Hypothalamic Endocrine Tumors: An Update



Abstract
1. Introduction
2. Clinical Manifestations of Hypothalamic Tumors
3. Tumor Classification
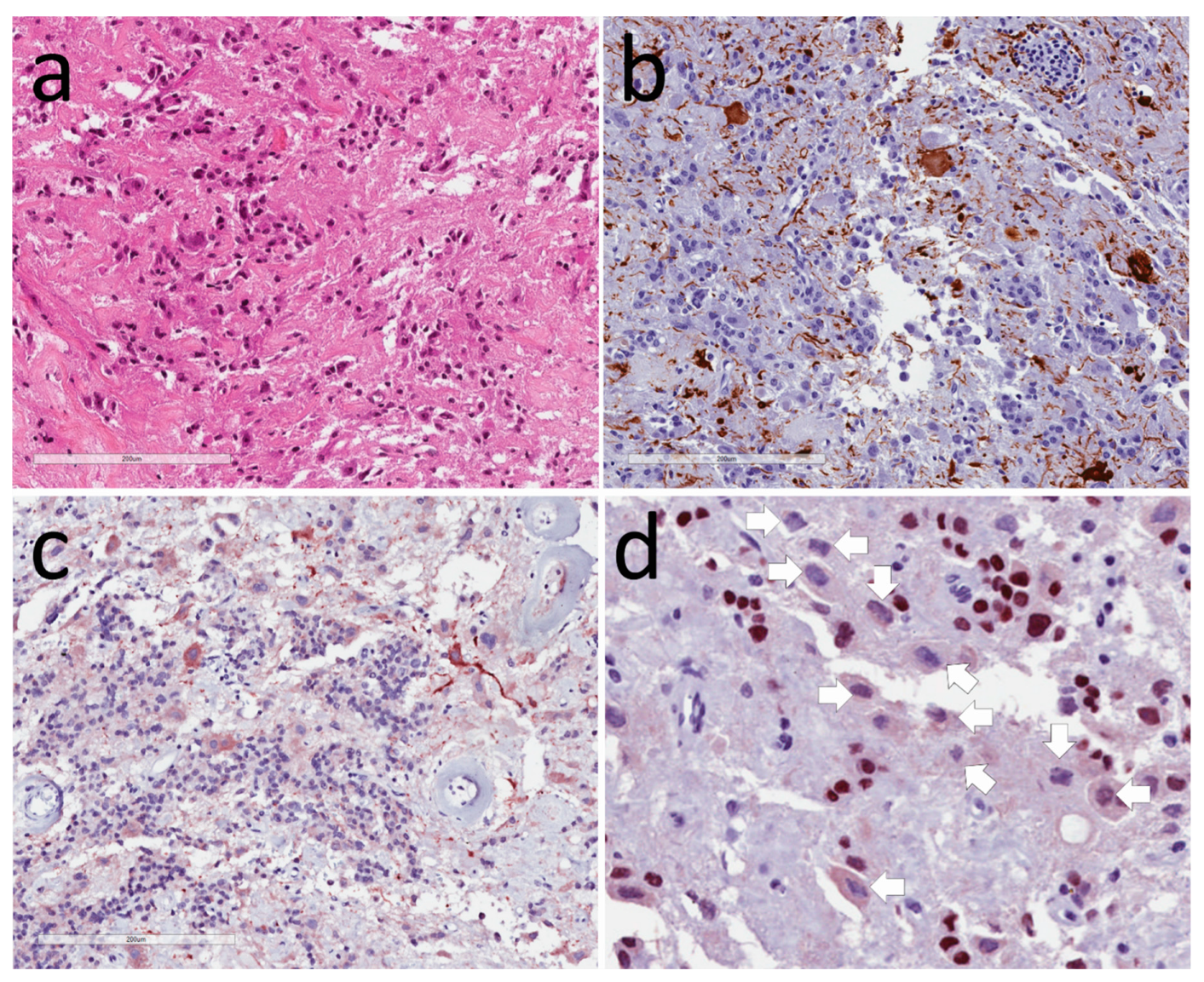
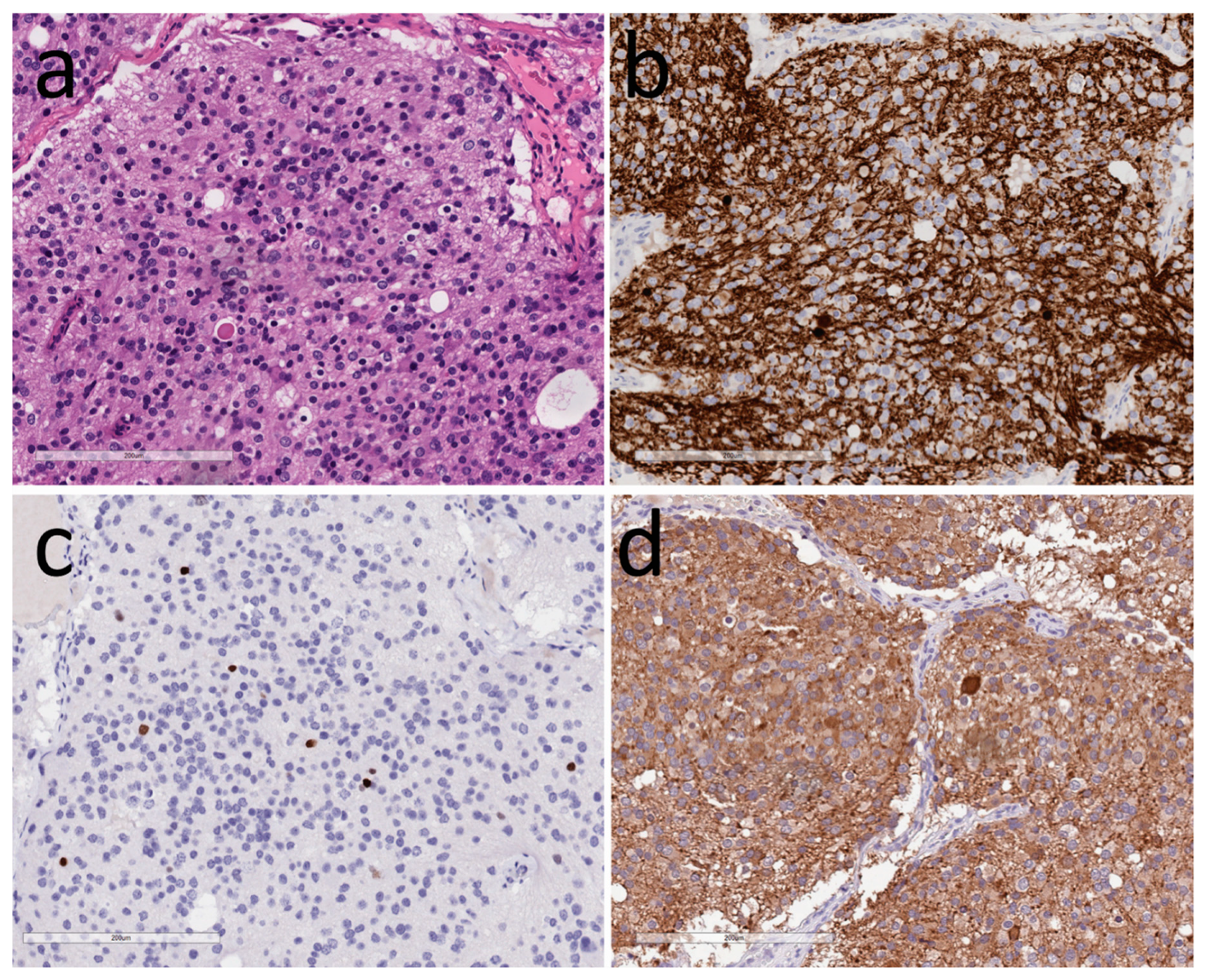
3.1. Neuronal Tumors
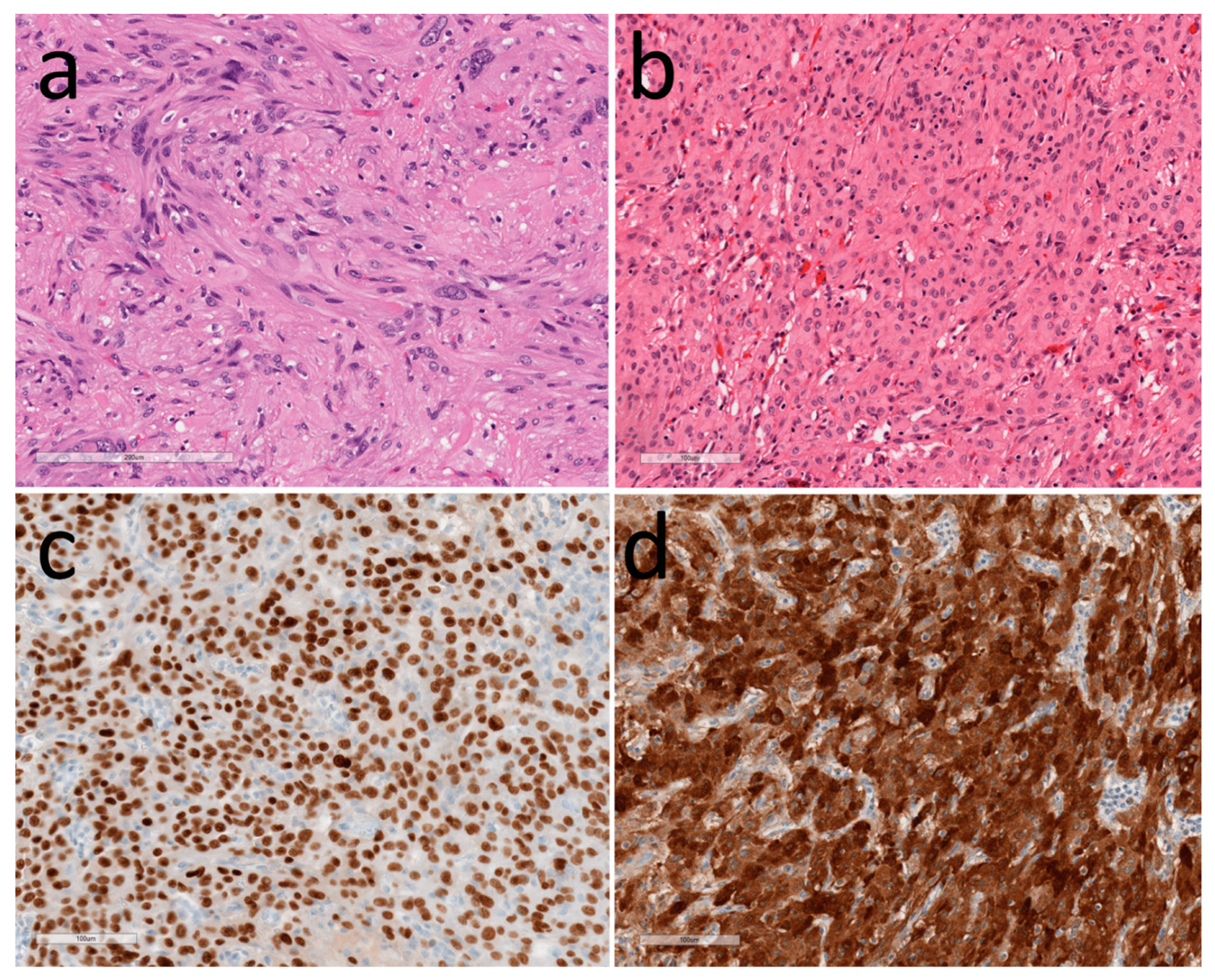
3.2. Tumors of Glia
3.3. Other Tumors in the Hypothalamus
4. Implications of Pathology Diagnosis
Author Contributions
Conflicts of Interest
References
- Asa, S.L.; Bilbao, J.M.; Kovacs, K.; Linfoot, J.A. Hypothalamic neuronal hamartoma associated with pituitary growth hormone cell adenoma and acromegaly. Acta Neuropathol. 1980, 52, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Scheithauer, B.W.; Kovacs, K.; Randall, R.V.; Horvath, E.; Okazaki, H.; Laws, E.R., Jr. Hypothalamic neuronal hamartoma and adenohypophyseal neuronal choristoma: Their association with growth hormone adenoma of the pituitary gland. J. Neuropathol. Exp. Neurol. 1983, 42, 648–663. [Google Scholar] [CrossRef] [PubMed]
- Asa, S.L.; Scheithauer, B.W.; Bilbao, J.M.; Horvath, E.V.A.; Ryan, N.; Kovacs, K.; Randall, R.V.; Laws, E.R., Jr.; Singer, W.; Linfoot, J.A.; et al. A case for hypothalamic acromegaly: A clinicopathological study of six patients with hypothalamic gangliocytomas producing growth hormone-releasing factor. J. Clin. Endocrinol. Metab. 1984, 58, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Asa, S.L.; Kovacs, K.; Tindall, G.T.; Barrow, D.L.; Horvath, E.; Vecsei, P. Cushing’s disease associated with an intrasellar gangliocytoma producing corticotrophin-releasing factor. Ann. Int. Med. 1984, 101, 789–793. [Google Scholar] [CrossRef]
- Asa, S.L.; Ezzat, S.; Kelly, D.F.; Cohan, P.; Takasumi, Y.; Barkhoudarian, G.; Heaney, A.P.; Ridout, R.; Chik, C.L.; Thompson, L.D.; et al. Hypothalamic Vasopressin-producing Tumors: Often Inappropriate Diuresis But Occasionally Cushing Disease. Am. J. Surg. Pathol. 2018, 42, 251–260. [Google Scholar] [CrossRef]
- Li, J.Y.; Racadot, O.; Kujas, M.; Kouadri, M.; Peillon, F.; Racadot, J. Immunocytochemistry of four mixed pituitary adenomas and intrasellar gangliocytomas associated with different clinical syndromes: Acromegaly, amenorrhea-galactorrhea, Cushing’s disease and isolated tumoral syndrome. Acta Neuropathol. 1989, 77, 320–328. [Google Scholar] [CrossRef]
- Mikami, S.; Kameyama, K.; Takahashi, S.; Yoshida, K.; Kawase, T.; Sano, T.; Mukai, M. Combined gangliocytoma and prolactinoma of the pituitary gland. Endocr. Pathol. 2008, 19, 117–121. [Google Scholar] [CrossRef]
