NK Cell-Based Immunotherapy for Hematological Malignancies

 ,
,  ,
, 

Abstract
1. Introduction
2. NK Cell Receptors
2.1. HLA-Specific NK Receptors
2.2. Non-HLA Specific Activating NK Receptors
2.3. Inhibitory Checkpoints Expressed on Human NK Cells
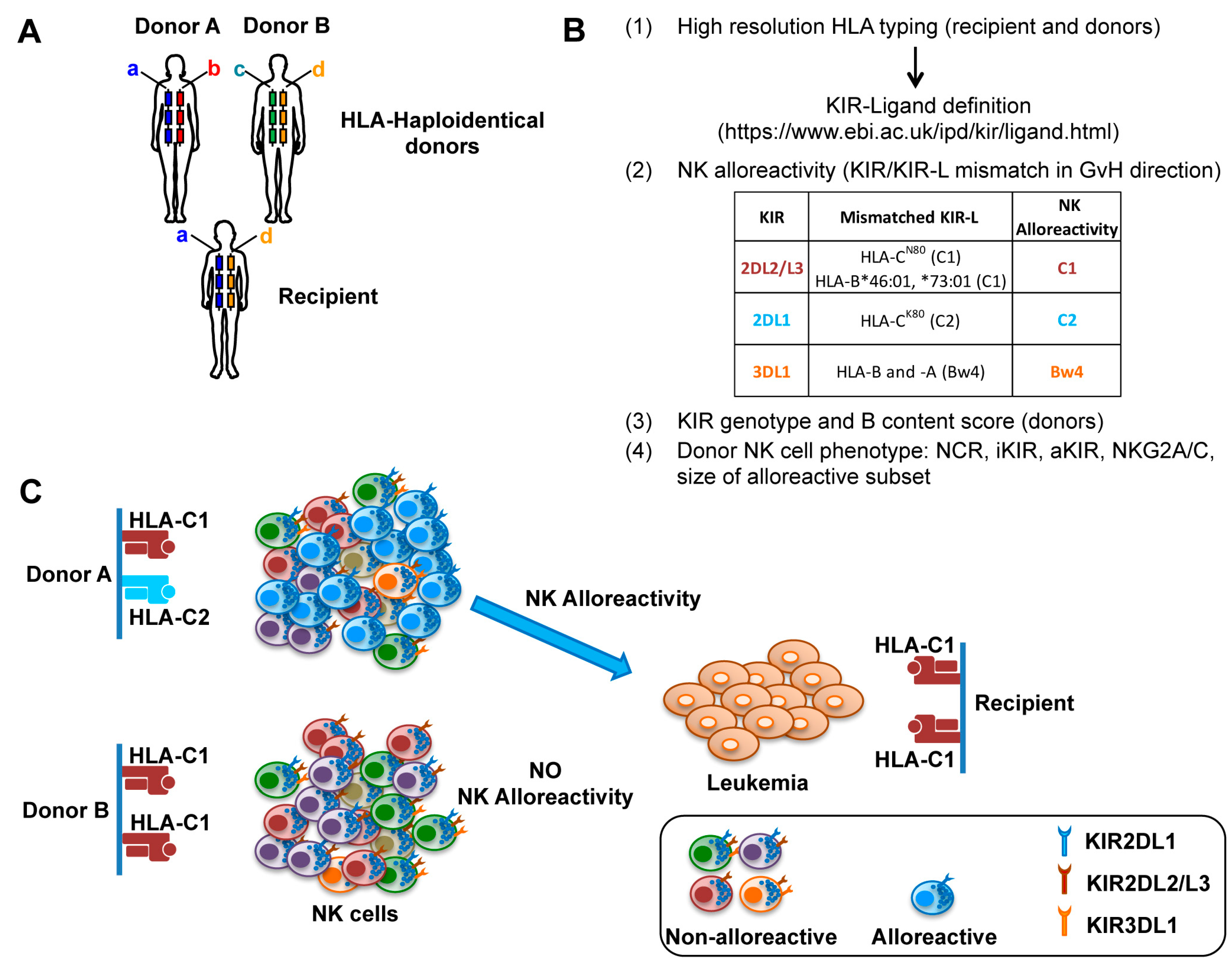
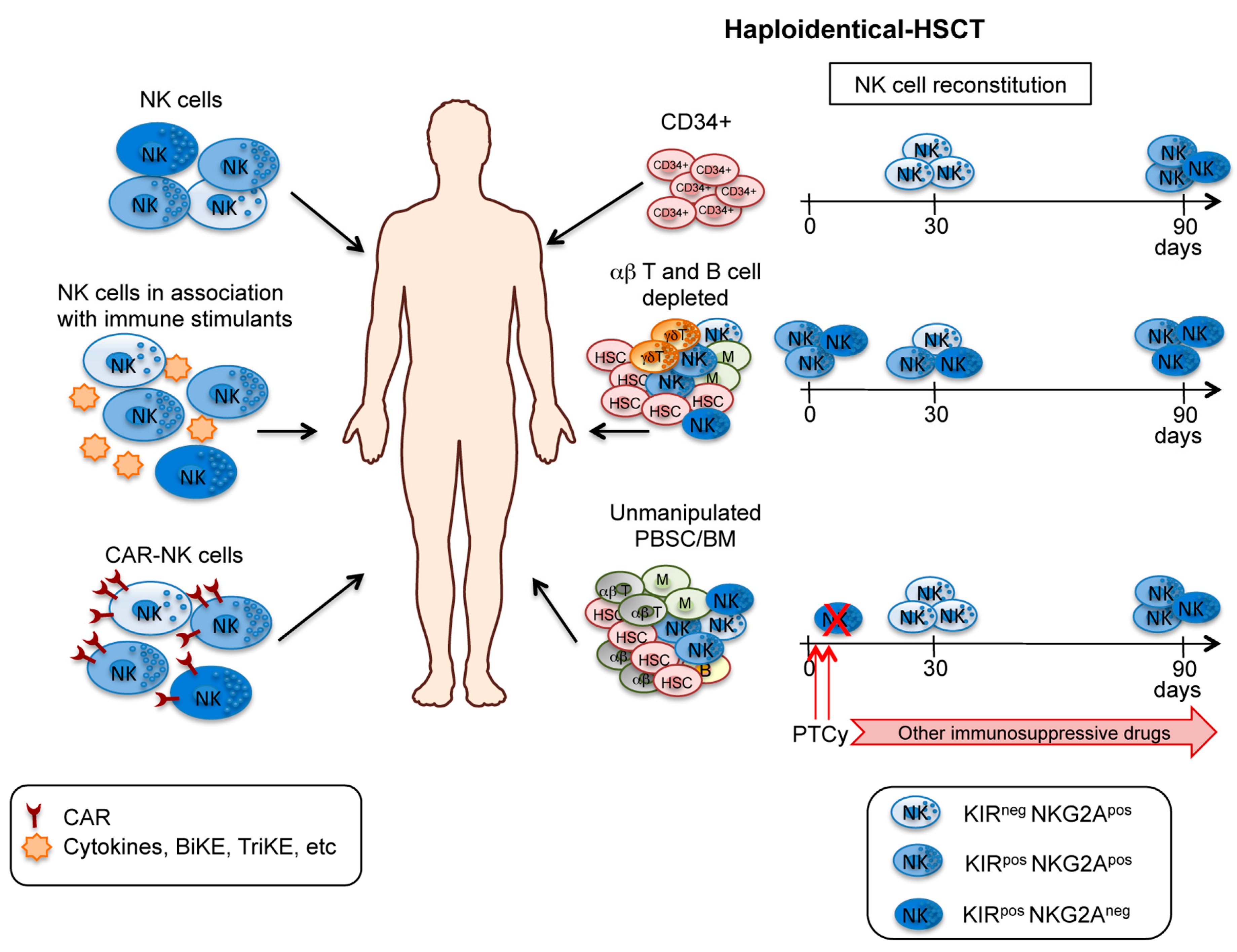
3. NK Cells in Haploidentical HSCT and Adoptive Immunotherapy
3.1. T Cell-Depleted and T Cell-Replete HSCT
3.2. Adoptive NK Cell Immunotherapy within Transplant Setting
3.3. Haploidentical HSCT after CD3/CD19 or TCRαβ/CD19 Depletion
3.4. Adoptive NK Cell Immunotherapy in Nontransplant Setting
4. Strategies to Induce NK Cell Activation, Persistence, and Expansion
4.1. Cytokine-Mediated NK cell Activation and Expansion to Improve Tumor Killing
4.2. CD16-Mediated Tumor Cell Killing to Cure Hematological Malignancies
4.3. Restoration of NK-Mediated Antitumor Responses by the Use of Antibodies Blocking Immune Checkpoints
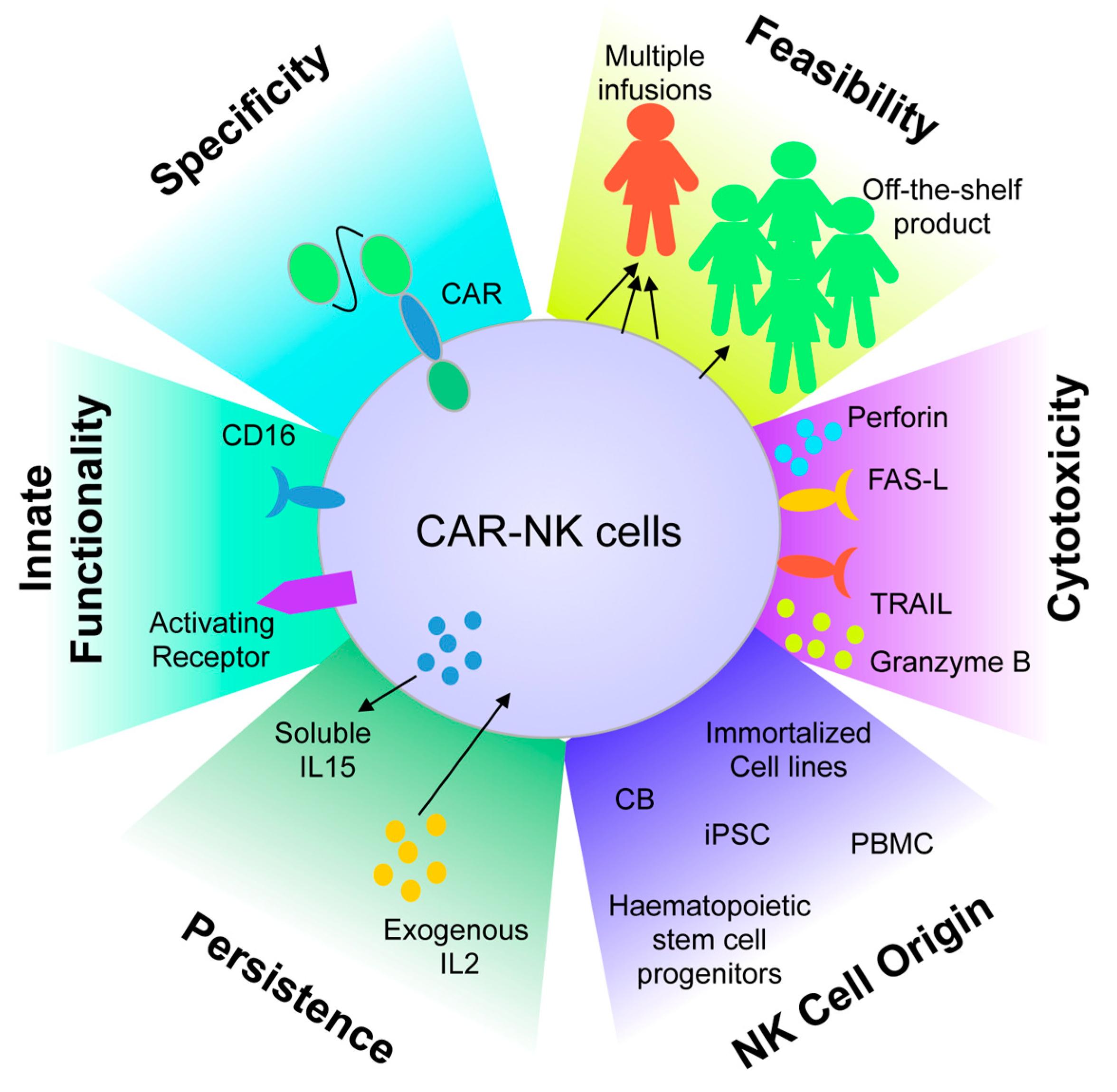
5. Adoptive Cell Therapy Using Chimeric Antigen Receptor-Engineered Natural Killer Cells
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef]
- Mariotti, F.R.; Quatrini, L.; Munari, E.; Vacca, P.; Moretta, L. Innate Lymphoid Cells: Expression of PD-1 and Other Checkpoints in Normal and Pathological Conditions. Front. Immunol. 2019, 10, 910. [Google Scholar] [CrossRef]
- Riggan, L.; Freud, A.G.; O’Sullivan, T.E. True Detective: Unraveling Group 1 Innate Lymphocyte Heterogeneity. Trends Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dhar, P.; Wu, J.D. NK Cell Plasticity in Cancer. J. Clin. Med. 2019, 8, 1492. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.M.; Yokoyama, W.M. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. 2011, 32, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Freud, A.G.; Mundy-Bosse, B.L.; Yu, J.; Caligiuri, M.A. The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity 2017, 47, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Parodi, M.; Pedrazzi, M.; Cantoni, C.; Averna, M.; Patrone, M.; Cavaletto, M.; Spertino, S.; Pende, D.; Balsamo, M.; Pietra, G.; et al. Natural Killer (NK)/melanoma cell interaction induces NK-mediated release of chemotactic High Mobility Group Box-1 (HMGB1) capable of amplifying NK cell recruitment. Oncoimmunology 2015, 4, e1052353. [Google Scholar] [CrossRef]
- Handgretinger, R.; Lang, P.; Andre, M.C. Exploitation of natural killer cells for the treatment of acute leukemia. Blood 2016, 127, 3341–3349. [Google Scholar] [CrossRef]
- Cooley, S.; Parham, P.; Miller, J.S. Strategies to activate NK cells to prevent relapse and induce remission following hematopoietic stem cell transplantation. Blood 2018, 131, 1053–1062. [Google Scholar] [CrossRef]
- Locatelli, F.; Pende, D.; Falco, M.; Della Chiesa, M.; Moretta, A.; Moretta, L. NK Cells Mediate a Crucial Graft-versus-Leukemia Effect in Haploidentical-HSCT to Cure High-Risk Acute Leukemia. Trends Immunol. 2018, 39, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Lanier, L.L. Natural Killer Cells in Cancer Immunotherapy. Annu. Rev. Cancer Biol. 2019, 3, 77–103. [Google Scholar] [CrossRef]
- Rea, D.; Henry, G.; Khaznadar, Z.; Etienne, G.; Guilhot, F.; Nicolini, F.; Guilhot, J.; Rousselot, P.; Huguet, F.; Legros, L.; et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: The IMMUNOSTIM study. Haematologica 2017, 102, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Ilander, M.; Olsson-Stromberg, U.; Schlums, H.; Guilhot, J.; Bruck, O.; Lahteenmaki, H.; Kasanen, T.; Koskenvesa, P.; Soderlund, S.; Hoglund, M.; et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia 2017, 31, 1108–1116. [Google Scholar] [CrossRef]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Biassoni, R.; Mingari, M.C.; Moretta, L. Receptors for HLA class-I molecules in human natural killer cells. Annu. Rev. Immunol. 1996, 14, 619–648. [Google Scholar] [CrossRef]
- Colonna, M.; Navarro, F.; Bellon, T.; Llano, M.; Garcia, P.; Samaridis, J.; Angman, L.; Cella, M.; Lopez-Botet, M. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J. Exp. Med. 1997, 186, 1809–1818. [Google Scholar] [CrossRef]
- Parham, P. MHC class I molecules and KIRs in human history, health and survival. Nat. Rev. Immunol. 2005, 5, 201–214. [Google Scholar] [CrossRef]
- Marsh, S.G.; Parham, P.; Dupont, B.; Geraghty, D.E.; Trowsdale, J.; Middleton, D.; Vilches, C.; Carrington, M.; Witt, C.; Guethlein, L.A.; et al. Killer-cell immunoglobulin-like receptor (KIR) nomenclature report, 2002. Hum. Immunol. 2003, 64, 648–654. [Google Scholar] [CrossRef]
- Biassoni, R.; Cantoni, C.; Falco, M.; Verdiani, S.; Bottino, C.; Vitale, M.; Conte, R.; Poggi, A.; Moretta, A.; Moretta, L. The human leukocyte antigen (HLA)-C-specific “activatory” or “inhibitory” natural killer cell receptors display highly homologous extracellular domains but differ in their transmembrane and intracytoplasmic portions. J. Exp. Med. 1996, 183, 645–650. [Google Scholar] [CrossRef]
- Olcese, L.; Cambiaggi, A.; Semenzato, G.; Bottino, C.; Moretta, A.; Vivier, E. Human killer cell activatory receptors for MHC class I molecules are included in a multimeric complex expressed by natural killer cells. J. Immunol. 1997, 158, 5083–5086. [Google Scholar]
- Lanier, L.L.; Corliss, B.C.; Wu, J.; Leong, C.; Phillips, J.H. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature 1998, 391, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Long, E.O. KIR2DL4 (CD158d): An activation receptor for HLA-G. Front. Immunol. 2012, 3, 258. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.M.; Kulski, J.K.; Gaudieri, S.; Witt, C.S.; Freitas, E.M.; Trowsdale, J.; Christiansen, F.T. Comparative genomic analysis, diversity and evolution of two KIR haplotypes A and B. Gene 2004, 335, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Pyo, C.W.; Guethlein, L.A.; Vu, Q.; Wang, R.; Abi-Rached, L.; Norman, P.J.; Marsh, S.G.; Miller, J.S.; Parham, P.; Geraghty, D.E. Different patterns of evolution in the centromeric and telomeric regions of group A and B haplotypes of the human killer cell Ig-like receptor locus. PLoS ONE 2010, 5, e15115. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Sivori, S.; Vitale, M.; Pende, D.; Morelli, L.; Augugliaro, R.; Bottino, C.; Moretta, L. Existence of both inhibitory (p58) and activatory (p50) receptors for HLA-C molecules in human natural killer cells. J. Exp. Med. 1995, 182, 875–884. [Google Scholar] [CrossRef]
- Pende, D.; Falco, M.; Vitale, M.; Cantoni, C.; Vitale, C.; Munari, E.; Bertaina, A.; Moretta, F.; Del Zotto, G.; Pietra, G.; et al. Killer Ig-Like Receptors (KIRs): Their Role in NK Cell Modulation and Developments Leading to Their Clinical Exploitation. Front. Immunol. 2019, 10, 1179. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Soderstrom, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Horowitz, A.; Djaoud, Z.; Nemat-Gorgani, N.; Blokhuis, J.; Hilton, H.G.; Beziat, V.; Malmberg, K.J.; Norman, P.J.; Guethlein, L.A.; Parham, P. Class I HLA haplotypes form two schools that educate NK cells in different ways. Sci. Immunol. 2016, 1, 1672. [Google Scholar] [CrossRef]
- Hallner, A.; Bernson, E.; Hussein, B.A.; Ewald Sander, F.; Brune, M.; Aurelius, J.; Martner, A.; Hellstrand, K.; Thoren, F.B. The HLA-B -21 dimorphism impacts on NK cell education and clinical outcome of immunotherapy in acute myeloid leukemia. Blood 2019, 133, 1479–1488. [Google Scholar] [CrossRef]
- Trinchieri, G. Biology of natural killer cells. Adv. Immunol. 1989, 47, 187–376. [Google Scholar]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Vitale, M.; Morelli, L.; Sanseverino, L.; Augugliaro, R.; Bottino, C.; Moretta, L.; Moretta, A. p46, a novel natural killer cell-specific surface molecule that mediates cell activation. J. Exp. Med. 1997, 186, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Pessino, A.; Sivori, S.; Bottino, C.; Malaspina, A.; Morelli, L.; Moretta, L.; Biassoni, R.; Moretta, A. Molecular Cloning of NKp46: A Novel Member of the Immunoglobulin Superfamily Involved in Triggering of Natural Cytotoxicity. J. Exp. Med. 1998, 188, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Bottino, C.; Sivori, S.; Sanseverino, L.; Castriconi, R.; Marcenaro, E.; Augugliaro, R.; Moretta, L.; Moretta, A. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med. 1998, 187, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, C.; Bottino, C.; Vitale, M.; Pessino, A.; Augugliaro, R.; Malaspina, A.; Parolini, S.; Moretta, L.; Moretta, A.; Biassoni, R. NKp44, A Triggering Receptor Involved in Tumor Cell Lysis by Activated Human Natural Killer Cells, Is a Novel Member of the Immunoglobulin Superfamily. J. Exp. Med. 1999, 189, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Pende, D.; Parolini, S.; Pessino, A.; Sivori, S.; Augugliaro, R.; Morelli, L.; Marcenaro, E.; Accame, L.; Malaspina, A.; Biassoni, R.; et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 1999, 190, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.S.; Yusa, S.; Kikuchi-Maki, A.; Catina, T.L. NKp44 triggers NK cell activation through DAP12 association that is not influenced by a putative cytoplasmic inhibitory sequence. J. Immunol. 2004, 172, 899–906. [Google Scholar] [CrossRef]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef]
