
| p90RSK | A serine/threonine kinase associated with EC dysfunction in diabetes mellitus-induced cardiovascular disorders | Phosphorylation and consequent inhibition of SENP2 deSUMOylating enzymes which blocks activation of ERK5 and p53 genes | [33,70] |
| KLF | A family of transcription factors that play an important role in inflammatory and cardiovascular disorders KLF4 is a member of this family which acts as a negative regulator of cell proliferation via p21 interaction in non-SUMOylated state | SUMOylation of KLF4 leads to recruitment of co-repressors to the p21 promoter and increases the proliferation of VSMCs | [68,75,80,81] |
| MAGI-1 | A scaffold protein that is associated with adherence junctions and modulates crucial molecular events in atherogenesis including the activation of ECs and endoplasmic reticulum stress-induced apoptosis. | DeSUMOylation of MAGI-1-K931 is required for nuclear translocation of p90RSK | [52,87] |
| p53 | An important tumor suppressor that plays a role in determining the fate of cell for apoptosis or cell cycle arrest in response to various stresses | SUMOylated p53 exports from nucleus to the cytoplasm and induces ECs apoptosis via direct interaction by pro-apoptotic proteins | [33,90,91,102,103] |
| AMPK | A stress-activated kinase that orchestrates cellular responses to different stresses | AMPKα2 activation and protection from UQ destruction via SUMO2 modification AMPKα1 inhibition via SUMOylation | [106,107,108,110] |
| LRH-1 | A member of NR5A subfamily for nuclear receptors with various functions such as cholesterol and bile acid hemostasis | Association of K289 LRH-1 SUMOylation and atherosclerosis pathogenesis | [111,112] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dehnavi, S.; Sadeghi, M.; Penson, P.E.; Banach, M.; Jamialahmadi, T.; Sahebkar, A. The Role of Protein SUMOylation in the Pathogenesis of Atherosclerosis. J. Clin. Med. 2019, 8, 1856. https://doi.org/10.3390/jcm8111856
Dehnavi S, Sadeghi M, Penson PE, Banach M, Jamialahmadi T, Sahebkar A. The Role of Protein SUMOylation in the Pathogenesis of Atherosclerosis. Journal of Clinical Medicine. 2019; 8(11):1856. https://doi.org/10.3390/jcm8111856
Chicago/Turabian StyleDehnavi, Sajad, Mahvash Sadeghi, Peter E. Penson, Maciej Banach, Tannaz Jamialahmadi, and Amirhossein Sahebkar. 2019. "The Role of Protein SUMOylation in the Pathogenesis of Atherosclerosis" Journal of Clinical Medicine 8, no. 11: 1856. https://doi.org/10.3390/jcm8111856
APA StyleDehnavi, S., Sadeghi, M., Penson, P. E., Banach, M., Jamialahmadi, T., & Sahebkar, A. (2019). The Role of Protein SUMOylation in the Pathogenesis of Atherosclerosis. Journal of Clinical Medicine, 8(11), 1856. https://doi.org/10.3390/jcm8111856