- Serri, O.; Berthelet, F.; Belair, M.; Vallette, S.; Asa, S.L. An unusual association of a sellar gangliocytoma with a prolactinoma. Pituitary 2008, 11, 85–87. [Google Scholar] [CrossRef]
- Culler, F.L.; James, H.E.; Simon, M.L.; Jones, K.L. Identification of gonadotropin-releasing hormone in neurons of a hypothalamic hamartoma in a boy with precocious puberty. Neurosurgery 1985, 17, 408–417. [Google Scholar] [CrossRef]
- Ilgren, E.; Briggs, M.; Aynsley-Green, M. Precocious puberty in a 3-year-old girl associated with a parasellar ganglionic hamartoma. Clin. Neuropathol. 1983, 2, 95–98. [Google Scholar]
- Culler, F.L.; Mathews Wray, M.J.; Jones, K.L. Identification of GNRH in neurons of a hypothalamic hamartoma in a male with precocious puberty. Clin. Res. 1982, 30, 113. [Google Scholar]
- Puchner, M.J.A.; Lüdecke, D.K.; Saeger, W.; Riedel, M.; Asa, S.L. Gangliocytomas of the sellar region—A review. Exp. Clin. Endocrinol. 1995, 103, 129–149. [Google Scholar] [CrossRef] [PubMed]
- Cossu, G.; Daniel, R.T.; Messerer, M. Gangliocytomas of the sellar region: A challenging diagnosis. Clin. Neurol. Neurosurg. 2016, 149, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavanee, W.K. World Health Organization Histological Classification of Tumours of the Central Nervous System. Fr. Int. Ag. Res. Cancer 2016. [Google Scholar]
- Felix, I.; Bilbao, J.M.; Asa, S.L.; Tyndel, F.; Kovacs, K.; Becker, L.E. Cerebral and cerebellar gangliocytomas: A morphological study of nine cases. Acta Neuropathol. 1994, 88, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Bevan, J.S.; Asa, S.L.; Rossi, M.L.; Esiri, M.M.; Adams, C.B.T.; Burke, C.W. Intrasellar gangliocytoma containing gastrin and growth hormone- releasing hormone associated with a growth hormone-secreting pituitary adenoma. Clin. Endocrinol. 1989, 30, 213–224. [Google Scholar] [CrossRef]
- Rhodes, R.H.; Dusseau, J.J.; Boyd, A.S.; Knigge, K.M. Intrasellar neural-adenohypophyseal choristoma. A morphological and immunocytochemical study. J. Neuropathol. Exp. Neurol. 1982, 41, 267–280. [Google Scholar] [CrossRef]
- Lopes, M.B.; Sloan, E.; Polder, J. Mixed Gangliocytoma-Pituitary Adenoma: Insights on the Pathogenesis of a Rare Sellar Tumor. Am. J. Surg. Pathol. 2017, 41, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Slowik, F.; Fazekas, I.; Balint, K.; Gazso, L.; Pasztor, E.; Czirjak, S.; Lapis, K. Intrasellar hamartoma associated with pituitary adenoma. Acta Neuropathol. 1990, 80, 328–333. [Google Scholar] [CrossRef]