- Parodi, M.; Favoreel, H.; Candiano, G.; Gaggero, S.; Sivori, S.; Mingari, M.C.; Moretta, L.; Vitale, M.; Cantoni, C. NKp44-NKp44 Ligand Interactions in the Regulation of Natural Killer Cells and Other Innate Lymphoid Cells in Humans. Front. Immunol. 2019, 10, 719. [Google Scholar] [CrossRef]
- Correia, D.V.; Fogli, M.; Hudspeth, K.; da Silva, M.G.; Mavilio, D.; Silva-Santos, B. Differentiation of human peripheral blood Vdelta1+ T cells expressing the natural cytotoxicity receptor NKp30 for recognition of lymphoid leukemia cells. Blood 2011, 118, 992–1001. [Google Scholar] [CrossRef]
- Pievani, A.; Borleri, G.; Pende, D.; Moretta, L.; Rambaldi, A.; Golay, J.; Introna, M. Dual-functional capability of CD3+CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood 2011, 118, 3301–3310. [Google Scholar] [CrossRef] [PubMed]
- Correia, M.P.; Stojanovic, A.; Bauer, K.; Juraeva, D.; Tykocinski, L.O.; Lorenz, H.M.; Brors, B.; Cerwenka, A. Distinct human circulating NKp30(+)FcepsilonRIγ(+)CD8(+) T cell population exhibiting high natural killer-like antitumor potential. Proc. Natl. Acad. Sci. USA 2018, 115, E5980–E5989. [Google Scholar] [CrossRef] [PubMed]
- Pende, D.; Spaggiari, G.M.; Marcenaro, S.; Martini, S.; Rivera, P.; Capobianco, A.; Falco, M.; Lanino, E.; Pierri, I.; Zambello, R.; et al. Analysis of the receptor-ligand interactions in the natural killer-mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: Evidence for the involvement of the Poliovirus receptor (CD155) and Nectin-2 (CD112). Blood 2005, 105, 2066–2073. [Google Scholar] [CrossRef] [PubMed]
- Barrow, A.D.; Martin, C.J.; Colonna, M. The Natural Cytotoxicity Receptors in Health and Disease. Front. Immunol. 2019, 10, 909. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Cantoni, C.; Della Chiesa, M.; Ferlazzo, G.; Carlomagno, S.; Pende, D.; Falco, M.; Pessino, A.; Muccio, L.; De Maria, A.; et al. An Historical Overview: The Discovery of How NK Cells Can Kill Enemies, Recruit Defense Troops, and More. Front. Immunol. 2019, 10, 1415. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.S.; Baratin, M.; Yi, E.C.; Kennedy, J.; Gao, Z.; Fox, B.; Haldeman, B.; Ostrander, C.D.; Kaifu, T.; Chabannon, C.; et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 2009, 206, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Baychelier, F.; Sennepin, A.; Ermonval, M.; Dorgham, K.; Debre, P.; Vieillard, V. Identification of a cellular ligand for the natural cytotoxicity receptor NKp44. Blood 2013, 122, 2935–2942. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, A.; Garcia-Beltran, W.F.; Norman, P.J.; Watson, G.M.; Holzemer, A.; Chapel, A.; Richert, L.; Pommerening-Roser, A.; Korner, C.; Ozawa, M.; et al. A subset of HLA-DP molecules serve as ligands for the natural cytotoxicity receptor NKp44. Nat. Immunol. 2019. [Google Scholar] [CrossRef]
- Rosental, B.; Brusilovsky, M.; Hadad, U.; Oz, D.; Appel, M.Y.; Afergan, F.; Yossef, R.; Rosenberg, L.A.; Aharoni, A.; Cerwenka, A.; et al. Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. J. Immunol. 2011, 187, 5693–5702. [Google Scholar] [CrossRef]
- Pogge von Strandmann, E.; Simhadri, V.R.; von Tresckow, B.; Sasse, S.; Reiners, K.S.; Hansen, H.P.; Rothe, A.; Boll, B.; Simhadri, V.L.; Borchmann, P.; et al. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity 2007, 27, 965–974. [Google Scholar] [CrossRef]
- Schlecker, E.; Fiegler, N.; Arnold, A.; Altevogt, P.; Rose-John, S.; Moldenhauer, G.; Sucker, A.; Paschen, A.; von Strandmann, E.P.; Textor, S.; et al. Metalloprotease-Mediated Tumor Cell Shedding of B7-H6, the Ligand of the Natural Killer Cell-Activating Receptor NKp30. Cancer Res. 2014, 74, 3429–3440. [Google Scholar] [CrossRef] [PubMed]
- Narni-Mancinelli, E.; Gauthier, L.; Baratin, M.; Guia, S.; Fenis, A.; Deghmane, A.E.; Rossi, B.; Fourquet, P.; Escaliere, B.; Kerdiles, Y.M.; et al. Complement factor P is a ligand for the natural killer cell-activating receptor NKp46. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Barrow, A.D.; Edeling, M.A.; Trifonov, V.; Luo, J.; Goyal, P.; Bohl, B.; Bando, J.K.; Kim, A.H.; Walker, J.; Andahazy, M.; et al. Natural Killer Cells Control Tumor Growth by Sensing a Growth Factor. Cell 2018, 172, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Gaggero, S.; Bruschi, M.; Petretto, A.; Parodi, M.; Del Zotto, G.; Lavarello, C.; Prato, C.; Santucci, L.; Barbuto, A.; Bottino, C.; et al. Nidogen-1 is a novel extracellular ligand for the NKp44 activating receptor. Oncoimmunology 2018, 7, e1470730. [Google Scholar] [CrossRef] [PubMed]
- Reiners, K.S.; Topolar, D.; Henke, A.; Simhadri, V.R.; Kessler, J.; Sauer, M.; Bessler, M.; Hansen, H.P.; Tawadros, S.; Herling, M.; et al. Soluble ligands for NK cell receptors promote evasion of chronic lymphocytic leukemia cells from NK cell anti-tumor activity. Blood 2013, 121, 3658–3665. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Pende, D.; Bottino, C.; Marcenaro, E.; Pessino, A.; Biassoni, R.; Moretta, L.; Moretta, A. NKp46 is the major triggering receptor involved in the natural cytotoxicity of fresh or cultured human NK cells. Correlation between surface density of NKp46 and natural cytotoxicity against autologous, allogeneic or xenogeneic target cells. Eur. J. Immunol. 1999, 29, 1656–1666. [Google Scholar] [CrossRef]
- Costello, R.T.; Sivori, S.; Marcenaro, E.; Lafage-Pochitaloff, M.; Mozziconacci, M.J.; Reviron, D.; Gastaut, J.A.; Pende, D.; Olive, D.; Moretta, A. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 2002, 99, 3661–3667. [Google Scholar] [CrossRef]
- Fauriat, C.; Just-Landi, S.; Mallet, F.; Arnoulet, C.; Sainty, D.; Olive, D.; Costello, R.T. Deficient expression of NCR in NK cells from acute myeloid leukemia: Evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood 2007, 109, 323–330. [Google Scholar] [CrossRef]
- Costello, R.T.; Knoblauch, B.; Sanchez, C.; Mercier, D.; Le Treut, T.; Sébahoun, G. Expression of natural killer cell activating receptors in patients with chronic lymphocytic leukaemia. Immunology 2012, 135, 151–157. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef]
- Castriconi, R.; Dondero, A.; Bellora, F.; Moretta, L.; Castellano, A.; Locatelli, F.; Corrias, M.V.; Moretta, A.; Bottino, C. Neuroblastoma-derived TGF-beta1 modulates the chemokine receptor repertoire of human resting NK cells. J. Immunol. 2013, 190, 5321–5328. [Google Scholar] [CrossRef] [PubMed]
- Spaggiari, G.M.; Capobianco, A.; Abdelrazik, H.; Becchetti, F.; Mingari, M.C.; Moretta, L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: Role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood 2008, 111, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Stabile, H.; Fionda, C.; Gismondi, A.; Santoni, A. Role of Distinct Natural Killer Cell Subsets in Anticancer Response. Front. Immunol. 2017, 8, 293. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Souza-Fonseca-Guimaraes, F.; Bald, T.; Ng, S.S.; Young, A.; Ngiow, S.F.; Rautela, J.; Straube, J.; Waddell, N.; Blake, S.J.; et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017, 18, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Bahram, S.; Bauer, S.; Herman, A.; Beauchamp, M.; Spies, T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc. Natl. Acad. Sci. USA 1996, 93, 12445–12450. [Google Scholar] [CrossRef]
- Pende, D.; Rivera, P.; Marcenaro, S.; Chang, C.C.; Biassoni, R.; Conte, R.; Kubin, M.; Cosman, D.; Ferrone, S.; Moretta, L.; et al. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: Analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. 2002, 62, 6178–6186. [Google Scholar]