- Puchner, M.J.; Lüdecke, D.K.; Valdueza, J.M.; Saeger, W.; Willig, R.P.; Stalla, G.K.; Odink, R.J. Cushing’s disease in a child caused by a corticotropin-releasing hormone-secreting intrasellar gangliocytoma associated with an adrenocorticotropic hormone-secreting pituitary adenoma. Neurosurgery 1993, 33, 920–925. [Google Scholar] [CrossRef]
- Judge, D.M.; Kulin, H.E.; Page, R.; Santen, R.; Trapukdi, S. Hypothalamic hamartoma. A source of luteinizing-hormone-releasing factor in precoucious puberty. N. Engl. J. Med. 1977, 296, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Hochman, H.I.; Judge, D.M.; Reichlin, S. Precocious puberty and hypothalamic hamartoma. Pediatrics 1981, 67, 236–244. [Google Scholar] [PubMed]
- Nishio, S.; Fujiwara, S.; Aiko, Y.; Takeshita, I.; Fukui, M. Hypothalamic hamartoma. Report of two cases. J. Neurosurg. 1989, 70, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Stefaneanu, L.; Kovacs, K.; Aiba, T.; Shishiba, Y.; Hara, M. Intrasellar gangliocytoma with multiple immunoreactivities. Endocr. Pathol. 1990, 1, 58–63. [Google Scholar] [CrossRef]
- Matsuno, A.; Nagashima, T. Prolactin-secreting gangliocytoma. J Neurosurg 2001, 95, 167–168. [Google Scholar] [PubMed]
- Horvath, E.; Kovacs, K.; Tran, A.; Scheithauer, B.W. Ganglion cells in the posterior pituitary: Result of ectopia or transdifferentiation? Acta Neuropathol. 2000, 100, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, C.; Jublanc, C.; Leclercq, D.; Boch, A.L.; Bielle, F.; Raverot, G.; Daly, A.F.; Trouillas, J.; Villa, C. Transdifferentiation of Neuroendocrine Cells: Gangliocytoma Associated With Two Pituitary Adenomas of Different Lineage in MEN1. Am. J. Surg. Pathol. 2017, 41, 849–853. [Google Scholar] [CrossRef]
- Geddes, J.F.; Jansen, G.H.; Robinson, S.F.D.; Gömöri, E.; Holton, J.L.; Monson, J.P.; Besser, G.M.; Revesz, T. ‘Gangliocytomas’ of the pituitary: A heterogeneous group of lesions with differing histogenesis. Am. J. Surg. Pathol. 2000, 24, 607–613. [Google Scholar] [CrossRef]
- Rades, D.; Schild, S.E.; Fehlauer, F. Prognostic value of the MIB-1 labeling index for central neurocytomas. Neurology 2004, 62, 987–989. [Google Scholar] [CrossRef]
- Myung, J.K.; Cho, H.J.; Park, C.K.; Chung, C.K.; Choi, S.H.; Kim, S.K.; Park, S.H. Clinicopathological and genetic characteristics of extraventricular neurocytomas. Neuropathology 2013, 33, 111–121. [Google Scholar] [CrossRef]
- Maguire, J.A.; Bilbao, J.M.; Kovacs, K.; Resch, L. Hypothalamic Neurocytoma with Vasopressin Immunoreactivity: Immunohistochemical and Ultrastructural Observations. Endocr. Pathol. 1992, 3, 99–104. [Google Scholar] [CrossRef]
- Hassoun, J.; Gambarelli, D.; Grisoli, F.; Pellet, W.; Salamon, G.; Pellissier, J.F.; Toga, M. Central neurocytoma. An electron-microscopic study of two cases. Acta Neuropathol. 1982, 56, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Sakai, N.; Andoh, T.; Yoshimura, S.; Yamada, H. Central neurocytoma presenting with gigantism: Case report. Surg. Neurol. 1992, 38, 141–145. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Kearney, T.; du Plessis, D.; Gnanalingham, K.K. Extraventricular neurocytoma of the sellar region. Br. J. Neurosurg. 2012, 26, 420–422. [Google Scholar] [CrossRef]
- Wang, J.; Song, D.L.; Deng, L.; Sun, S.Y.; Liu, C.; Gong, D.S.; Wang, Y.; Xu, Q.W. Extraventricular neurocytoma of the sellar region: Case report and literature review. Springerplus 2016, 5, 987. [Google Scholar] [CrossRef]
- Mariani, L.; Schaller, B.; Weis, J.; Ozdoba, C.; Seiler, R.W. Esthesioneuroblastoma of the pituitary gland: A clinicopathological entity? Case report and review of the literature. J. Neurosurg. 2004, 101, 1049–1052. [Google Scholar] [CrossRef]
- Sajko, T.; Rumboldt, Z.; Talan-Hranilovic, J.; Radic, I.; Gnjidic, Z. Primary sellar esthesioneuroblastoma. Acta Neurochir. 2005, 147, 447–448. [Google Scholar] [CrossRef]
- Oyama, K.; Yamada, S.; Usui, M.; Kovacs, K. Sellar neuroblastoma mimicking pituitary adenoma. Pituitary 2005, 8, 109–114. [Google Scholar] [CrossRef]
- Lin, J.H.; Tsai, D.H.; Chiang, Y.H. A primary sellar esthesioneuroblastomas with unusual presentations: A case report and reviews of literatures. Pituitary 2009, 12, 70–75. [Google Scholar] [CrossRef]
- Yumusakhuylu, A.C.; Binnetoglu, A.; Topuz, M.F.; Bozkurtlar, E.B.; Baglam, T.; Sari, M. Syndrome of inappropriate antidiuretic hormone secretion associated with olfactory neuroblastoma. J. Craniofac. Surg. 2013, 24, 2189–2193. [Google Scholar] [CrossRef]