- Hilpert, J.; Grosse-Hovest, L.; Grünebach, F.; Buechele, C.; Nuebling, T.; Raum, T.; Steinle, A.; Salih, H.R. Comprehensive Analysis of NKG2D Ligand Expression and Release in Leukemia: Implications for NKG2D-Mediated NK Cell Responses. J. Immunol. 2012, 189, 1360–1371. [Google Scholar] [CrossRef]
- Torelli, G.F.; Peragine, N.; Raponi, S.; Pagliara, D.; De Propris, M.S.; Vitale, A.; Bertaina, A.; Barberi, W.; Moretta, L.; Basso, G.; et al. Recognition of adult and pediatric acute lymphoblastic leukemia blasts by natural killer cells. Haematologica 2014, 99, 1248–1254. [Google Scholar] [CrossRef][Green Version]
- Shibuya, A.; Campbell, D.; Hannum, C.; Yssel, H.; Franz-Bacon, K.; McClanahan, T.; Kitamura, T.; Nicholl, J.; Sutherland, G.R.; Lanier, L.L.; et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity 1996, 4, 573–581. [Google Scholar] [CrossRef]
- Sivori, S.; Parolini, S.; Falco, M.; Marcenaro, E.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. 2B4 functions as a co-receptor in human NK cell activation. Eur. J. Immunol. 2000, 30, 787–793. [Google Scholar] [CrossRef]
- Bottino, C.; Falco, M.; Parolini, S.; Marcenaro, E.; Augugliaro, R.; Sivori, S.; Landi, E.; Biassoni, R.; Notarangelo, L.D.; Moretta, L.; et al. NTB-A [correction of GNTB-A], a novel SH2D1A-associated surface molecule contributing to the inability of natural killer cells to kill Epstein-Barr virus-infected B cells in X-linked lymphoproliferative disease. J. Exp. Med. 2001, 194, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Marcenaro, E.; Augugliaro, R.; Falco, M.; Castriconi, R.; Parolini, S.; Sivori, S.; Romeo, E.; Millo, R.; Moretta, L.; Bottino, C.; et al. CD59 is physically and functionally associated with natural cytotoxicity receptors and activates human NK cell-mediated cytotoxicity. Eur. J. Immunol. 2003, 33, 3367–3376. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Falco, M.; Castriconi, R.; Parolini, S.; Zambello, R.; Semenzato, G.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Identification of NKp80, a novel triggering molecule expressed by human NK cells. Eur. J. Immunol. 2001, 31, 233–242. [Google Scholar] [CrossRef]
- Bottino, C.; Castriconi, R.; Pende, D.; Rivera, P.; Nanni, M.; Carnemolla, B.; Cantoni, C.; Grassi, J.; Marcenaro, S.; Reymond, N.; et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med. 2003, 198, 557–567. [Google Scholar] [CrossRef] [PubMed]
- McArdel, S.L.; Terhorst, C.; Sharpe, A.H. Roles of CD48 in regulating immunity and tolerance. Clin. Immunol. 2016, 164, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Falco, M.; Carlomagno, S.; Romeo, E.; Moretta, L.; Moretta, A. Heterogeneity of TLR3 mRNA transcripts and responsiveness to poly (I:C) in human NK cells derived from different donors. Int. Immunol. 2007, 19, 1341–1348. [Google Scholar] [CrossRef]
- Sivori, S.; Falco, M.; Carlomagno, S.; Romeo, E.; Soldani, C.; Bensussan, A.; Viola, A.; Moretta, L.; Moretta, A. A novel KIR-associated function: Evidence that CpG DNA uptake and shuttling to early endosomes is mediated by KIR3DL2. Blood 2010, 116, 1637–1647. [Google Scholar] [CrossRef]
- Chiossone, L.; Vienne, M.; Kerdiles, Y.M.; Vivier, E. Natural killer cell immunotherapies against cancer: Checkpoint inhibitors and more. Semin. Immunol. 2017, 31, 55–63. [Google Scholar] [CrossRef]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef]
- Guan, J.; Wang, R.; Hasan, S.; Tao, L.; Wazir, M.; Jain, A.G.; Zhu, X.; Perkins, S.; Mohamed, S.; Chang, C.C.; et al. Prognostic Significance of the Dynamic Change of Programmed Death-ligand 1 Expression in Patients with Multiple Myeloma. Cureus 2019, 11, e4401. [Google Scholar] [CrossRef] [PubMed]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Pesce, S.; Greppi, M.; Grossi, F.; Del Zotto, G.; Moretta, L.; Sivori, S.; Genova, C.; Marcenaro, E. PD/1-PD-Ls Checkpoint: Insight on the Potential Role of NK Cells. Front. Immunol. 2019, 10, 1242. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Hobo, W.; Hutten, T.J.A.; Schaap, N.P.M.; Dolstra, H. Immune checkpoint molecules in acute myeloid leukaemia: Managing the double-edged sword. Br. J. Haematol. 2018, 181, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD96: New checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef]
- Zhou, X.M.; Li, W.Q.; Wu, Y.H.; Han, L.; Cao, X.G.; Yang, X.M.; Wang, H.F.; Zhao, W.S.; Zhai, W.J.; Qi, Y.M.; et al. Intrinsic Expression of Immune Checkpoint Molecule TIGIT Could Help Tumor Growth in vivo by Suppressing the Function of NK and CD8(+) T Cells. Front. Immunol. 2018, 9, 2821. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- da Silva, I.P.; Gallois, A.; Jimenez-Baranda, S.; Khan, S.; Anderson, A.C.; Kuchroo, V.K.; Osman, I.; Bhardwaj, N. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol. Res. 2014, 2, 410–422. [Google Scholar] [CrossRef]
- Huard, B.; Tournier, M.; Triebel, F. LAG-3 does not define a specific mode of natural killing in human. Immunol. Lett. 1998, 61, 109–112. [Google Scholar] [CrossRef]
- Miyazaki, T.; Dierich, A.; Benoist, C.; Mathis, D. Independent modes of natural killing distinguished in mice lacking Lag3. Science 1996, 272, 405–408. [Google Scholar] [CrossRef]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Akiba, H.; Takeda, K.; Kojima, Y.; Hashiguchi, M.; Azuma, M.; Yagita, H.; Okumura, K. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 2009, 113, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Bialoszewska, A.; Malejczyk, J. Biological and Clinical Significance of Human NKRP1A/LLT1 Receptor/Ligand Interactions. Crit. Rev. Immunol. 2018, 38, 479–489. [Google Scholar] [CrossRef]
- Germain, C.; Guillaudeux, T.; Galsgaard, E.D.; Hervouet, C.; Tekaya, N.; Gallouet, A.S.; Fassy, J.; Bihl, F.; Poupon, G.; Lazzari, A.; et al. Lectin-like transcript 1 is a marker of germinal center-derived B-cell non-Hodgkin’s lymphomas dampening natural killer cell functions. Oncoimmunology 2015, 4, e1026503. [Google Scholar] [CrossRef]
- Poggi, A.; Costa, P.; Tomasello, E.; Moretta, L. IL-12-induced up-regulation of NKRP1A expression in human NK cells and consequent NKRP1A-mediated down-regulation of NK cell activation. Eur. J. Immunol. 1998, 28, 1611–1616. [Google Scholar] [CrossRef]
- Azzoni, L.; Zatsepina, O.; Abebe, B.; Bennett, I.M.; Kanakaraj, P.; Perussia, B. Differential transcriptional regulation of CD161 and a novel gene, 197/15a, by IL-2, IL-15, and IL-12 in NK and T cells. J. Immunol. 1998, 161, 3493–3500. [Google Scholar]
- Rocha, V.; Locatelli, F. Searching for alternative hematopoietic stem cell donors for pediatric patients. Bone Marrow Transplant. 2007, 41, 207–214. [Google Scholar] [CrossRef]
- Aversa, F.; Tabilio, A.; Velardi, A.; Cunningham, I.; Terenzi, A.; Falzetti, F.; Ruggeri, L.; Barbabietola, G.; Aristei, C.; Latini, P.; et al. Treatment of high-risk acute leukemia with T-cell-depleted stem cells from related donors with one fully mismatched HLA haplotype. N. Engl. J. Med. 1998, 339, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, L.; Mancusi, A.; Capanni, M.; Urbani, E.; Carotti, A.; Aloisi, T.; Stern, M.; Pende, D.; Perruccio, K.; Burchielli, E.; et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: Challenging its predictive value. Blood 2007, 110, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Reisner, Y.; Hagin, D.; Martelli, M.F. Haploidentical hematopoietic transplantation: Current status and future perspectives. Blood 2011, 118, 6006–6017. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Dhedin, N.; Vernant, J.P.; Kuentz, M.; Al Jijakli, A.; Rouas-Freiss, N.; Carosella, E.D.; Boudifa, A.; Debre, P.; Vieillard, V. NK-cell reconstitution after haploidentical hematopoietic stem-cell transplantations: Immaturity of NK cells and inhibitory effect of NKG2A override GvL effect. Blood 2005, 105, 4135–4142. [Google Scholar] [CrossRef]
- Pende, D.; Marcenaro, S.; Falco, M.; Martini, S.; Bernardo, M.E.; Montagna, D.; Romeo, E.; Cognet, C.; Martinetti, M.; Maccario, R.; et al. Anti-leukemia activity of alloreactive NK cells in KIR ligand-mismatched haploidentical HSCT for pediatric patients: Evaluation of the functional role of activating KIR and redefinition of inhibitory KIR specificity. Blood 2009, 113, 3119–3129. [Google Scholar] [CrossRef]
- Luznik, L.; O’Donnell, P.V.; Fuchs, E.J. Post-transplantation cyclophosphamide for tolerance induction in HLA-haploidentical bone marrow transplantation. Semin. Oncol. 2012, 39, 683–693. [Google Scholar] [CrossRef]