- Lee, B.J.; Cho, G.J.; Norgren, R.B., Jr.; Junier, M.P.; Hill, D.F.; Tapia, V.; Costa, M.E.; Ojeda, S.R. TTF-1, a homeodomain gene required for diencephalic morphogenesis, is postnatally expressed in the neuroendocrine brain in a developmentally regulated and cell-specific fashion. Mol. Cell Neurosci. 2001, 17, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kimura, S.; Yamazaki, M.; Kawaguchi, A.; Inoue, K.; Sakai, T. Immunohistochemical analyses of thyroid-specific enhancer-binding protein in the fetal and adult rat hypothalami and pituitary glands. Brain Res. Dev. Brain Res. 2001, 130, 159–166. [Google Scholar] [CrossRef]
- Takei, Y.; Seyama, S.; Pearl, G.S.; Tindall, G.T. Ultrastructural study of the human neurohypophysis. II. Cellular elements of neural parenchyma, the pituicytes. Cell Tissue Res. 1980, 205, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Kamil, Z.; Sinson, S.; Gucer, H.; Asa, S.L.; Mete, O. TTF-1 expressing sellar neoplasm with ependymal rosettes and oncocytic change: Mixed ependymal and oncocytic variant pituicytoma. Endocr. Pathol. 2013, 25, 436–438. [Google Scholar] [CrossRef]
- Mete, O.; Lopes, M.B.; Asa, S.L. Spindle cell oncocytomas and granular cell tumors of the pituitary are variants of pituicytoma. Am. J. Surg. Pathol. 2013, 37, 1694–1699. [Google Scholar] [CrossRef]
- Lee, E.B.; Tihan, T.; Scheithauer, B.W.; Zhang, P.J.; Gonatas, N.K. Thyroid transcription factor 1 expression in sellar tumors: A histogenetic marker? J. Neuropathol. Exp. Neurol. 2009, 68, 482–488. [Google Scholar] [CrossRef]
- Roncaroli, F.; Scheithauer, B.W.; Cenacchi, G.; Horvath, E.; Kovacs, K.; Lloyd, R.V.; Abell-Aleff, P.; Santi, M.; Yates, A.J. ‘Spindle cell oncocytoma’ of the adenohypophysis: A tumor of folliculostellate cells? Am. J. Surg. Pathol. 2002, 26, 1048–1055. [Google Scholar] [CrossRef]
- Dahiya, S.; Sarkar, C.; Hedley-Whyte, E.T.; Sharma, M.C.; Zervas, N.T.; Sridhar, E.; Louis, D.N. Spindle cell oncocytoma of the adenohypophysis: Report of two cases. Acta Neuropathol. 2005, 110, 97–99. [Google Scholar] [CrossRef]
- Kloub, O.; Perry, A.; Tu, P.H.; Lipper, M.; Lopes, M.B. Spindle cell oncocytoma of the adenohypophysis: Report of two recurrent cases. Am. J. Surg. Pathol. 2005, 29, 247–253. [Google Scholar] [CrossRef]
- Vajtai, I.; Sahli, R.; Kappeler, A. Spindle cell oncocytoma of the adenohypophysis: Report of a case with a 16-year follow-up. Pathol. Res. Pract. 2006, 202, 745–750. [Google Scholar] [CrossRef]
- Policarpio-Nicolas, M.L.; Le, B.H.; Mandell, J.W.; Lopes, M.B. Granular cell tumor of the neurohypophysis: Report of a case with intraoperative cytologic diagnosis. Diagn. Cytopathol. 2008, 36, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Shanklin, W.M. The origin, histology and senescence of tumorettes in the human neurohypophysis. Cells Tissues Organs 1953, 18, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Luse, S.A.; Kernohan, J.W. Granular cell tumors of the stalk and posterior lobe of the pituitary gland. Cancer 1955, 8, 616–622. [Google Scholar] [CrossRef]
- Vaquero, J.; Leunda, G.; Cabezudo, J.M.; Salazar, A.R.; de Miguel, J. Granular pituicytomas of the pituitary stalk. Acta Neurochir. 1981, 59, 209–215. [Google Scholar] [CrossRef]
- Landolt, A.M. Granular cell tumors of the neurohypophysis. Acta Neurochir. 1975, (Suppl. 22), 120–128. [Google Scholar]
- Ulrich, J.; Heitz, P.U.; Fischer, T.; Obrist, E.; Gullotta, F. Granular cell tumors: Evidence for heterogeneous tumor cell differentiation. An immunocytochemical study. Virchows Arch. 1987, 53, 52–57. [Google Scholar] [CrossRef]
- Rodriguez, F.J.; Scheithauer, B.W.; Roncaroli, F.; Silva, A.I.; Kovacs, K.; Brat, D.J.; Jin, L. Galectin-3 expression is ubiquitous in tumors of the sellar region, nervous system, and mimics: An immunohistochemical and RT-PCR study. Am. J. Surg. Pathol. 2008, 32, 1344–1352. [Google Scholar] [CrossRef]
- Nishioka, H.; Ii, K.; Llena, J.F.; Hirano, A. Immunohistochemical study of granular cell tumors of the neurohypophysis. Virchows Arch. 1991, 60, 413–417. [Google Scholar] [CrossRef]
- Rossi, M.L.; Bevan, J.S.; Esiri, M.M.; Hughes, J.T.; Adams, C.B.T. Pituicytoma (pilocytic astrocytoma). J. Neurosurg. 1987, 67, 768–772. [Google Scholar] [CrossRef]
- Winer, J.B.; Lidov, H.; Scaravilli, F. An ependymoma involving the pituitary fossa. J. Neurol. Neurosurg. Psychiatry 1989, 52, 1443–1444. [Google Scholar] [CrossRef]
- Liwnicz, B.H.; Berger, T.S.; Liwnicz, R.G.; Aron, B.S. Radiation-associated gliomas: A report of four cases and analysis of postradiation tumors of the central nervous system. Neurosurgery 1985, 17, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Hufnagel, T.J.; Kim, J.H.; Lesser, R.; Miller, J.M.; Abrahams, J.J.; Piepmeier, J.; Manuelidis, E.E. Malignant glioma of the optic chiasm eight years after radiotherapy for prolactinoma. Arch. Ophthalmol. 1988, 106, 1701–1705. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-I.; Chiou, W.-H.; Ho, D.M. Oligodendroglioma occurring after radiation therapy for pituitary adenoma. J. Neurol. Neurosurg. Psychiatry 1987, 50, 1619–1624. [Google Scholar] [CrossRef] [PubMed]
- Dierssen, G.; Figols, J.; Trigueros, F.; Alvarez, G. Gliomas astrocitarios asociados a radioterapia previa. Arch. Neurobiol. 1987, 50, 303–308. [Google Scholar]