- Russo, A.; Oliveira, G.; Berglund, S.; Greco, R.; Gambacorta, V.; Cieri, N.; Toffalori, C.; Zito, L.; Lorentino, F.; Piemontese, S.; et al. NK cell recovery after haploidentical HSCT with posttransplant cyclophosphamide: Dynamics and clinical implications. Blood 2018, 131, 247–262. [Google Scholar] [CrossRef]
- Shimoni, A.; Labopin, M.; Lorentino, F.; Van Lint, M.T.; Koc, Y.; Gulbas, Z.; Tischer, J.; Bruno, B.; Blaise, D.; Pioltelli, P.; et al. Killer cell immunoglobulin-like receptor ligand mismatching and outcome after haploidentical transplantation with post-transplant cyclophosphamide. Leukemia 2019, 33, 230–239. [Google Scholar] [CrossRef]
- Veluchamy, J.P.; Kok, N.; van der Vliet, H.J.; Verheul, H.M.W.; de Gruijl, T.D.; Spanholtz, J. The Rise of Allogeneic Natural Killer Cells as a Platform for Cancer Immunotherapy: Recent Innovations and Future Developments. Front. Immunol. 2017, 8, 631. [Google Scholar] [CrossRef]
- Van Elssen, C.; Ciurea, S.O. NK cell therapy after hematopoietic stem cell transplantation: Can we improve anti-tumor effect? Int. J. Hematol. 2018, 107, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Lupo, K.B.; Matosevic, S. Natural Killer Cells as Allogeneic Effectors in Adoptive Cancer Immunotherapy. Cancers 2019, 11, 769. [Google Scholar] [CrossRef] [PubMed]
- Passweg, J.R.; Tichelli, A.; Meyer-Monard, S.; Heim, D.; Stern, M.; Kühne, T.; Favre, G.; Gratwohl, A. Purified donor NK-lymphocyte infusion to consolidate engraftment after haploidentical stem cell transplantation. Leukemia 2004, 18, 1835–1838. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.; Passweg, J.R.; Meyer-Monard, S.; Esser, R.; Tonn, T.; Soerensen, J.; Paulussen, M.; Gratwohl, A.; Klingebiel, T.; Bader, P.; et al. Pre-emptive immunotherapy with purified natural killer cells after haploidentical SCT: A prospective phase II study in two centers. Bone Marrow Transplant. 2013, 48, 433–438. [Google Scholar] [CrossRef]
- Killig, M.; Friedrichs, B.; Meisig, J.; Gentilini, C.; Bluthgen, N.; Loddenkemper, C.; Labopin, M.; Basara, N.; Pfrepper, C.; Niederwieser, D.W.; et al. Tracking in vivo dynamics of NK cells transferred in patients undergoing stem cell transplantation. Eur. J. Immunol. 2014, 44, 2822–2834. [Google Scholar] [CrossRef]
- Jaiswal, S.R.; Zaman, S.; Nedunchezhian, M.; Chakrabarti, A.; Bhakuni, P.; Ahmed, M.; Sharma, K.; Rawat, S.; O’Donnell, P.; Chakrabarti, S. CD56-enriched donor cell infusion after post-transplantation cyclophosphamide for haploidentical transplantation of advanced myeloid malignancies is associated with prompt reconstitution of mature natural killer cells and regulatory T cells with reduced incidence of acute graft versus host disease: A pilot study. Cytotherapy 2017, 19, 531–542. [Google Scholar] [CrossRef]
- Ciurea, S.O.; Schafer, J.R.; Bassett, R.; Denman, C.J.; Cao, K.; Willis, D.; Rondon, G.; Chen, J.; Soebbing, D.; Kaur, I.; et al. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood 2017, 130, 1857–1868. [Google Scholar] [CrossRef]
- Brehm, C.; Huenecke, S.; Quaiser, A.; Esser, R.; Bremm, M.; Kloess, S.; Soerensen, J.; Kreyenberg, H.; Seidl, C.; Becker, P.S.; et al. IL-2 stimulated but not unstimulated NK cells induce selective disappearance of peripheral blood cells: Concomitant results to a phase I/II study. PLoS ONE 2011, 6, e27351. [Google Scholar] [CrossRef]
- Koehl, U.; Kalberer, C.; Spanholtz, J.; Lee, D.A.; Miller, J.S.; Cooley, S.; Lowdell, M.; Uharek, L.; Klingemann, H.; Curti, A.; et al. Advances in clinical NK cell studies: Donor selection, manufacturing and quality control. Oncoimmunology 2016, 5, e1115178. [Google Scholar] [CrossRef]
- Handgretinger, R. New approaches to graft engineering for haploidentical bone marrow transplantation. Semin. Oncol. 2012, 39, 664–673. [Google Scholar] [CrossRef]
- Bethge, W.A.; Haegele, M.; Faul, C.; Lang, P.; Schumm, M.; Bornhauser, M.; Handgretinger, R.; Kanz, L. Haploidentical allogeneic hematopoietic cell transplantation in adults with reduced-intensity conditioning and CD3/CD19 depletion: Fast engraftment and low toxicity. Exp. Hematol. 2006, 34, 1746–1752. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.; Teltschik, H.M.; Feuchtinger, T.; Muller, I.; Pfeiffer, M.; Schumm, M.; Ebinger, M.; Schwarze, C.P.; Gruhn, B.; Schrauder, A.; et al. Transplantation of CD3/CD19 depleted allografts from haploidentical family donors in paediatric leukaemia. Br. J. Haematol. 2014, 165, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Bertaina, A.; Merli, P.; Rutella, S.; Pagliara, D.; Bernardo, M.E.; Masetti, R.; Pende, D.; Falco, M.; Handgretinger, R.; Moretta, F.; et al. HLA-haploidentical stem cell transplantation after removal of αβ+ T and B cells in children with nonmalignant disorders. Blood 2014, 124, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Li Pira, G.; Malaspina, D.; Girolami, E.; Biagini, S.; Cicchetti, E.; Conflitti, G.; Broglia, M.; Ceccarelli, S.; Lazzaro, S.; Pagliara, D.; et al. Selective Depletion of αβ T Cells and B Cells for Human Leukocyte Antigen-Haploidentical Hematopoietic Stem Cell Transplantation. A Three-Year Follow-Up of Procedure Efficiency. Biol. Blood Marrow Transplant. 2016, 22, 2056–2064. [Google Scholar] [CrossRef]
- Airoldi, I.; Bertaina, A.; Prigione, I.; Zorzoli, A.; Pagliara, D.; Cocco, C.; Meazza, R.; Loiacono, F.; Lucarelli, B.; Bernardo, M.E.; et al. gammadelta T cell reconstitution after HLA-haploidentical hematopoietic transplantation depleted of TCR-αβ+/CD19+ lymphocytes. Blood 2015. [Google Scholar] [CrossRef]
- Locatelli, F.; Merli, P.; Pagliara, D.; Li Pira, G.; Falco, M.; Pende, D.; Rondelli, R.; Lucarelli, B.; Brescia, L.P.; Masetti, R.; et al. Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood 2017, 130, 677–685. [Google Scholar] [CrossRef]
- Cooley, S.; Weisdorf, D.J.; Guethlein, L.A.; Klein, J.P.; Wang, T.; Le, C.T.; Marsh, S.G.; Geraghty, D.; Spellman, S.; Haagenson, M.D.; et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood 2010, 116, 2411–2419. [Google Scholar] [CrossRef]
- Moretta, L.; Locatelli, F.; Pende, D.; Marcenaro, E.; Mingari, M.C.; Moretta, A. Killer Ig-like receptor-mediated control of natural killer cell alloreactivity in haploidentical hematopoietic stem cell transplantation. Blood 2011, 117, 764–771. [Google Scholar] [CrossRef]
- Oevermann, L.; Michaelis, S.U.; Mezger, M.; Lang, P.; Toporski, J.; Bertaina, A.; Zecca, M.; Moretta, L.; Locatelli, F.; Handgretinger, R. KIR B haplotype donors confer a reduced risk for relapse after haploidentical transplantation in children with ALL. Blood 2014, 124, 2744–2747. [Google Scholar] [CrossRef]
- Venstrom, J.M.; Pittari, G.; Gooley, T.A.; Chewning, J.H.; Spellman, S.; Haagenson, M.; Gallagher, M.M.; Malkki, M.; Petersdorf, E.; Dupont, B.; et al. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N. Engl. J. Med. 2012, 367, 805–816. [Google Scholar] [CrossRef]
- Ciurea, S.O.; Al Malki, M.M.; Kongtim, P.; Fuchs, E.J.; Luznik, L.; Huang, X.J.; Ciceri, F.; Locatelli, F.; Aversa, F.; Castagna, L.; et al. The European Society for Blood and Marrow Transplantation (EBMT) consensus recommendations for donor selection in haploidentical hematopoietic cell transplantation. Bone Marrow Transplant. 2019. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed]
- Cooley, S.; He, F.; Bachanova, V.; Vercellotti, G.M.; DeFor, T.E.; Curtsinger, J.M.; Robertson, P.; Grzywacz, B.; Conlon, K.C.; Waldmann, T.A.; et al. First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv. 2019, 3, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Rubnitz, J.E.; Inaba, H.; Ribeiro, R.C.; Pounds, S.; Rooney, B.; Bell, T.; Pui, C.H.; Leung, W. NKAML: A pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Ruggeri, L.; D’Addio, A.; Bontadini, A.; Dan, E.; Motta, M.R.; Trabanelli, S.; Giudice, V.; Urbani, E.; Martinelli, G.; et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood 2011, 118, 3273–3279. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Ruggeri, L.; Parisi, S.; Bontadini, A.; Dan, E.; Motta, M.R.; Rizzi, S.; Trabanelli, S.; Ocadlikova, D.; Lecciso, M.; et al. Larger Size of Donor Alloreactive NK Cell Repertoire Correlates with Better Response to NK Cell Immunotherapy in Elderly Acute Myeloid Leukemia Patients. Clin. Cancer Res. 2016, 22, 1914–1921. [Google Scholar] [CrossRef]