- Marus, G.; Levin, C.V.; Rutherfoord, G.S. Malignant glioma following radiotherapy for unrelated primary tumors. Cancer 1986, 58, 886–894. [Google Scholar] [CrossRef]
- Okamoto, S.; Handa, H.; Yamashita, J.; Tokuriki, Y.; Abe, M. Post-irradiation brain tumors. Neurol. Med. Chir. 1985, 25, 528–533. [Google Scholar] [CrossRef]
- Piatt, J.H.; Blue, J.M.; Schold, S.C.; Burger, P.C. Glioblastoma multiforme after radiotherapy for acromegaly. Neurosurgery 1983, 13, 85–89. [Google Scholar] [CrossRef]
- Zampieri, P.; Zorat, P.L.; Mingrino, S.; Soattin, G.B. Radiation-associated cerebral gliomas. A report of two cases and review of the literature. J. Neursurg. Sci. 1989, 33, 271–279. [Google Scholar]
- Kitanaka, C.; Shitara, N.; Nakagomi, T.; Nakamura, H.; Genka, S.; Nakagawa, K.; Akanuma, A.; Aoyama, H.; Takakura, K. Postradiation astrocytoma. Report of two cases. J. Neurosurg. 1989, 70, 469–474. [Google Scholar] [CrossRef]
- Ushio, Y.; Arita, N.; Yoshimine, T.; Nagatani, M.; Mogami, H. Glioblastoma after radiotherapy for craniopharyngioma: Case report. Neurosurgery 1987, 21, 33–38. [Google Scholar] [CrossRef]
- Maat-Schieman, M.L.C.; Bots, G.T.A.M.; Thomeer, R.T.W.M.; Vielvoye, G.J. Malignant astrocytoma following radiotherapy for craniopharyngioma. Br. J. Radiol. 1985, 58, 480–482. [Google Scholar] [CrossRef] [PubMed]
- Alvord, E.C., Jr.; Lofton, S. Gliomas of the optic nerve or chiasm. Outcome by patients’ age, tumor site, and treatment. J. Neurosurg. 1988, 68, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Rush, J.A.; Younge, B.R.; Campbell, R.J.; MacCarty, C.S. Optic glioma. Long-term follow-up of 85 histopathologically verified cases. Ophthalmology 1982, 89, 1213–1219. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. WHO Classification of Tumours of the Central Nervous System; IARC: Lyon, France, 2007. [Google Scholar]
- Riccardi, V.M. Neurofibromatosis. In Neurocutaneous Syndromes—A Practical Approach; Gomez, M.R., Ed.; Butterworths: Boston, MA, USA, 1987; pp. 11–29. [Google Scholar]
- Byrne, S.; Connor, S.; Lascelles, K.; Siddiqui, A.; Hargrave, D.; Ferner, R.E. Clinical presentation and prognostic indicators in 100 adults and children with neurofibromatosis 1 associated non-optic pathway brain gliomas. J. Neurooncol. 2017, 133, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Tihan, T.; Fisher, P.G.; Kepner, J.L.; Godfraind, C.; McComb, R.D.; Goldthwaite, P.T.; Burger, P.C. Pediatric astrocytomas with monomorphous pilomyxoid features and a less favorable outcome. J. Neuropathol. Exp. Neurol. 1999, 58, 1061–1068. [Google Scholar] [CrossRef]