- Parisi, S.; Lecciso, M.; Ocadlikova, D.; Salvestrini, V.; Ciciarello, M.; Forte, D.; Corradi, G.; Cavo, M.; Curti, A. The More, The Better: “Do the Right Thing” For Natural Killer Immunotherapy in Acute Myeloid Leukemia. Front. Immunol. 2017, 8, 1330. [Google Scholar] [CrossRef]
- Floros, T.; Tarhini, A.A. Anticancer Cytokines: Biology and Clinical Effects of Interferon-α2, Interleukin (IL)-2, IL-15, IL-21, and IL-12. Semin. Oncol. 2015, 42, 539–548. [Google Scholar] [CrossRef]
- Srivastava, S.; Pelloso, D.; Feng, H.; Voiles, L.; Lewis, D.; Haskova, Z.; Whitacre, M.; Trulli, S.; Chen, Y.J.; Toso, J.; et al. Effects of interleukin-18 on natural killer cells: Costimulation of activation through Fc receptors for immunoglobulin. Cancer Immunol. Immunother. 2013, 62, 1073–1082. [Google Scholar] [CrossRef]
- Davis, Z.B.; Felices, M.; Verneris, M.R.; Miller, J.S. Natural Killer Cell Adoptive Transfer Therapy: Exploiting the First Line of Defense against Cancer. Cancer J. 2015, 21, 486–491. [Google Scholar] [CrossRef]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Mule, J.J.; Spiess, P.J.; Reichert, C.M.; Schwarz, S.L. Regression of established pulmonary metastases and subcutaneous tumor mediated by the systemic administration of high-dose recombinant interleukin 2. J. Exp. Med. 1985, 161, 1169–1188. [Google Scholar] [CrossRef] [PubMed]
- Delespine-Carmagnat, M.; Bouvier, G.; Bertoglio, J. Association of STAT1, STAT3 and STAT5 proteins with the IL-2 receptor involves different subdomains of the IL-2 receptor beta chain. Eur. J. Immunol. 2000, 30, 59–68. [Google Scholar] [CrossRef]
- Kovanen, P.E.; Leonard, W.J. Cytokines and immunodeficiency diseases: Critical roles of the γ(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol. Rev. 2004, 202, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Chang, A.E.; Avis, F.P.; Leitman, S.; Linehan, W.M.; Robertson, C.N.; Lee, R.E.; Rubin, J.T.; et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N. Engl. J. Med. 1987, 316, 889–897. [Google Scholar] [CrossRef]
- Dutcher, J.P.; Gaynor, E.R.; Boldt, D.H.; Doroshow, J.H.; Bar, M.H.; Sznol, M.; Mier, J.; Sparano, J.; Fisher, R.I.; Weiss, G.; et al. A phase II study of high-dose continuous infusion interleukin-2 with lymphokine-activated killer cells in patients with metastatic melanoma. J. Clin. Oncol. 1991, 9, 641–648. [Google Scholar] [CrossRef]
- Chang, A.E.; Hyatt, C.L.; Rosenberg, S.A. Systemic administration of recombinant human interleukin-2 in mice. J. Biol. Response Mod. 1984, 3, 561–572. [Google Scholar]
- Kruit, W.H.; Punt, C.J.; Goey, S.H.; de Mulder, P.H.; Gratama, J.W.; Eggermont, A.M.; Bolhuis, R.L.; Stoter, G. Dose efficacy study of two schedules of high-dose bolus administration of interleukin 2 and interferon α in metastatic melanoma. Br. J. Cancer 1996, 74, 951–955. [Google Scholar] [CrossRef]
- Kruit, W.H.; Punt, K.J.; Goey, S.H.; de Mulder, P.H.; van Hoogenhuyze, D.C.; Henzen-Logmans, S.C.; Stoter, G. Cardiotoxicity as a dose-limiting factor in a schedule of high dose bolus therapy with interleukin-2 and α-interferon. An unexpectedly frequent complication. Cancer 1994, 74, 2850–2856. [Google Scholar] [CrossRef]
- Lotze, M.T.; Matory, Y.L.; Ettinghausen, S.E.; Rayner, A.A.; Sharrow, S.O.; Seipp, C.A.; Custer, M.C.; Rosenberg, S.A. In vivo administration of purified human interleukin 2. II. Half life, immunologic effects, and expansion of peripheral lymphoid cells in vivo with recombinant IL 2. J. Immunol. 1985, 135, 2865–2875. [Google Scholar]
- Ghiringhelli, F.; Menard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, G.; Hemmers, S.; Firth, M.A.; Le Floc’h, A.; Huse, M.; Sun, J.C.; Rudensky, A.Y. IL-2-dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J. Exp. Med. 2013, 210, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Sitrin, J.; Ring, A.; Garcia, K.C.; Benoist, C.; Mathis, D. Regulatory T cells control NK cells in an insulitic lesion by depriving them of IL-2. J. Exp. Med. 2013, 210, 1153–1165. [Google Scholar] [CrossRef]
- Miller, J.S.; Tessmer-Tuck, J.; Pierson, B.A.; Weisdorf, D.; McGlave, P.; Blazar, B.R.; Katsanis, E.; Verfaillie, C.; Lebkowski, J.; Radford, J., Jr.; et al. Low dose subcutaneous interleukin-2 after autologous transplantation generates sustained in vivo natural killer cell activity. Biol. Blood Marrow Transplant. 1997, 3, 34–44. [Google Scholar]
- Bachanova, V.; Burns, L.J.; McKenna, D.H.; Curtsinger, J.; Panoskaltsis-Mortari, A.; Lindgren, B.R.; Cooley, S.; Weisdorf, D.; Miller, J.S. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol. Immunother. 2010, 59, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Geller, M.A.; Miller, J.S. Use of allogeneic NK cells for cancer immunotherapy. Immunotherapy 2011, 3, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.M.; Bates, D.L.; Ring, A.M.; Krieg, C.; Lin, J.T.; Su, L.; Moraga, I.; Raeber, M.E.; Bowman, G.R.; Novick, P.; et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature 2012, 484, 529–533. [Google Scholar] [CrossRef]
- Sim, G.C.; Liu, C.; Wang, E.; Liu, H.; Creasy, C.; Dai, Z.; Overwijk, W.W.; Roszik, J.; Marincola, F.; Hwu, P.; et al. IL2 Variant Circumvents ICOS+ Regulatory T-cell Expansion and Promotes NK Cell Activation. Cancer Immunol. Res. 2016, 4, 983–994. [Google Scholar] [CrossRef]
- Carmenate, T.; Pacios, A.; Enamorado, M.; Moreno, E.; Garcia-Martinez, K.; Fuente, D.; Leon, K. Human IL-2 mutein with higher antitumor efficacy than wild type IL-2. J. Immunol. 2013, 190, 6230–6238. [Google Scholar] [CrossRef]
- Waldmann, T.A. The biology of interleukin-2 and interleukin-15: Implications for cancer therapy and vaccine design. Nat. Rev. Immunol. 2006, 6, 595–601. [Google Scholar] [CrossRef]
- Grabstein, K.H.; Eisenman, J.; Shanebeck, K.; Rauch, C.; Srinivasan, S.; Fung, V.; Beers, C.; Richardson, J.; Schoenborn, M.A.; Ahdieh, M.; et al. Cloning of a T cell growth factor that interacts with the beta chain of the interleukin-2 receptor. Science 1994, 264, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Giri, J.G.; Kumaki, S.; Ahdieh, M.; Friend, D.J.; Loomis, A.; Shanebeck, K.; DuBose, R.; Cosman, D.; Park, L.S.; Anderson, D.M. Identification and cloning of a novel IL-15 binding protein that is structurally related to the α chain of the IL-2 receptor. EMBO J. 1995, 14, 3654–3663. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Kumaki, S.; Ahdieh, M.; Bertles, J.; Tometsko, M.; Loomis, A.; Giri, J.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; et al. Functional characterization of the human interleukin-15 receptor α chain and close linkage of IL15RA and IL2RA genes. J. Biol. Chem. 1995, 270, 29862–29869. [Google Scholar] [CrossRef] [PubMed]
- Mortier, E.; Advincula, R.; Kim, L.; Chmura, S.; Barrera, J.; Reizis, B.; Malynn, B.A.; Ma, A. Macrophage- and dendritic-cell-derived interleukin-15 receptor alpha supports homeostasis of distinct CD8+ T cell subsets. Immunity 2009, 31, 811–822. [Google Scholar] [CrossRef]
- Rubinstein, M.P.; Kovar, M.; Purton, J.F.; Cho, J.H.; Boyman, O.; Surh, C.D.; Sprent, J. Converting IL-15 to a superagonist by binding to soluble IL-15R{α}. Proc. Natl. Acad. Sci. USA 2006, 103, 9166–9171. [Google Scholar] [CrossRef]
- Wong, H.C.; Jeng, E.K.; Rhode, P.R. The IL-15-based superagonist ALT-803 promotes the antigen-independent conversion of memory CD8(+) T cells into innate-like effector cells with antitumor activity. Oncoimmunology 2013, 2, e26442. [Google Scholar] [CrossRef]
- Han, K.P.; Zhu, X.; Liu, B.; Jeng, E.; Kong, L.; Yovandich, J.L.; Vyas, V.V.; Marcus, W.D.; Chavaillaz, P.A.; Romero, C.A.; et al. IL-15:IL-15 receptor alpha superagonist complex: High-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine 2011, 56, 804–810. [Google Scholar] [CrossRef]
- Romee, R.; Cooley, S.; Berrien-Elliott, M.M.; Westervelt, P.; Verneris, M.R.; Wagner, J.E.; Weisdorf, D.J.; Blazar, B.R.; Ustun, C.; DeFor, T.E.; et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood 2018, 131, 2515–2527. [Google Scholar] [CrossRef]
- Liu, B.; Kong, L.; Han, K.; Hong, H.; Marcus, W.D.; Chen, X.; Jeng, E.K.; Alter, S.; Zhu, X.; Rubinstein, M.P.; et al. A Novel Fusion of ALT-803 (Interleukin (IL)-15 Superagonist) with an Antibody Demonstrates Antigen-specific Antitumor Responses. J. Biol. Chem. 2016, 291, 23869–23881. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Yi, P.; Dong, W.; Nalin, A.P.; Zhang, J.; Zhu, Z.; Chen, L.; Benson, D.M.; Mundy-Bosse, B.L.; et al. The IL-15-AKT-XBP1s signaling pathway contributes to effector functions and survival in human NK cells. Nat. Immunol. 2019, 20, 10–17. [Google Scholar] [CrossRef]