- Komotar, R.J.; Mocco, J.; Jones, J.E.; Zacharia, B.E.; Tihan, T.; Feldstein, N.A.; Anderson, R.C. Pilomyxoid astrocytoma: Diagnosis, prognosis, and management. Neurosurg. Focus 2005, 18, E7. [Google Scholar] [CrossRef]
- Asa, S.L. Tumors of the Pituitary Gland. In AFIP Atlas of Tumor Pathology; Series 4, Fascicle 15; Silverberg, S.G., Ed.; ARP Press: Silver Spring, MD, USA, 2011. [Google Scholar]
- Asa, S.L.; Casar-Borota, O.; Chanson, P.; Delgrange, E.; Earls, P.; Ezzat, S.; Grossman, A.; Ikeda, H.; Inoshita, N.; Karavitaki, N.; et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): An International Pituitary Pathology Club proposal. Endocr. Relat. Cancer 2017, 24, C5–C8. [Google Scholar] [CrossRef]
- Asa, S.L.; Kovacs, K.; Horvath, E.; Ezrin, C.; Weiss, M.H. Sellar glomangioma. Ultrastruct. Pathol. 1984, 7, 49–54. [Google Scholar] [CrossRef]
- Asa, S.L.; Kovacs, K.; Horvath, E.; Ezrin, C.; Weiss, M.H. Sellar glomangioma. Endocr. Pathol. 2011, 22, 218–221. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Neuronal Neoplasms | Gangliocytomas |
|---|---|
| Neurocytomas | |
| Glial Neoplasms | Gliomas |
| Pituicytomas (including oncocytic, ependymal, and granular cell variants) Hypothalamic and optic gliomas | |
| Neural Stromal Neoplasms | Schwannomas |
| Meningiomas | |
| Chordomas | |
| Other Stromal Neoplasms | Vascular and mesenchymal tumors |
| Lymphomas | |
| Germ cell tumors | |
| Infiltrating Neoplasms | PitNETs (Pituitary neuroendocrine tumors) |
| Craniopharyngiomas | |
| Germ cell tumors, including teratomas | |
| Metastatic Neoplasms |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asa, S.L.; Mete, O. Hypothalamic Endocrine Tumors: An Update. J. Clin. Med. 2019, 8, 1741. https://doi.org/10.3390/jcm8101741
Asa SL, Mete O. Hypothalamic Endocrine Tumors: An Update. Journal of Clinical Medicine. 2019; 8(10):1741. https://doi.org/10.3390/jcm8101741
Chicago/Turabian StyleAsa, Sylvia L., and Ozgur Mete. 2019. "Hypothalamic Endocrine Tumors: An Update" Journal of Clinical Medicine 8, no. 10: 1741. https://doi.org/10.3390/jcm8101741
APA StyleAsa, S. L., & Mete, O. (2019). Hypothalamic Endocrine Tumors: An Update. Journal of Clinical Medicine, 8(10), 1741. https://doi.org/10.3390/jcm8101741