- Ni, J.; Miller, M.; Stojanovic, A.; Garbi, N.; Cerwenka, A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J. Exp. Med. 2012, 209, 2351–2365. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Schneider, S.E.; Leong, J.W.; Chase, J.M.; Keppel, C.R.; Sullivan, R.P.; Cooper, M.A.; Fehniger, T.A. Cytokine activation induces human memory-like NK cells. Blood 2012, 120, 4751–4760. [Google Scholar] [CrossRef] [PubMed]
- Berrien-Elliott, M.M.; Wagner, J.A.; Fehniger, T.A. Human Cytokine-Induced Memory-Like Natural Killer Cells. J. Innate Immun. 2015, 7, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Terren, I.; Mikelez, I.; Odriozola, I.; Gredilla, A.; Gonzalez, J.; Orrantia, A.; Vitalle, J.; Zenarruzabeitia, O.; Borrego, F. Implication of Interleukin-12/15/18 and Ruxolitinib in the Phenotype, Proliferation, and Polyfunctionality of Human Cytokine-Preactivated Natural Killer Cells. Front. Immunol. 2018, 9, 737. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [PubMed]
- Kasaian, M.T.; Whitters, M.J.; Carter, L.L.; Lowe, L.D.; Jussif, J.M.; Deng, B.; Johnson, K.A.; Witek, J.S.; Senices, M.; Konz, R.F.; et al. IL-21 limits NK cell responses and promotes antigen-specific T cell activation: A mediator of the transition from innate to adaptive immunity. Immunity 2002, 16, 559–569. [Google Scholar] [CrossRef]
- Sivori, S.; Cantoni, C.; Parolini, S.; Marcenaro, E.; Conte, R.; Moretta, L.; Moretta, A. IL-21 induces both rapid maturation of human CD34+ cell precursors towards NK cells and acquisition of surface killer Ig-like receptors. Eur. J. Immunol. 2003, 33, 3439–3447. [Google Scholar] [CrossRef]
- Watanabe, M.; Kono, K.; Kawaguchi, Y.; Mizukami, Y.; Mimura, K.; Maruyama, T.; Fujii, H. Interleukin-21 can efficiently restore impaired antibody-dependent cell-mediated cytotoxicity in patients with oesophageal squamous cell carcinoma. Br. J. Cancer 2010, 102, 520–529. [Google Scholar] [CrossRef]
- Wagner, J.; Pfannenstiel, V.; Waldmann, A.; Bergs, J.W.J.; Brill, B.; Huenecke, S.; Klingebiel, T.; Rodel, F.; Buchholz, C.J.; Wels, W.S.; et al. A Two-Phase Expansion Protocol Combining Interleukin (IL)-15 and IL-21 Improves Natural Killer Cell Proliferation and Cytotoxicity against Rhabdomyosarcoma. Front. Immunol. 2017, 8, 676. [Google Scholar] [CrossRef]
- Denman, C.J.; Senyukov, V.V.; Somanchi, S.S.; Phatarpekar, P.V.; Kopp, L.M.; Johnson, J.L.; Singh, H.; Hurton, L.; Maiti, S.N.; Huls, M.H.; et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE 2012, 7, e30264. [Google Scholar] [CrossRef]
- Lim, S.H.; Beers, S.A.; French, R.R.; Johnson, P.W.; Glennie, M.J.; Cragg, M.S. Anti-CD20 monoclonal antibodies: Historical and future perspectives. Haematologica 2010, 95, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, G.G.; Queva, C.; Tabrizi, M.; van Abbema, A.; Chavez, C.; Wang, P.; Foord, O.; Ahluwalia, K.; Laing, N.; Raja, S.; et al. Development of a new fully human anti-CD20 monoclonal antibody for the treatment of B-cell malignancies. Investig. New Drugs 2010, 28, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Goede, V.; Fischer, K.; Busch, R.; Engelke, A.; Eichhorst, B.; Wendtner, C.M.; Chagorova, T.; de la Serna, J.; Dilhuydy, M.S.; Illmer, T.; et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N. Engl. J. Med. 2014, 370, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Herter, S.; Herting, F.; Mundigl, O.; Waldhauer, I.; Weinzierl, T.; Fauti, T.; Muth, G.; Ziegler-Landesberger, D.; Van Puijenbroek, E.; Lang, S.; et al. Preclinical activity of the type II CD20 antibody GA101 (obinutuzumab) compared with rituximab and ofatumumab in vitro and in xenograft models. Mol. Cancer Ther. 2013, 12, 2031–2042. [Google Scholar] [CrossRef] [PubMed]
- Terszowski, G.; Klein, C.; Stern, M. KIR/HLA interactions negatively affect rituximab- but not GA101 (obinutuzumab)-induced antibody-dependent cellular cytotoxicity. J. Immunol. 2014, 192, 5618–5624. [Google Scholar] [CrossRef]
- Capuano, C.; Pighi, C.; Molfetta, R.; Paolini, R.; Battella, S.; Palmieri, G.; Giannini, G.; Belardinilli, F.; Santoni, A.; Galandrini, R. Obinutuzumab-mediated high-affinity ligation of FcγRIIIA/CD16 primes NK cells for IFNγ production. Oncoimmunology 2017, 6, e1290037. [Google Scholar] [CrossRef]
- Cartron, G.; Dacheux, L.; Salles, G.; Solal-Celigny, P.; Bardos, P.; Colombat, P.; Watier, H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood 2002, 99, 754–758. [Google Scholar] [CrossRef]
- Veeramani, S.; Wang, S.Y.; Dahle, C.; Blackwell, S.; Jacobus, L.; Knutson, T.; Button, A.; Link, B.K.; Weiner, G.J. Rituximab infusion induces NK activation in lymphoma patients with the high-affinity CD16 polymorphism. Blood 2011, 118, 3347–3349. [Google Scholar] [CrossRef]
- Koerner, S.P.; Andre, M.C.; Leibold, J.S.; Kousis, P.C.; Kubler, A.; Pal, M.; Haen, S.P.; Buhring, H.J.; Grosse-Hovest, L.; Jung, G.; et al. An Fc-optimized CD133 antibody for induction of NK cell reactivity against myeloid leukemia. Leukemia 2017, 31, 459–469. [Google Scholar] [CrossRef]
- von Stackelberg, A.; Locatelli, F.; Zugmaier, G.; Handgretinger, R.; Trippett, T.M.; Rizzari, C.; Bader, P.; O’Brien, M.M.; Brethon, B.; Bhojwani, D.; et al. Phase I/Phase II Study of Blinatumomab in Pediatric Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2016, 34, 4381–4389. [Google Scholar] [CrossRef]
- Ribera, J.M. Efficacy and safety of bispecific T-cell engager blinatumomab and the potential to improve leukemia-free survival in B-cell acute lymphoblastic leukemia. Expert Rev. Hematol. 2017, 10, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.L.; Ellerman, D.; Mathieu, M.; Hristopoulos, M.; Chen, X.; Li, Y.; Yan, X.; Clark, R.; Reyes, A.; Stefanich, E.; et al. Anti-CD20/CD3 T cell-dependent bispecific antibody for the treatment of B cell malignancies. Sci. Transl. Med. 2015, 7, 287ra270. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Franklin, J.L.; Chia, V.M.; Arrindell, D.; Kormany, W.; Wright, J.; Parson, M.; Amouzadeh, H.R.; Choudhry, J.; Joseph, G. Benefit-Risk Assessment of Blinatumomab in the Treatment of Relapsed/Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Drug Saf. 2019, 42, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.L.; Bautista, C.; Pong, E.; Nguyen, D.H.; Jacinto, J.; Eivazi, A.; Muchhal, U.S.; Karki, S.; Chu, S.Y.; Lazar, G.A. A novel bispecific antibody format enables simultaneous bivalent and monovalent co-engagement of distinct target antigens. MAbs 2011, 3, 546–557. [Google Scholar] [CrossRef]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and Trispecific Killer Cell Engagers Directly Activate Human NK Cells through CD16 Signaling and Induce Cytotoxicity and Cytokine Production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef]
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 2014, 123, 3016–3026. [Google Scholar] [CrossRef]
- Wiernik, A.; Foley, B.; Zhang, B.; Verneris, M.R.; Warlick, E.; Gleason, M.K.; Ross, J.A.; Luo, X.; Weisdorf, D.J.; Walcheck, B.; et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 × 33 bispecific killer cell engager and ADAM17 inhibition. Clin. Cancer Res. 2013, 19, 3844–3855. [Google Scholar] [CrossRef]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef]
- Sarhan, D.; Brandt, L.; Felices, M.; Guldevall, K.; Lenvik, T.; Hinderlie, P.; Curtsinger, J.; Warlick, E.; Spellman, S.R.; Blazar, B.R.; et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. 2018, 2, 1459–1469. [Google Scholar] [CrossRef]
- Felices, M.; Kodal, B.; Hinderlie, P.; Kaminski, M.F.; Cooley, S.; Weisdorf, D.J.; Vallera, D.A.; Miller, J.S.; Bachanova, V. Novel CD19-targeted TriKE restores NK cell function and proliferative capacity in CLL. Blood Adv. 2019, 3, 897–907. [Google Scholar] [CrossRef]
- Mejstríková, E.; Hrusak, O.; Borowitz, M.J.; Whitlock, J.A.; Brethon, B.; Trippett, T.M.; Zugmaier, G.; Gore, L.; von Stackelberg, A.; Locatelli, F. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Arvindam, U.S.; van Hauten, P.; Hallstrom, C.; Vallera, D.A.; Dolstra, H.; Miller, J.S.; Felices, M. CD16-IL15-CLEC12A Trispecific Killer Engager (TriKE) Drives NK Cell Expansion, Activation, and Antigen Specific Killing of Cancer Stem Cells in Acute Myeloid Leukemia. Blood 2018, 132, 1454. [Google Scholar] [CrossRef]
- Ellwanger, K.; Reusch, U.; Fucek, I.; Wingert, S.; Ross, T.; Müller, T.; Schniegler-Mattox, U.; Haneke, T.; Rajkovic, E.; Koch, J.; et al. Redirected optimized cell killing (ROCK®): A highly versatile multispecific fit-for-purpose antibody platform for engaging innate immunity. mAbs 2019, 11, 899–918. [Google Scholar] [CrossRef] [PubMed]
- Rothe, A.; Sasse, S.; Topp, M.S.; Eichenauer, D.A.; Hummel, H.; Reiners, K.S.; Dietlein, M.; Kuhnert, G.; Kessler, J.; Buerkle, C.; et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood 2015, 125, 4024–4031. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Busch, L.K.; Salazar-Fontana, L.I.; Bellosillo, B.; Morte, C.; Garcia, P.; Lopez-Botet, M. The CD94/NKG2C killer lectin-like receptor constitutes an alternative activation pathway for a subset of CD8+ T cells. Eur. J. Immunol. 2005, 35, 2071–2080. [Google Scholar] [CrossRef]
- Muntasell, A.; Vilches, C.; Angulo, A.; Lopez-Botet, M. Adaptive reconfiguration of the human NK-cell compartment in response to cytomegalovirus: A different perspective of the host-pathogen interaction. Eur. J. Immunol. 2013, 43, 1133–1141. [Google Scholar] [CrossRef]
- Muccio, L.; Bertaina, A.; Falco, M.; Pende, D.; Meazza, R.; Lopez-Botet, M.; Moretta, L.; Locatelli, F.; Moretta, A.; Della Chiesa, M. Analysis of memory-like natural killer cells in human cytomegalovirus-infected children undergoing αβ+T and B cell-depleted hematopoietic stem cell transplantation for hematological malignancies. Haematologica 2016, 101, 371–381. [Google Scholar] [CrossRef]
- Cichocki, F.; Cooley, S.; Davis, Z.; DeFor, T.E.; Schlums, H.; Zhang, B.; Brunstein, C.G.; Blazar, B.R.; Wagner, J.; Diamond, D.J.; et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia 2016, 30, 456–463. [Google Scholar] [CrossRef]
- Cichocki, F.; Taras, E.; Chiuppesi, F.; Wagner, J.E.; Blazar, B.R.; Brunstein, C.; Luo, X.; Diamond, D.J.; Cooley, S.; Weisdorf, D.J.; et al. Adaptive NK cell reconstitution is associated with better clinical outcomes. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Schlums, H.; Cichocki, F.; Tesi, B.; Theorell, J.; Beziat, V.; Holmes, T.D.; Han, H.; Chiang, S.C.; Foley, B.; Mattsson, K.; et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 2015, 42, 443–456. [Google Scholar] [CrossRef]
- Capuano, C.; Battella, S.; Pighi, C.; Franchitti, L.; Turriziani, O.; Morrone, S.; Santoni, A.; Galandrini, R.; Palmieri, G. Tumor-Targeting Anti-CD20 Antibodies Mediate In Vitro Expansion of Memory Natural Killer Cells: Impact of CD16 Affinity Ligation Conditions and In Vivo Priming. Front. Immunol. 2018, 9, 1031. [Google Scholar] [CrossRef] [PubMed]
- Chiu, E.; Cichocki, F.; Felices, M.; Cooley, S.; Chu, H.; Ge, M.Q.; Bjordahl, R.; Kaufman, D.S.; Malmberg, K.-J.; Valamehr, B.; et al. Induced Pluripotent Stem Cell-Derived NK Cells Genetically Modified to Express NKG2C/DAP12 Mediate Potent Function When Targeted through an NKG2C/IL-15/CD33 Tri-Specific Killer Engager (TriKE). Blood 2018, 132, 729. [Google Scholar] [CrossRef]
- Liu, L.L.; Beziat, V.; Oei, V.Y.S.; Pfefferle, A.; Schaffer, M.; Lehmann, S.; Hellstrom-Lindberg, E.; Soderhall, S.; Heyman, M.; Grander, D.; et al. Ex Vivo Expanded Adaptive NK Cells Effectively Kill Primary Acute Lymphoblastic Leukemia Cells. Cancer Immunol. Res. 2017, 5, 654–665. [Google Scholar] [CrossRef]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713 e1716. [Google Scholar] [CrossRef] [PubMed]
- Romagne, F.; Andre, P.; Spee, P.; Zahn, S.; Anfossi, N.; Gauthier, L.; Capanni, M.; Ruggeri, L.; Benson, D.M., Jr.; Blaser, B.W.; et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood 2009, 114, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- Vey, N.; Bourhis, J.H.; Boissel, N.; Bordessoule, D.; Prebet, T.; Charbonnier, A.; Etienne, A.; Andre, P.; Romagne, F.; Benson, D.; et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood 2012, 120, 4317–4323. [Google Scholar] [CrossRef]
- Benson, D.M., Jr.; Hofmeister, C.C.; Padmanabhan, S.; Suvannasankha, A.; Jagannath, S.; Abonour, R.; Bakan, C.; Andre, P.; Efebera, Y.; Tiollier, J.; et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood 2012, 120, 4324–4333. [Google Scholar] [CrossRef]
- Benson, D.M., Jr.; Cohen, A.D.; Jagannath, S.; Munshi, N.C.; Spitzer, G.; Hofmeister, C.C.; Efebera, Y.A.; Andre, P.; Zerbib, R.; Caligiuri, M.A. A Phase I Trial of the Anti-KIR Antibody IPH2101 and Lenalidomide in Patients with Relapsed/Refractory Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4055–4061. [Google Scholar] [CrossRef]
- Andre, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Blery, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Senechal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Daher, M.; Rezvani, K. Next generation natural killer cells for cancer immunotherapy: The promise of genetic engineering. Curr. Opin. Immunol. 2018, 51, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Moretta, L.; Locatelli, F.; Pende, D.; Sivori, S.; Falco, M.; Bottino, C.; Mingari, M.C.; Moretta, A. Human NK receptors: From the molecules to the therapy of high risk leukemias. FEBS Lett. 2011, 585, 1563–1567. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994, 8, 652–658. [Google Scholar] [PubMed]
- Mehta, R.S.; Rezvani, K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front. Immunol. 2018, 9, 283. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192. [Google Scholar] [CrossRef]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.J. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin. Immunopathol. 2019, 41, 59–68. [Google Scholar] [CrossRef]
- Tam, Y.K.; Martinson, J.A.; Doligosa, K.; Klingemann, H.G. Ex vivo expansion of the highly cytotoxic human natural killer-92 cell-line under current good manufacturing practice conditions for clinical adoptive cellular immunotherapy. Cytotherapy 2003, 5, 259–272. [Google Scholar] [CrossRef]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Romanski, A.; Uherek, C.; Bug, G.; Seifried, E.; Klingemann, H.; Wels, W.S.; Ottmann, O.G.; Tonn, T. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J. Cell. Mol. Med. 2016, 20, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Uherek, C.; Tonn, T.; Uherek, B.; Becker, S.; Schnierle, B.; Klingemann, H.G.; Wels, W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood 2002, 100, 1265–1273. [Google Scholar] [CrossRef]
- Imai, C.; Iwamoto, S.; Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005, 106, 376–383. [Google Scholar] [CrossRef]
- Muller, T.; Uherek, C.; Maki, G.; Chow, K.U.; Schimpf, A.; Klingemann, H.G.; Tonn, T.; Wels, W.S. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol. Immunother. 2008, 57, 411–423. [Google Scholar] [CrossRef]
- Mihara, K.; Bhattacharyya, J.; Kitanaka, A.; Yanagihara, K.; Kubo, T.; Takei, Y.; Asaoku, H.; Takihara, Y.; Kimura, A. T-cell immunotherapy with a chimeric receptor against CD38 is effective in eliminating myeloma cells. Leukemia 2012, 26, 365–367. [Google Scholar] [CrossRef]
- You, F.; Wang, Y.; Jiang, L.; Zhu, X.; Chen, D.; Yuan, L.; An, G.; Meng, H.; Yang, L. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am. J. Cancer Res. 2019, 9, 64–78. [Google Scholar]
- Chen, K.H.; Wada, M.; Firor, A.E.; Pinz, K.G.; Jares, A.; Liu, H.; Salman, H.; Golightly, M.; Lan, F.; Jiang, X.; et al. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget 2016, 7, 56219–56232. [Google Scholar] [CrossRef]
- Chen, K.H.; Wada, M.; Pinz, K.G.; Liu, H.; Lin, K.W.; Jares, A.; Firor, A.E.; Shuai, X.; Salman, H.; Golightly, M.; et al. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia 2017, 31, 2151–2160. [Google Scholar] [CrossRef]
- Esser, R.; Muller, T.; Stefes, D.; Kloess, S.; Seidel, D.; Gillies, S.D.; Aperlo-Iffland, C.; Huston, J.S.; Uherek, C.; Schonfeld, K.; et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J. Cell. Mol. Med. 2012, 16, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Tassev, D.V.; Cheng, M.; Cheung, N.K. Retargeting NK92 cells using an HLA-A2-restricted, EBNA3C-specific chimeric antigen receptor. Cancer Gene Ther. 2012, 19, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chu, J.; Keung Chan, W.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.H.; Grandi, P.; et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef] [PubMed]
- Sahm, C.; Schonfeld, K.; Wels, W.S. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol. Immunother. 2012, 61, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K.; et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014, 28, 917–927. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar]
- Kloess, S.; Oberschmidt, O.; Dahlke, J.; Vu, X.K.; Neudoerfl, C.; Kloos, A.; Gardlowski, T.; Matthies, N.; Heuser, M.; Meyer, J.; et al. Preclinical Assessment of Suitable Natural Killer Cell Sources for Chimeric Antigen Receptor Natural Killer-Based “Off-the-Shelf” Acute Myeloid Leukemia Immunotherapies. Hum. Gene Ther. 2019, 30, 381–401. [Google Scholar] [CrossRef]
- Maki, G.; Klingemann, H.G.; Martinson, J.A.; Tam, Y.K. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J. Hematother. Stem Cell Res. 2001, 10, 369–383. [Google Scholar] [CrossRef]
- Suck, G.; Odendahl, M.; Nowakowska, P.; Seidl, C.; Wels, W.S.; Klingemann, H.G.; Tonn, T. NK-92: An ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol. Immunother. 2016, 65, 485–492. [Google Scholar] [CrossRef]
- Sarvaria, A.; Jawdat, D.; Madrigal, J.A.; Saudemont, A. Umbilical Cord Blood Natural Killer Cells, Their Characteristics, and Potential Clinical Applications. Front. Immunol. 2017, 8, 329. [Google Scholar] [CrossRef]
- Shah, N.; Martin-Antonio, B.; Yang, H.; Ku, S.; Lee, D.A.; Cooper, L.J.; Decker, W.K.; Li, S.; Robinson, S.N.; Sekine, T.; et al. Antigen presenting cell-mediated expansion of human umbilical cord blood yields log-scale expansion of natural killer cells with anti-myeloma activity. PLoS ONE 2013, 8, e76781. [Google Scholar] [CrossRef] [PubMed]
- Ayello, J.; Hochberg, J.; Flower, A.; Chu, Y.; Baxi, L.V.; Quish, W.; van de Ven, C.; Cairo, M.S. Genetically re-engineered K562 cells significantly expand and functionally activate cord blood natural killer cells: Potential for adoptive cellular immunotherapy. Exp. Hematol. 2017, 46, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, R.; Handgretinger, R.; Babarin-Dorner, A.; Leimig, T.; Otto, M.; Geiger, T.L.; Holladay, M.S.; Houston, J.; Leung, W. Purification of human natural killer cells using a clinical-scale immunomagnetic method. Cytotherapy 2003, 5, 479–484. [Google Scholar] [CrossRef]
- Koehl, U.; Esser, R.; Zimmermann, S.; Tonn, T.; Kotchetkov, R.; Bartling, T.; Sorensen, J.; Gruttner, H.P.; Bader, P.; Seifried, E.; et al. Ex vivo expansion of highly purified NK cells for immunotherapy after haploidentical stem cell transplantation in children. Klinische Padiatrie 2005, 217, 345–350. [Google Scholar] [CrossRef]
- Sutlu, T.; Stellan, B.; Gilljam, M.; Quezada, H.C.; Nahi, H.; Gahrton, G.; Alici, E. Clinical-grade, large-scale, feeder-free expansion of highly active human natural killer cells for adoptive immunotherapy using an automated bioreactor. Cytotherapy 2010, 12, 1044–1055. [Google Scholar] [CrossRef]
- Leung, W. Use of NK cell activity in cure by transplant. Br. J. Haematol. 2011, 155, 14–29. [Google Scholar] [CrossRef]
- Leung, W. Infusions of allogeneic natural killer cells as cancer therapy. Clin. Cancer Res. 2014, 20, 3390–3400. [Google Scholar] [CrossRef]
- Kweon, S.; Phan, M.T.; Chun, S.; Yu, H.; Kim, J.; Kim, S.; Lee, J.; Ali, A.K.; Lee, S.H.; Kim, S.K.; et al. Expansion of Human NK Cells Using K562 Cells Expressing OX40 Ligand and Short Exposure to IL-21. Front. Immunol. 2019, 10, 879. [Google Scholar] [CrossRef]
- Fujisaki, H.; Kakuda, H.; Shimasaki, N.; Imai, C.; Ma, J.; Lockey, T.; Eldridge, P.; Leung, W.H.; Campana, D. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009, 69, 4010–4017. [Google Scholar] [CrossRef]
- Quintarelli, C.; Sivori, S.; Caruso, S.; Carlomagno, S.; Falco, M.; Boffa, I.; Orlando, D.; Guercio, M.; Abbaszadeh, Z.; Sinibaldi, M.; et al. Efficacy of third-party chimeric antigen receptor modified peripheral blood natural killer cells for adoptive cell therapy of B Cell Precursor Acute Lymphoblastic Leukemia. Leukemia 2019, in press. [Google Scholar]
- De Oliveira, S.N.; Ryan, C.; Giannoni, F.; Hardee, C.L.; Tremcinska, I.; Katebian, B.; Wherley, J.; Sahaghian, A.; Tu, A.; Grogan, T.; et al. Modification of hematopoietic stem/progenitor cells with CD19-specific chimeric antigen receptors as a novel approach for cancer immunotherapy. Hum. Gene Ther. 2013, 24, 824–839. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wang, X.; Zhang, H.; Zhi, L.; Lv, T.; Li, M.; Lu, C.; Zhu, W. Structure-based rational design of a novel chimeric PD1-NKG2D receptor for natural killer cells. Mol. Immunol. 2019, 114, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Topfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Muller, N.; Lindemann, D.; Bachmann, M.; Fussel, M.; Schackert, G.; Temme, A. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef]
- Xiao, L.; Cen, D.; Gan, H.; Sun, Y.; Huang, N.; Xiong, H.; Jin, Q.; Su, L.; Liu, X.; Wang, K.; et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol. Ther. 2019, 27, 1114–1125. [Google Scholar] [CrossRef]
- Ingegnere, T.; Mariotti, F.R.; Pelosi, A.; Quintarelli, C.; De Angelis, B.; Tumino, N.; Besi, F.; Cantoni, C.; Locatelli, F.; Vacca, P.; et al. Human CAR NK Cells: A New Non-viral Method Allowing High Efficient Transfection and Strong Tumor Cell Killing. Front. Immunol. 2019, 10, 957. [Google Scholar] [CrossRef]
- Oberschmidt, O.; Morgan, M.; Huppert, V.; Kessler, J.; Gardlowski, T.; Matthies, N.; Aleksandrova, K.; Arseniev, L.; Schambach, A.; Koehl, U.; et al. Development of Automated Separation, Expansion, and Quality Control Protocols for Clinical-Scale Manufacturing of Primary Human NK Cells and Αretroviral Chimeric Antigen Receptor Engineering. Hum. Gene Ther. Methods 2019, 30, 102–120. [Google Scholar] [CrossRef]
- Boissel, L.; Betancur, M.; Lu, W.; Wels, W.S.; Marino, T.; Van Etten, R.A.; Klingemann, H. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk Lymphoma 2012, 53, 958–965. [Google Scholar] [CrossRef]
- Veerman, R.E.; Gucluler Akpinar, G.; Eldh, M.; Gabrielsson, S. Immune Cell-Derived Extracellular Vesicles—Functions and Therapeutic Applications. Trends Mol. Med. 2019, 25, 382–394. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identifier | NK Type/Source | CAR Target | Conditions | Phase | Status | Last Up-Date | Location |
|---|---|---|---|---|---|---|---|
| NCT03415100 | Autologous or allogeneic NK cells | NKG2D-Ligand | Metastatic Solid Tumours | I | Recruiting | August, 2018 | China |
| NCT03056339 NCT03579927 | Primary NK/CB | CD19-IL15 | B Lymphoid Malignancies | I/II I/II | Recruiting Not yet recruiting | July, 2019/October, 2019 | USA |
| NCT03692767 | ND | CD22 | Relapsed Refractory B cell Lymphoma | I | Not yet recruiting | January, 2019 | ND |
| NCT03690310 | ND | CD19 | Refractory B Cell Lymphoma | I | Not yet recruiting | January, 2019 | ND |
| NCT03692663 | ND | PSMA | Castration-Resistant Prostate Cancer | I | Not yet recruiting | October, 2018 | ND |
| NCT03824964 | ND | CD19/CD22 | Relapsed and Refractory B Cell Lymphoma | I | Not yet recruiting | January, 2019 | ND |
| NCT03692637 | ND | Mesothelin | Epithelial Ovarian Cancer | I | Not yet recruiting | January, 2019 | ND |
| NCT03940833 | NK-92 | BCMA | Relapsed/Refractory MM | I | Recruiting | May, 2019 | China |
| NCT03940820 NCT03931720 | ND | ROBO1 | Metastatic Solid Tumours/Malignant Tumors | I I/II | Recruiting | May, 2019 | China |
| NCT03941457 | ND | ROBO1 | Pancreatic Cancer | I/II | Recruiting | May, 2019 | China |
| NCT03383978 | NK-92 | HER-2 | Glioblastoma | I | Recruiting | May, 2019 | Germany |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivori, S.; Meazza, R.; Quintarelli, C.; Carlomagno, S.; Della Chiesa, M.; Falco, M.; Moretta, L.; Locatelli, F.; Pende, D. NK Cell-Based Immunotherapy for Hematological Malignancies. J. Clin. Med. 2019, 8, 1702. https://doi.org/10.3390/jcm8101702
Sivori S, Meazza R, Quintarelli C, Carlomagno S, Della Chiesa M, Falco M, Moretta L, Locatelli F, Pende D. NK Cell-Based Immunotherapy for Hematological Malignancies. Journal of Clinical Medicine. 2019; 8(10):1702. https://doi.org/10.3390/jcm8101702
Chicago/Turabian StyleSivori, Simona, Raffaella Meazza, Concetta Quintarelli, Simona Carlomagno, Mariella Della Chiesa, Michela Falco, Lorenzo Moretta, Franco Locatelli, and Daniela Pende. 2019. "NK Cell-Based Immunotherapy for Hematological Malignancies" Journal of Clinical Medicine 8, no. 10: 1702. https://doi.org/10.3390/jcm8101702
APA StyleSivori, S., Meazza, R., Quintarelli, C., Carlomagno, S., Della Chiesa, M., Falco, M., Moretta, L., Locatelli, F., & Pende, D. (2019). NK Cell-Based Immunotherapy for Hematological Malignancies. Journal of Clinical Medicine, 8(10), 1702. https://doi.org/10.3390/jcm8101702